
Abstract
BACKGROUND:
Rho-kinase, an effector of the small GTPase RhoA, is known to be a novel inhibitory regulator of eNOS in endothelial cells under basal conditions and disease states. However, although RBC possesses active RhoA/Rho-kinase pathway, Rho-kinase mediated eNOS regulation has not been investigated in RBC, so far.
OBJECTIVE:
The aim of the present study is to investigate whether eNOS activity is regulated by Rho-kinase under basal conditions and to evaluate whether inhibition of this enzyme causes eNOS activation and intracellular NO production in RBC.
METHODS:
RBC packeds were isolated from healthy volunteers and resuspended in Hepes solution at a hematocrit of 0.01 l/l. Intracellular NO and Ca+2 levels and eNOS activation measured by flow cytometry in response to Rho-kinase inhibitors, fasudil and Y-27632, in the absence and presence of NOS, and PI3K inhibitors.
RESULTS:
Rho-kinase inhibitors fasudil and Y-27632 found to increase intracellular NO concentrations. These inhibitors also cause enhancement of intracellular Ca+2 and serine 1177 phosphorylated eNOS levels. Besides, although these responses have shown to be suppressed by NOS enzyme, PI3K inhibition had no effect on this mechanism.
CONCLUSIONS:
The results of the present study demonstrated that RBC eNOS enzyme activity is regulated by inhibitory Rho-kinase pathway under basal conditions and inhibition of this pathway enhances the activity of eNOS in RBC. This activation is mediated by both intracellular Ca+2 and Serine 1177 phosphorylated eNOS increment, with no contribution of AKT activation, in RBC. The mechanism we described here gives first evidences about Rho-kinase mediated eNOS regulation in RBC under basal conditions. This pathway could also be more important under disease states.
Introduction
Red blood cells (RBC) possess nitric oxide (NO) synthase enzyme which has similar properties with endothelial type nitric oxide synthase enzyme (eNOS) based on structural and functional properties [1]. RBC eNOS activity has been demonstrated to be induced by applied shear stress and is critical to maintain RBC deformability under the conditions of exercise [2–7]. Besides, important roles of RBC eNOS enzyme activity on the physiological regulation of blood flow, blood pressure, and myocardial function, has been demonstrated by previous studies [8–13].
Rho-kinase (ROCK) is an ubiquitously expressed serine/threonine protein kinase which acts as a downstream effectors of small G-proteins, mainly RhoA [14]. First descried role of ROCK is vascular contraction via phosphorylation of both myosin light chain (MLC) and myosine phosphatase target subunit 1 (MYPT1) in muscle cells [14–16]. However, in non-muscle cells ROCK activation has shown to cause stress fiber formation, actin cytoskeleton organization and cell migration [14, 18]. Activation of ROCK also decreases deformability of cells, including RBC, by altering the properties of the actin cytoskeleton and inhibition of this pathway has been shown to decrease the stiffness of the cell membrane and deformability [19]. On the other hand, ROCK activity effectively regulates gene expression and enzyme activity. Increased ROCK activity has been found to contribute to pathogenesis of pulmonary and systemic hypertension, myocardial hypertrophy and diabetes related complications [20–23]. For these reasons ROCK inhibitors are important drug targets for such diseases [24].
ROCK has acute and long term regulatory roles on NO availability via effecting phosphorylation or expression status of the enzyme, respectively [25, 26]. Under basal conditions, ROCK decreases eNOS activity by two independent mechanism: First, by inhibiting serine1177 phosphorylation and second, by causing dephosphorylation of threonin495 [25–27]. Moreover, ROCK also increases intracellular arginase activity which leads to degradation of eNOS substrate, L-arginine [28]. Besides, ROCK activation leads to decreased eNOS mRNA stability and eNOS expression [26]. For these reasons, inhibition of ROCK activity leads to increased NO availability under basal and disease conditions [24–26]. Considering; important negative regulatory role of ROCK on eNOS activity and NO bioavailability, we investigated the importance of ROCK pathway on RBC eNOS activity, under basal conditions.
The aim of this study is to investigate the possible negative regulatory role of ROCK on RBC eNOS enzyme activity under basal conditions and to further explore whether this effect, if exists is act though intracellular calcium increment and ser1177 phosphorylation of the enzyme.
Material and methods
Blood samples and preparation of red blood cell suspensions
Venous blood samples were obtained from healthy human male volunteers and treated with sodium heparin as the anticoagulant (15 IU/ml). Sampling protocols were approved by local ethical committees (24.02.2016/150) and performed following oral informed consent of the volunteers. Leukocytes were removed by centrifuging 4 ml of blood through 2 ml of polysucrose (60 g/l) and sodium diatrizoate (167 g/l) solution (Histopaque 1119, Sigma Chemical Co., St. Louis, MO, USA) in a 10 ml polypropylene tube at 700 g for 30 min. The RBC pellet was washed three times with calcium-and magnesium-free phosphate buffered saline (PBS, 290 mOsm/kg, pH = 7.4) and resuspended in HEPES buffer (125 mmol/l NaCl, 3 mmol/L KCl, 1 mmol/l MgCl2, 2 mmol/l CaCl2, 16 mmol/l HEPES, 1.2 mmol/l sodium phosphate, and 10 mmol/l glucose, pH 7.4) at a hematocrit of 0.01 l/l. All procedures were conducted at room temperature (20±2°C), unless otherwise indicated.
