
Abstract
Polycarbonate (PC) substrate is well suited for culturing human mesenchymal stem cells (MSCs) with high proliferation rate, low cell apoptosis rate and negligible cytotoxic effects. However, little is known about the influence of PC on MSC activity including senescence, differentiation and secretion. In this study, the PC cell culture insert was applied for human MSC culture and was compared with polystyrene (PS) and standard tissue culture plate (TCP). The results showed that MSCs were able to adhere on PC surface, exhibiting a spindle-shaped morphology. The size and distribution of focal adhesions of MSCs were similar on PC and TCP. The senescence level of MSCs on PC was comparable to that on TCP, but was significantly lower than that on PS. MSCs on PC were capable of self-renewal and differentiation into multiple cell lineages, including osteogenic and adipogenic lineages. MSCs cultured on PC secreted a higher level inflammatory cytokines and pro-angiogenic factors including FGF2 and VEGF. Conclusively, PC represents a promising cell culture material for human MSCs.
Introduction
Mesenchymal stem cells (MSCs) attracted interest because of their promising therapeutic potential in regenerative medicine due to their self-renewal, multi-lineages differentiation potential and the secretion of a variety of functional cytokines [1–3]. MSCs can be obtained from different tissues, such as bone marrow, adipose tissue and umbilical cord tissue [4–6]. However, the quantity of MSCs that separated from those tissues can not satisfy the needs of clinic cell therapy (10–400 million MSCs per treatment) [7]. Therefore, the isolated MSCs need to be expanded in vitro before implantation. It has been shown that MSCs are very sensitive to the substrate properties, such as the extracellular matrix (ECM) stiffness [8], surface topography [9] and chemical property of the culture surface [10]. The functions and behavior of MSCs can be influenced during the period of cell expansion in vitro [11].
Polycarbonate (PC) has technological significance in life science applications, such as intravenous connection components or cell culture devices [12, 13]. Favorable properties of PC include the high light transmittance, impact toughness, dimensional stability, creep resistance and thermoplastic process ability [14]. In addition, PC can be sterilized by a variety of methods including ethylene oxide (EtO), irradiation and steam autoclaving. As cell culture devices, PC exhibited excellent cell compatibility, favoring cell adhesion and proliferation [15]. However, there are few reports about the influence of PC on the most important behaviors of MSCs such as senescence, differentiation and secretion.
Here we hypothesized that PC is a suitable substrate for cultivation of human bone marrow mesenchymal stem cells (hBMSCs) without further modification. In this study we compare a PC-based substrate with polystyrene-based tissue culture plate (TCP), whose surface is modified in order to support adherent cells, and a polystyrene (PS)-based substrate without surface modification. Those substrates serve as positive and negative reference materials. The cell adhesion, morphology, senescence, cytokine secretion and differentiation of hBMSCs on PC, PS and TCP were compared.
Materials and methods
Materials for cell culture
The polycarbonate (PC, trade name Makrolon® 2805, Bayer, Germany) and polystyrene (PS, Type 158K, BASF, Germany) cell culture inserts were fabricated via injection molding [16]. The surfaces of PS and PC inserts showed the similarity in both mechanical properties and surface profile, as reported previously [15]. The Young’s modulus of PC and PS was 13±2 and 16±4 GPa respectively, as measured with AFM indentation. Profilometry analysis at an area of 7×7 mm2 indicated that the bottom roughness of the inserts is at a similar level (root arithmetic mean square roughness (Rq) = 0.12±0.04 μm for PS and 0.34±0.13 μm for PC). In addition, the PS and PC presented comparable surface wettability (water advancing contact angle 85±8° for PC and 99±5° for PS), as determined via water contact angle measurement using the captive bubble method [15]. Prior to cell culture, the PC and PS inserts were sterilized via gas sterilization (gas phase: 10% (v/v) ethylene oxide, 54 °C, 65% relative humidity, 1.7 bar, 3 hours of gas exposure time and 21 hours of aeration phase). In addition, the tissue culture plate (TCP, Coring, New York, USA) was used in this study as a positive control material.