Demonstration of NO generation
Measurements were performed using the 4-amino-5-methylamino-20, 70-difluorofluorescein diacetate (DAF-FM DA) fluorescent NO probe. Firstly a stock solution was prepared by dissolving the DAF-FM DA in DMSO. Then aliquots were stored – 20°C until the day of experiment. RBC suspensions used for monitoring changes in intracellular NO concentration were incubated at room temperature with 10μM DAF-FM DA for 30 min, then washed three times with HEPES buffer following which they were re-suspended with the same buffer and incubated for 15 min to allow de-esterification of DAF-FM. Specific ROCK inhibitors were added to the suspensions: Fasudil was added to the suspensinon at doses of 5, 10, 30 and 100μM and incubated for 60 minutes at room temperature and Y-27632 was added to the suspensions at doses of 1, 3, 10, 30μM and incubated at room temperature for 30 minutes. Based on the aim of the experiment L-N-acetyl-methyl-arginine (L-NAME, 1 mM) or PI3K inhibitor wortmannin (1μM) were added to the suspensions and incubated for 30 minutes prior to ROCK inhibitor addition. Lastly, all resuspensions were proceeded to flow cytometric analysis.
Demonstration of eNOS and Akt activity
10μl of RBC suspension at a hematocrit value of 0.1 l/l fixed in 1 ml cold 0.05% glutaraldehyde for 10 minutes at room temperature and then washed once with 5 ml staining buffer (PBS with 1% FBS, 0.09% NaN3). Following centrifugation at 200×g for 5 minutes, RBCs were resuspended in 0.5 ml 0.1% Triton X-100 and incubated for 10 minutes at room temperature. After centrifugation and aspiration of the supernatant, RBCs were resuspended in 0.5 ml staining buffer which contains primary antibody for peNOS (cell signaling, 9571) or peAkt (abcam, 18206) and incubated for 20 minutes at room temperature (in the dark). Following washing with 2 ml of staining buffer at 200×g for 5 minutes, secondary antibody was (invitrogen) added to the suspension and incubated at RT for 15–20 minutes. Then all resuspensions were proceeded to flow cytometric analysis.
Demonstration of intracellular Ca+2 levels
Fluo-4/AM (F1241, Invitrogen, Carlsbad CA, USA)was added to RBC suspensions at a dose of 1μM and incubated with mild shaking for 30 min at 37°C. RBC were washed three times with PBS following the incubation period and then re-suspended in HEPES buffer was used at 0.01 l/l hematocrit value. ROCK inhibitor fasudil was added to suspensions at a dose of 5μM and incubated for 60 minutes at room temperature. Based on the aim of the experiment L-N-acetyl-methyl-arginine (L-NAME, 1 mM) or PI3K inhibitor wortmanin (1μM) were added to suspensions and incubated for 30 minutes prior to Fasudil addition. Then all resuspensions were subjected to flow cytometric analysis.
Results
Intracellular NO concentrations: Dose-response
Figure 1 shows percent increase in DAF-FM DA fluorescence intensity (indicative of NO production) in response to incubation of RBC with two different ROCK inhibitors, Fasudil (Fig. 1a) and Y-27632 (Fig. 1b). Both inhibitors caused significant increase in intracellular NO levels at all doses studied, demonstrating negative regulatory role of ROCK on RBC eNOS, under basal conditions. Since 5μM Fasudil and 1μM Y-27632 doses were found to increase intracellular NO levels, these doses were used for the rest of the experiments.
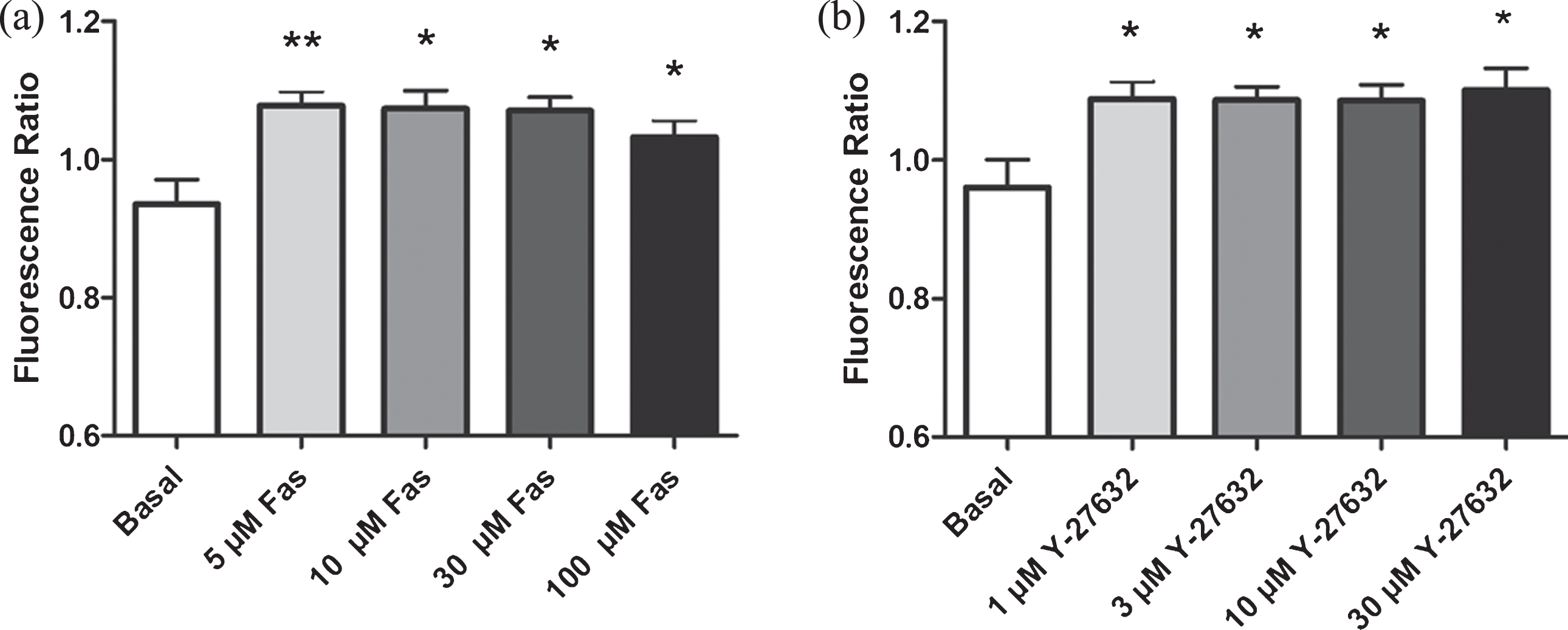
Effects of ROCK inhibition (with two different ROCK inhibitors) on intracellular NO levels in RBC. a) Effect of Fasudil and b) Effect of Y-27632. Values are presented as mean±SEM. *P < 0.05 and **P < 0.01, Difference from Basal (n = 12).
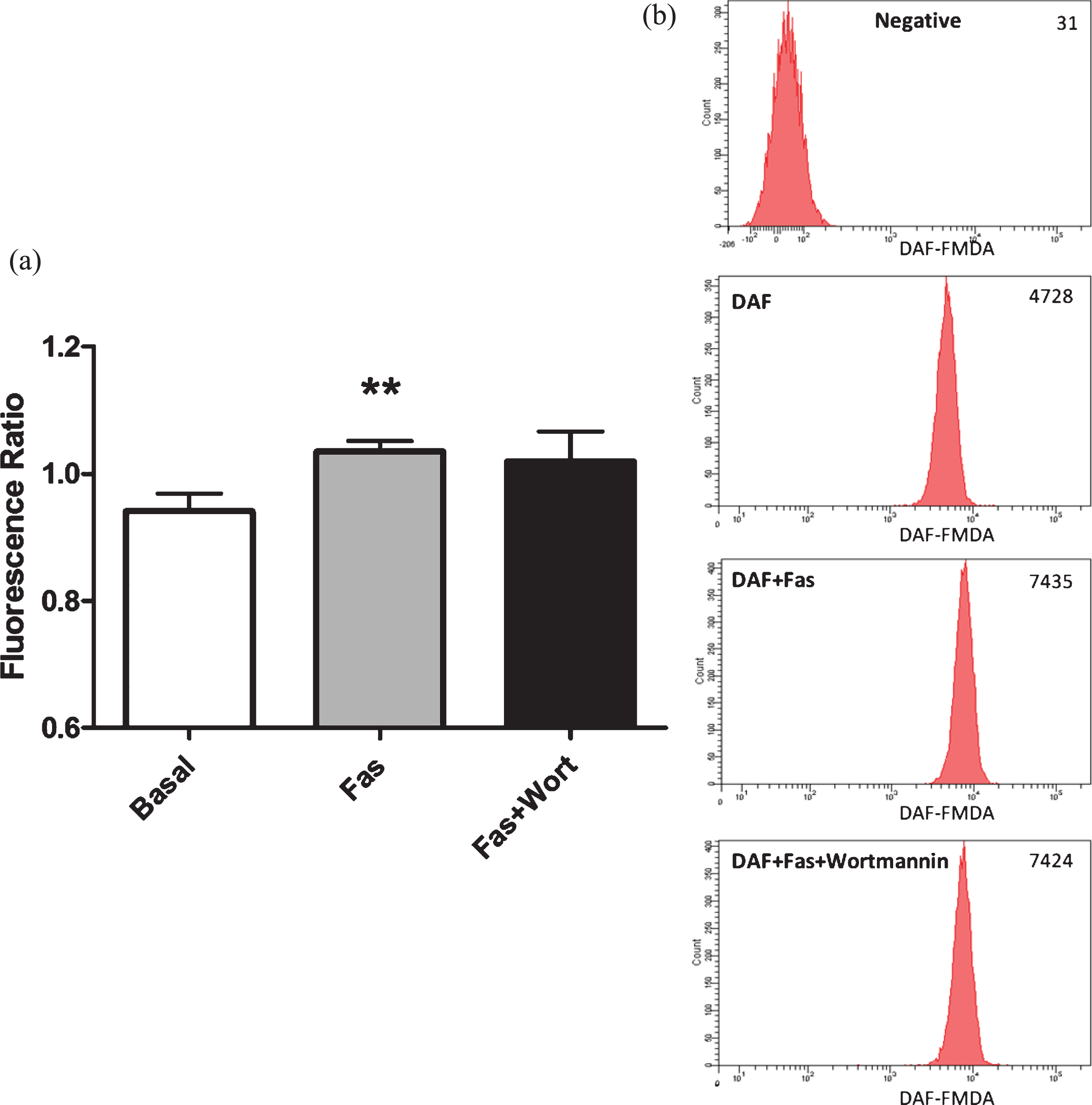
For the purpose of demonstrating that the increased DAF-FM DA fluorescence was resulted from enhanced eNOS activation in RBC, we added non-specific NOS inhibitor L-N-acetyl-methyl-arginine (L-NAME, 1 mM) into the RBC suspensions, 30 minutes prior to incubation with ROCK inhibitors. As presented in Fig. 2a, L-NAME significantly blocked Fasudil mediated increment in intracellular NO generation in RBC. Similarly, L-NAME also abolished Y-27632 