Human bone marrow mesenchymal stem cells (hBMSCs)
The hBMSCs were purchased from Merck Millipore (SCC034, Merk Millipore, Darmstadt, Germany). The cells were maintained in MesenPRO RS™ growth medium (ThermoFisher Scientific, Waltham, USA), in a humidified atmosphere containing 5% (v/v) CO2. The medium was changed in every 3 days. For hBMSC phenotype characterization, the cells were harvested and stained with a Human MSC Analysis Kit (StemflowTM, BD Biosciences, Heidelberg, Germany) according to the manufacturer’s instruction. A flow cytometer (MACSQuant®, Miltenyi Biotec, Bergisch Gladbach, Germany) was used to analyze the cells and the data was processed using Flowjo software (Tree Star Inc., Ashland OR, USA).
Immunostaining
Cells were fixed with 4% (w/v) paraformaldehyde (Sigma–Aldrich, St. Louis, MO, USA) and permeabilized with 0.1% (v/v) Triton X-100 (Sigma–Aldrich, St. Louis, MO, USA), then blocked with 3% (w/v) BSA buffer. Vinculin was stained with mouse anti-human vinculin monoclonal antibody (Merck Millipore, Darmstadt, Germany) and Alexa Fluor® 488 labeled IgG antibody (Life Technologies, Darmstadt, Germany). F-actin was stained with ActinRed™ 555 (Life Technologies, Darmstadt, Germany). After washing, the cells were visualized with a confocal laser scanning microscope (LSM 780 META, Carl Zeiss, Jena Germany).
Senescence assay
The senescence-associated β-galactosidase activity of hBMSCs cultured on different materials was quantified using a cellular senescence assay kit (Cell Biolabs, Inc., San Diego, CA, USA) according to the manual introduction at different time points (day 1, 8, 15 and 22). The concentration of the total protein in the cell extraction was determined using a BCA protein assay kit (Thermo Fisher Scientific, Bonn, Germany). The cellular senescence level was expressed as the fluorescent intensity normalized with the amount of total protein.
Secretion assay
The cytokines in the conditioned medium and the cell number were quantified at day 4, 7, 10, 13, 16 and 19. The cytokines in the conditioned medium were quantified using a Bio-Plex® 200 system (BioRad, Munich, Germany) according to the manufacturer’s instructions. The cell numbers at different time points were determined using a Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Munich, Germany) as described before [15]. To evaluate the activity of cell secretion, the cytokine amount was normalized with the cell number.
Differentiation assay
For the differentiation study, the hBMSCs were cultured in osteogenic (StemPro Osteogenesis differentiation kit) and adipogenic (StemPro Adipogenesis differentiation kit) induction media, respectively. The induction media were changed in every 3 days. To evaluate the adipogenic differentiation, the cells were cultured in adipogenic induction medium for 14 days. Then, the cells were fixed with 4% (w/v) paraformaldehyde and permeabilized with 0.1% (v/v)Triton X-100 solution. After blocking with 3% (w/v) BSA, staining was performed using rabbit anti-human FABP-4 monoclonal antibody (Merck Millipore, Darmstadt, Germany) and Alexa Fluor® 488 labeled IgG antibodies (Life Technologies, Darmstadt, Germany). Cell nuclei were stained with SYTOX Green Nucleic Acid Stain (Life Technologies, Darmstadt, Germany). The stained samples were scanned with a confocal laser scanning microscope (LSM 780, Carl Zeiss, Jena, Germany). Osteogenic differentiation was evaluated after culturing the cells in osteogenic induction medium for 21 days. The cells were fixed with 4% (w/v) paraformaldehyde, and Alizarin red S (Sigma–Aldrich, St. Louis, MO, USA) staining was performed to detect the mineral deposition.
Statistics
The number of replication for quantitative experiments was three as indicated in figure legend, and the data were expressed as arithmetic mean±standard deviation (SD). Statistical analysis was performed by one-way ANOVA with post hoc Tukey HSD test. A p value less than 0.05 was considered statistically significant.