related increment in intracellular NO generation (Fig. 2c). Through using two different inhibitors, the results obtained in these experiments clearly demonstrate ROCK inhibition enhances intracellular NO generation. Taken together, these results indicate that ROCK negatively controls eNOS activation and intracellular NO generation in RBC under basal conditions.
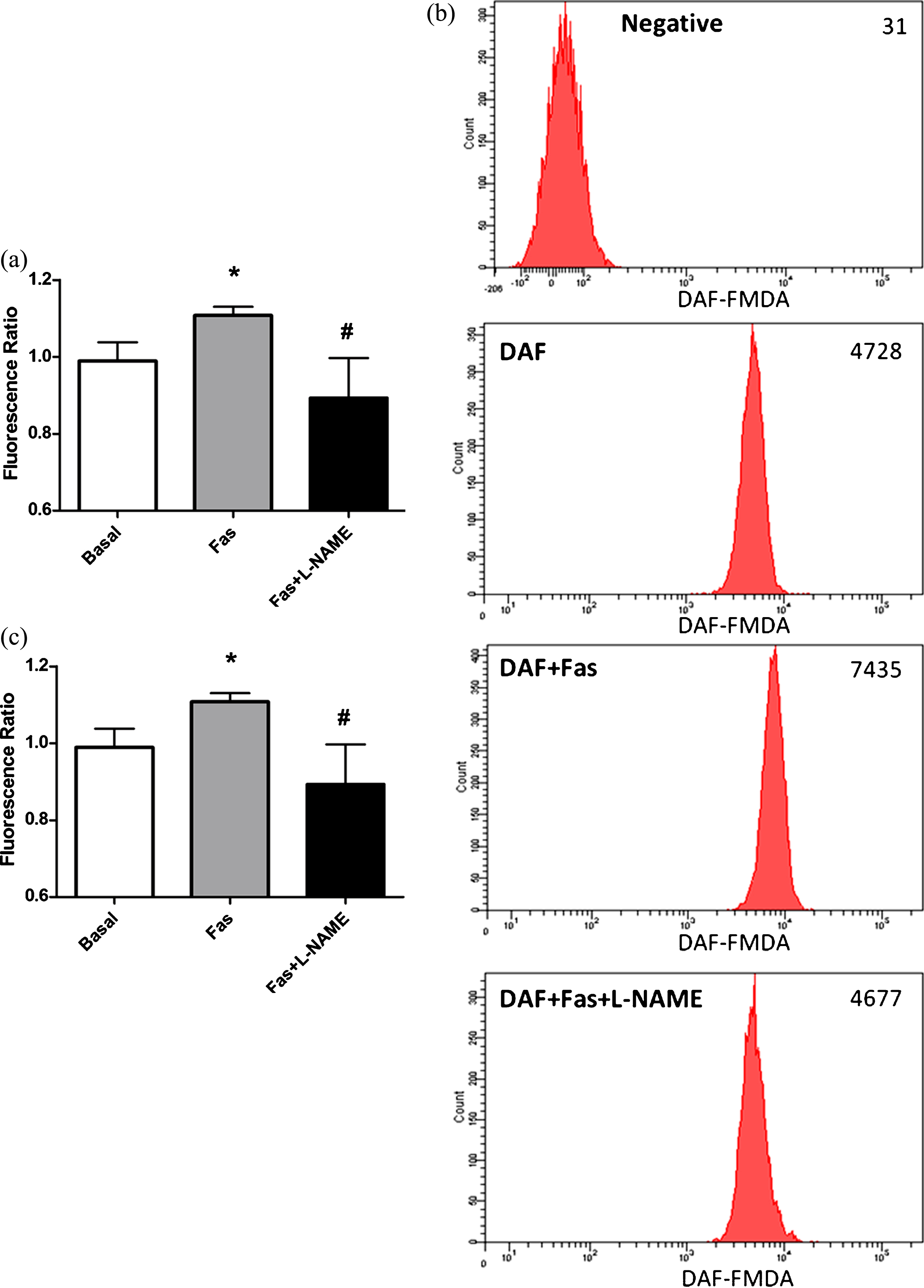
Effects of Fasudil (Fig. 2a) and Y-27632 (Fig. 2c) on intracellular NO generation in RBC, in the presence of L-NAME. Representative examples of flow cytometric results of Fasudil (Fig. 2b). Values are presented as mean±SEM. *P < 0.05 and **P < 0.01, Difference from Basal; #P < 0.05, Difference from Fasudil; # #P < 0.01, Difference from Y-27632 (n = 12).