Results
hBMSC phenotype
To analyze the phenotype of the cells, the cell morphology and surface markers were characterized. The cells presented the typical MSC’s spindle-shaped morphology (Fig. 1A). Furthermore, the cell surface markers were analyzed with flow cytometry. The results indicated that the typical MSC markers (CD44, CD73, CD90 and CD105) were well preserved in the cells and the non-MSC markers (CD45, CD34, CD11b, CD19 and HLA-DR) were not detected (Fig. 1 B-F).
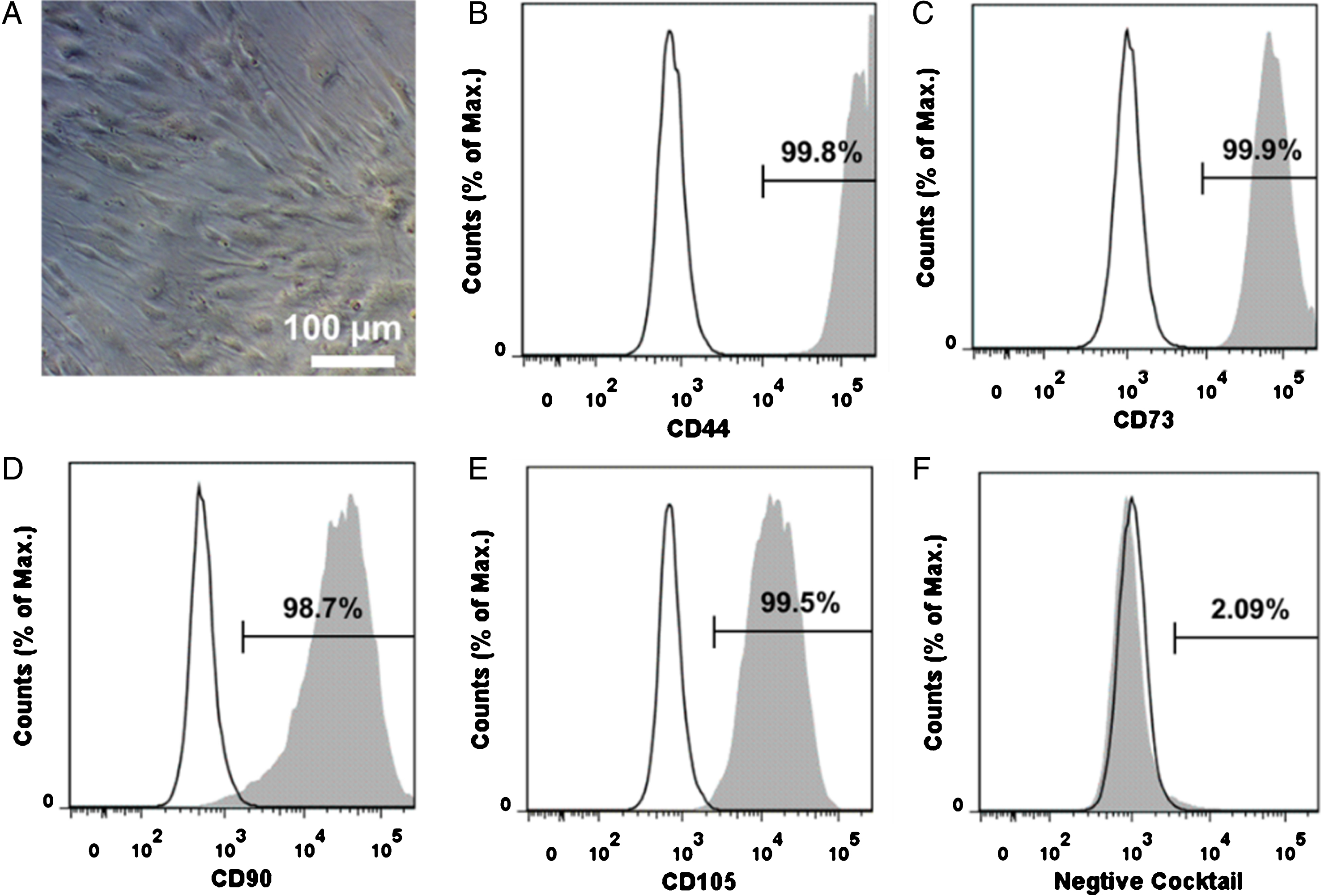
Characterization of hBMSCs. A: The image of hBMSCs growing on TCP. B, C, D and E: characterization of MSC phenotypic markers (CD44, CD73, CD90 and CD105). F: characterization of non-MSC markers (CD45, CD34, CD11b, CD19 and HLA-DR).
To study the cell-material interaction, the formation of focal adhesions (FAs) was examined by immunofluorescent staining of vinculin and F-actin (Fig. 2). As hBMSCs were difficult to attach and spread on PS surface, the FA formation of cells on PC and TCP surfaces were characterized. After 4 days of cultivation in growth medium, the cells spread out on the PC and TCP. The F-actin cytoskeleton organization and FA complex formation were similar in cells growing on PC and TCP.
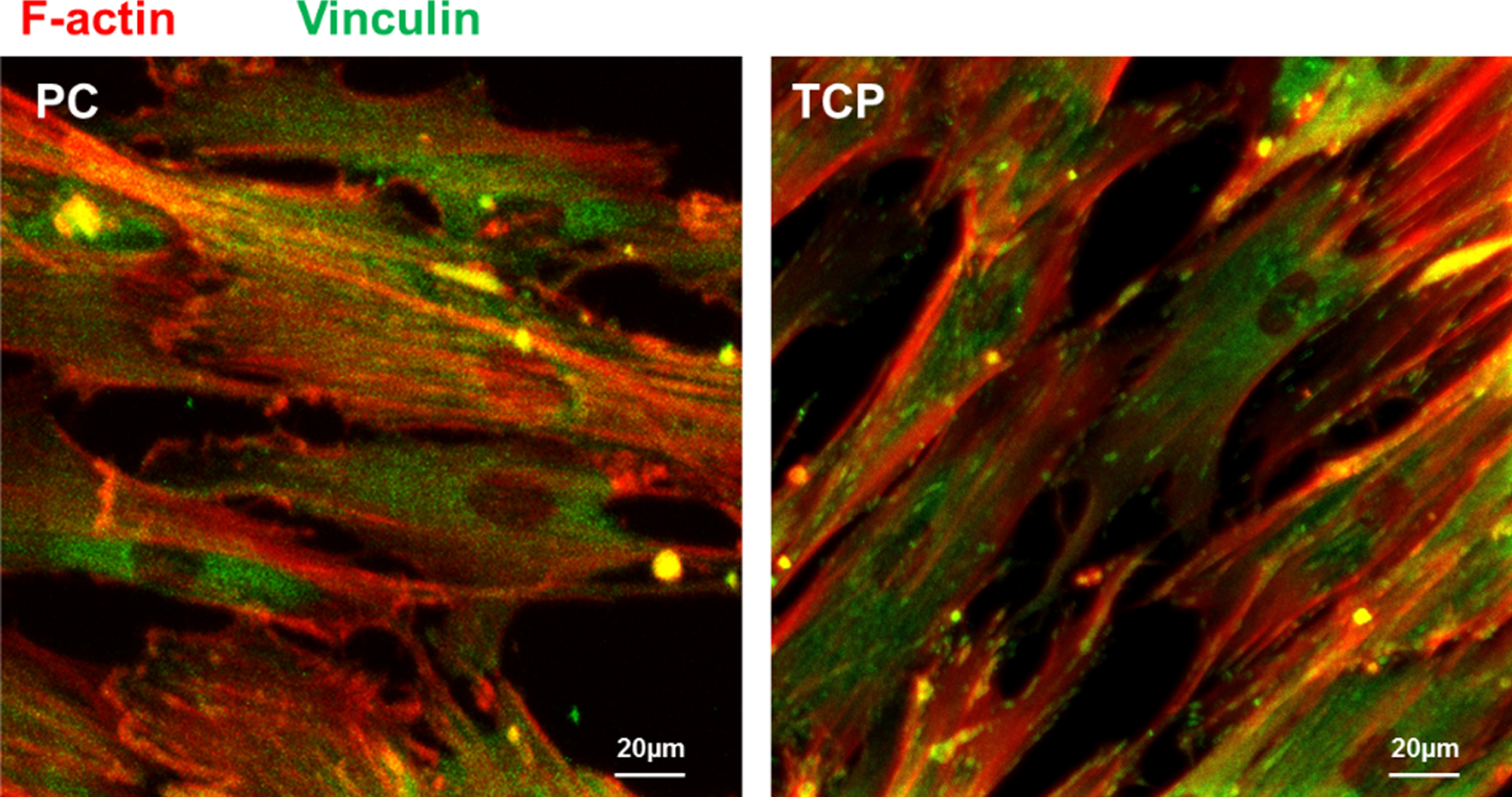
Representative laser scanning microscopic images of hBMSCs cultured on PC and TCP for 4 days. F-actin (red) and vinculin (green) were fluorescently stained. (bar = 20 μm). The reader is referred to the web version of this article, where the coloured version of the figure is provided.