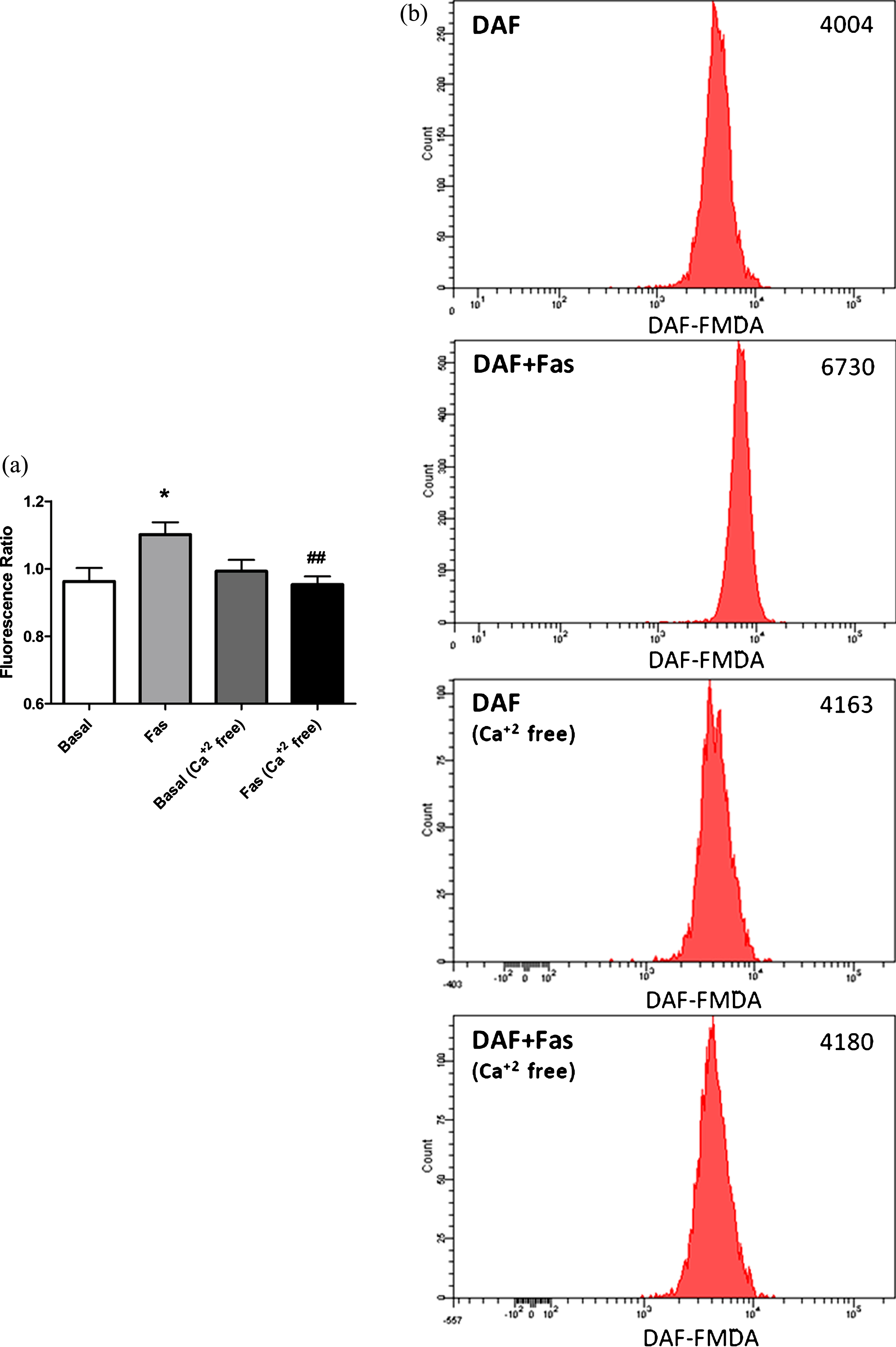
eNOS activity is regulated by calcium dependent and independent mechanisms. Intracellular calcium increment has shown to be essential for eNOS activation in response to several types of stimuli. Ca+2 regulates eNOS activity through causing calmodulin binding to eNOS and detaching it from the membrane [14]. In order to investigate the role of intracellular Ca+2 increment in RBC-eNOS activation in response to ROCK inhibition, we measured intracellular NO concentrations in RBC suspended in Ca+2 containing and Ca+2 free media. We found that intracellular Ca+2 increment is essential for ROCK inhibition induced eNOS activation in RBC.