The influence of culture materials on hBMSC senescence level was evaluated by measuring the activity of senescence-associated β-galactosidase (SA-β-gal). The results indicated that in the early stage (day 1) the senescence level of hBMSCs on TCP surface was significantly higher than that on PS. With the elongation of cultivation, the senescence level increased on all of the tested surfaces. The senescence level of cells on PS was higher than that on PC and TCP at day 8 and day 22. There was no significant difference of the senescence level between the cells cultured on PC and TCP (Fig. 3).

The senescence level of hBMSCs cultured on different materials at the indicated time points. (n = 3; *p < 0.05).
To analyze the paracrine factors secreted by hBMSCs on different materials, the conditioned media of hBMSC cultures were collected and the factors were quantified (Fig. 4). The paracrine factors secretion level of cells on PS is higher than that on TCP and PC. In the early stage (day 4) the cells on PC showed a higher secretion level than the cells on TCP except for the MCAF secretion. Compared to TCP, the cells on PC secreted the higher level of inflammatory cytokines such as IL-6, IL-17, IFN-γ and TNF-α as well as pro-angiogenic factors including FGF2 and VEGF.
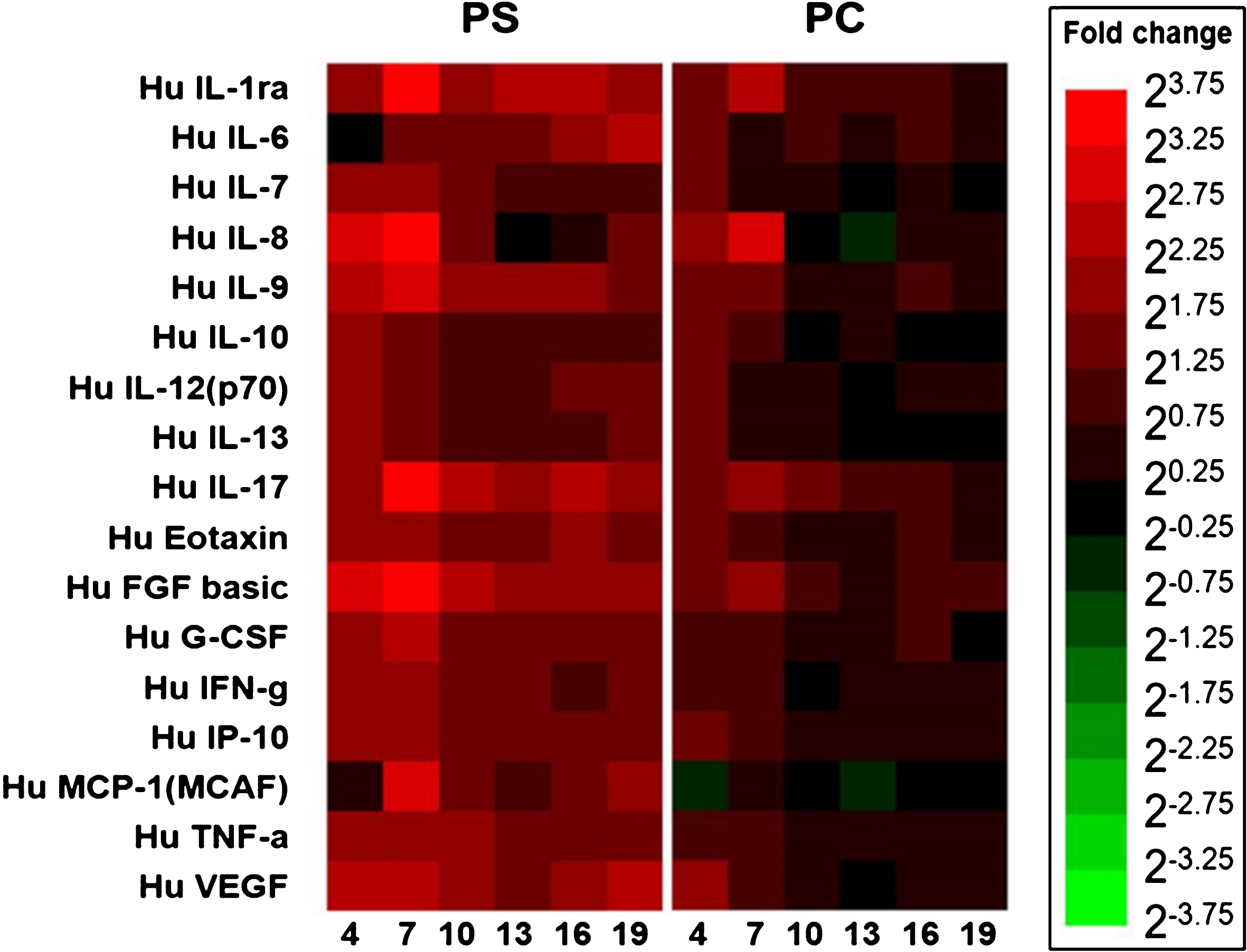