In order the strengthen our previous findings about calcium dependency of ROCK inhibition mediated eNOS activation in RBC, we measured intracellular Ca+2 levels in response to ROCK inhibition, in the RBC loaded with an intracellular calcium dye Fluo-4. As demonstrated in Fig. 4, ROCK inhibition with 5μM Fasudil caused significantly increment in intracellular Ca+2 levels.
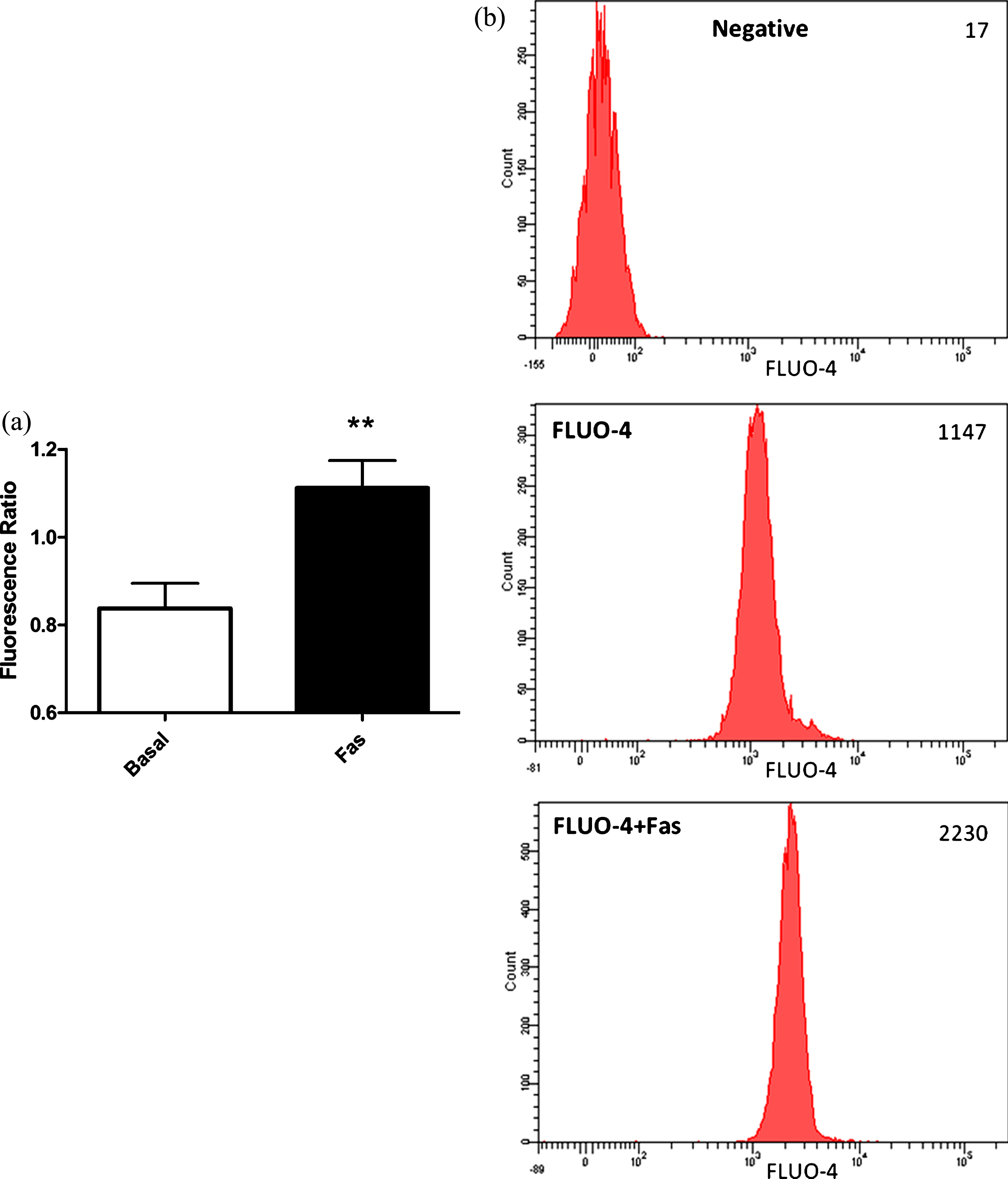
Calcium independent mechanisms of eNOS activation are mainly composed of phosphorylation of the enzyme from the serine, threonine and/or tyrosine rezidues by several kinases [29]. AKT is one of those kinases and is known to phosphorylate eNOS protein at the serine amino acid group at position 1177 leading to activation of the enzyme. In order to evaluate the role of AKT on ROCK dependent eNOS activation in RBC, we measured Ser1177 phosphorylated eNOS levels in RBC in response to Fasudil treatment, in the presence and/or absence of PI3/AKT-kinase inhibitor wortmannin. As shown in Fig. 5, application of 5μM Fasudil caused increment in Ser1177 phosphorylation levels of RBC-eNOS. However, AKT inhibition did not cause any significant change in ROCK inhibition induced enhanced eNOS phosphorylation.