The heatmap showed the fold change (compared to TCP) of protein secretion over time by hBMSCs seeded on different materials. The reader is referred to the web version of this article, where the coloured version of the figure is provided.
The osteogenic and adipogenic differentiation of hBMSCs on different materials were examined by Alizarin Red S (ARS) staining and FABP-4 immunostaining, respectively. The results indicated that hBMSCs did not differentiate in the non-induced medium on all of the tested surfaces (Fig. 5). When cultured in osteogenic induction medium, the cells could differentiate into osteoblasts (Fig. 5A). In addition, the immunofluorescence staining of FABP-4 indicated that the hBMSCs could be induced for adipogenic differentiation when adipogenic induction medium was applied (Fig. 5B).
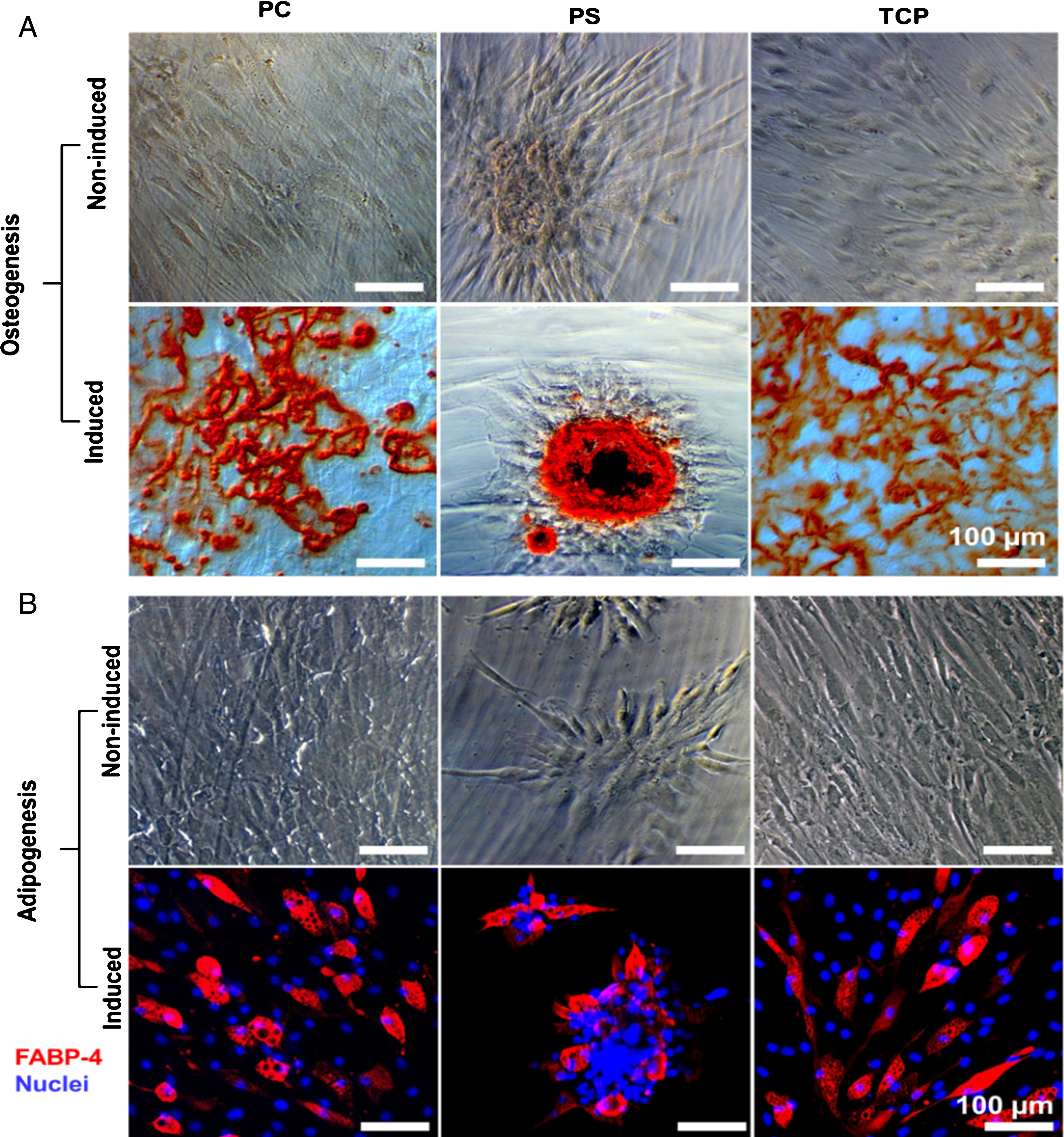
Differentiation of hBMSCs. A: The representative images of alizarin red S (ARS) staining for characterization of hBMSC osteogenic differentiation on different materials (bar = 100 μm). B: The representative laser scanning microscopic images of FABP-4 immunostaining for characterization of hBMSC adipogenic differentiation (red: FABP-4; blue: nuclei; bar = 100 μm). The reader is referred to the web version of this article, where the coloured version of the figure is provided.
The cell behavior can be regulated by substrate properties, such as the surface free energy, the surface polarity [17], the presence of functional groups [18], the stiffness of substrate [19] and the surface topography [20]. The cells actively sense and react to the substrates through the activation of cell surface integrin and then the focal adhesion (FA) complex formation [21–23]. The cell surface integrin activation can further activate the focal adhesion kinase (FAK), which is indispensable for the focal adhesion complex formation [24]. The FA complex consists of many signaling molecules, including the vinculin, talin, paxillin, Src and Cas [25–27]. These signaling molecules can be regulated by chemical and physical properties of the substrates, and play a key role for remolding the size, shape and distribution of FAs [28]. The FAs can be found at the peripheral and central areas of the cells. They are linked with the terminal of F-actin to regulate cells via transfer the signaling from the outside to inside [29]. Here, the formation and distribution of FAs were characterized by staining F-actin and vinculin. The images showed that the distribution and size of FAs on PC were comparable to that on TCP, indicating the excellent cell compatibility of PC for hBMSC adhesion. This observation was in line with our previous results [15].