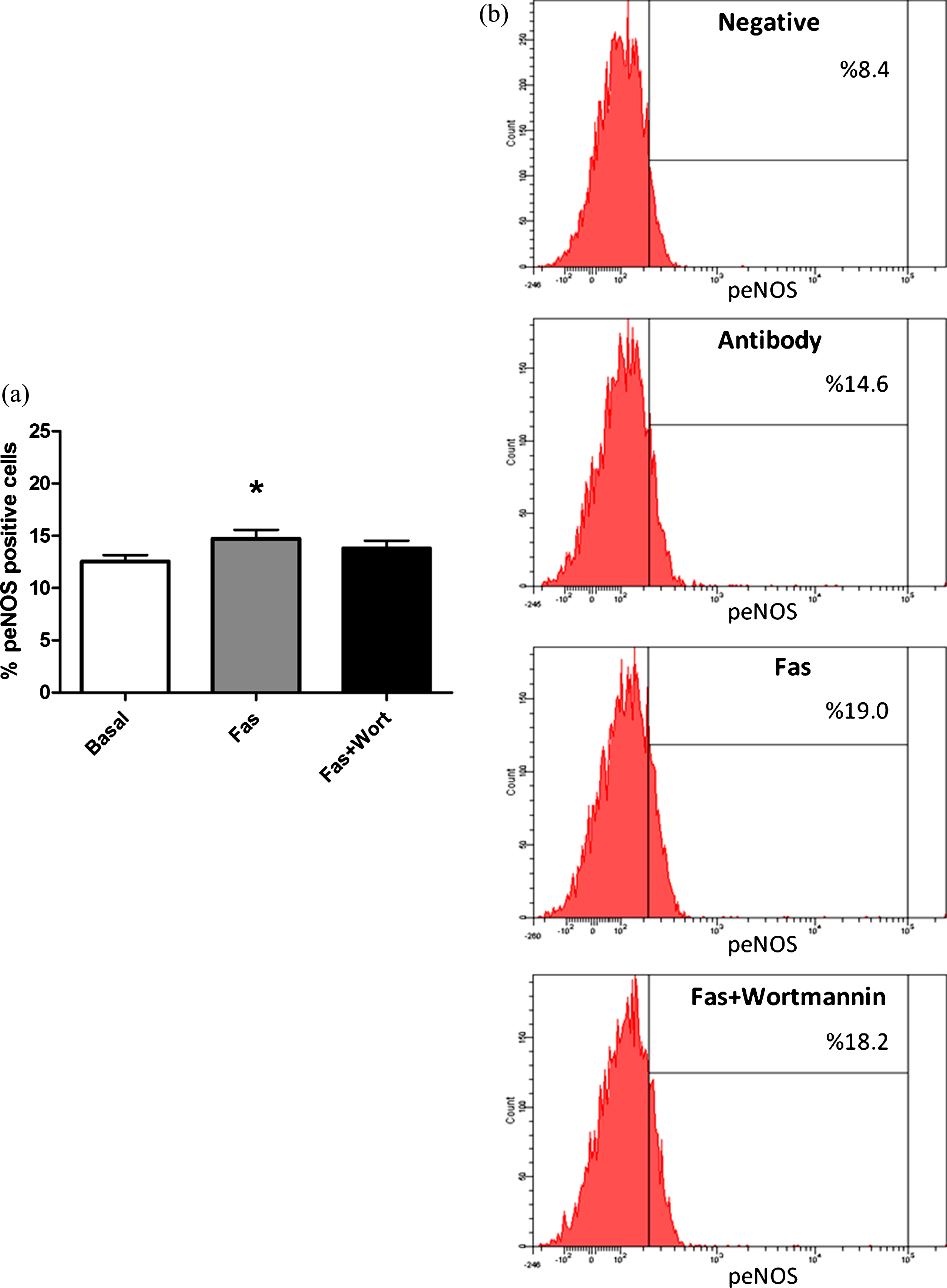
Contribution of the PI3-kinase/AKT pathway to the effect of Fasudil on serine 1177 phosphorylation of eNOS in RBC (Fig. 5a). Pictures show representative flow cytometry results indicating RBC NOS ser1177 positive cells (Fig. 5b). Values are presented as mean±SEM. *P < 0.05, Difference from Basal (n = 13).