MSCs are multipotent cells, which are capable of extensively replicating and maintaining their multi-potential during culture. However, the long-term cell cultivation in vitro may lead to cellular aging, alteration of genetic and epigenetic features as well as spontaneous cell transformation [30]. The primary cells do not grow infinitely, but undergo only a limited number of cell division, in a process called cellular senescence [31]. The MSCs cease growth in vitro at about 40 to 50 cell doublings [32]. The stem cell senescence could result in impaired regenerative capacity and reduced tissue function, which might consequently lead to organismal aging in vivo [33–36].
The senescent cells undergo irreversible growth arrest, but the metabolism is active. They normally present a large and flat morphology and display the characteristic changes in gene expression. Senescent cells typically exhibit an increase of SA-β-gal activity [37, 38]. In this study, the result showed that with the elongation of cultivation the senescence level of hBMSCs increased on all of the tested materials, which could be explained by the regulation from the genetic factors. The senescence level of hBMSCs on PS was significantly higher than that on PC and TCP, which might be caused by the poor cell adhesion and spreading of cells on PS [15].
In addition to growth arresting, morphology change and accumulation of lipofuscin in cell plasma, senescence in stem cells could cause the damage of DNA and the alteration of functions [37]. In senescent MSCs, the potential of differentiation decreased with the disruption of the balance between osteogenic and adipogenic differentiation [39–41]. Moreover, long-term cultivation of MSCs could result in the complete loss of osteogenic potential and the decrease of adipogenic potential [42]. In this study, similar to hBMSCs on TCP, cells cultured on PC could be induced to differentiate towards osteogenic and adipogenic lineages by induction medium, and kept in undifferentiated status in growth medium. These results demonstrated that the hBMSCs were capable of maintaining their self-renewing and differentiation potential on PC.
MSCs can secrete a broad range of paracrine factors [1, 44]. These factors could exert the anti-inflammatory, anti-apoptotic, anti-fibrotic and pro-angiogenic functions [45]. The secretion of MSCs could be modulated by various cues from the microenvironment, such as the extracellular matrix (ECM) stiffness [8], surface topography [9] and the chemical property of substrate surface [10]. In our study, the hBMSCs on PS showed higher secretion activity compared to that on PC and TCP, which might be due to the poor cell adhesion and low cell number on PS. Notably, the higher secretion activity of inflammatory cytokines and pro-angiogenic factors including FGF2 and VEGF was observed in cells on PC, as compared to those on TCP. Since VEGF is one of the most important grow factors for promoting angiogenesis [46], this observation suggested that the pro-angiogenic capacity of MSCs might be improved by cultivation on PC.
In summary, our results demonstrated the high cell biocompatibility of PC for stem cell cultivation. The hBMSCs could attach on PC, forming FAs and cytoskeleton similar to those on TCP. Their self-renewal and differentiation potential could be maintained. Compared to TCP, hBMSCs on PC presented a comparable level of senescence and a higher secretion activity.
Conclusion
In this study, the hBMSC senescence level, secretion behavior and differentiation potential on PC were evaluated comparing cell culture on TCP and PS. The PC showed high cell compatibility for hBMSC adhesion and spreading. Cells on PC presented the similar focal adhesion and cytoskeletal structure, as compared to TCP. The cellular senescence of hBMSCs on PC was at a comparable level to that on TCP. Cells on PC showed a higher secretion activity than on TCP, with the upregulated inflammatory cytokines and pro-angiogenic factors. The hBMSCs could maintain their stemness in growth medium on PC, and could undergo osteogenic and adipogenic differentiation. Conclusively, PC represents a promising culture material for stem cells.
Footnotes
Acknowledgments
The authors acknowledge Robert Jeziorski and Mario Rettschlag for preparation of sterilized PS and PC inserts. This work was financially supported by the Helmholtz Association (Helmholtz-Portfolio Topic “Technology and Medicine”, Helmholtz Virtual Institute “Multifunctional Biomaterials for Medicine” (VH-VI-423), Helmholtz Graduate School of Macromolecular Bioscience (MacroBio), and programme-oriented funding).