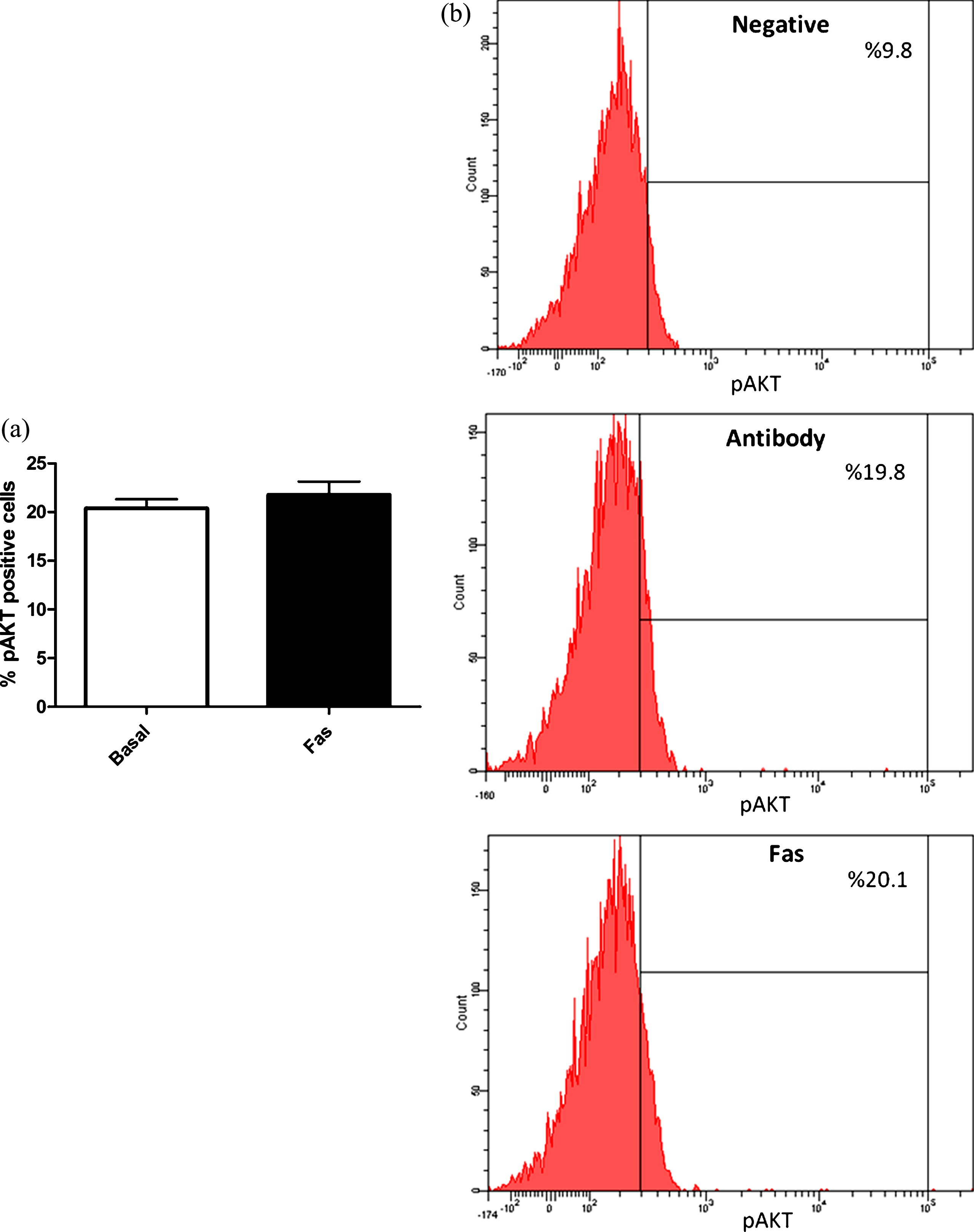
In order to further evaluate the role of PI3-kinase/Akt pathway in ROCK inhibition induced eNOS phosphorylation and activation, we measured active AKT (ser473 phosphorylated) levels in response to ROCK inhibition in RBC. As presented in Fig. 6, AKT is not activated upon ROCK inhibition, demonstrating AKT does not play significant role in ROCK inhibition mediated eNOS activation in RBC.
In order to ensure Akt does not play significant role in the ROCK mediated eNOS activation mechanism in RBC, we next pursed the changes in intracellular NO levels in response to ROCK inhibition in the presence of AKT inhibitor. Addition of AKT inhibitor prior to ROCK inhibition was not cause any significant change in intracellular NO levels strengthening previous results.
The results of the present study clearly demonstrate that basal RBC eNOS activity is under the repressive control of ROCK. Inhibition of ROCK activity by using two different selective ROCK inhibitors resulted in increased intracellular NO generation and this response was blunted in the presence non-specific NOS inhibitor, L-NAME. ROCK inhibition induced increased NO generation was also inhibited in the presence of calcium-free media. Moreover, intracellular Ca+2 concentrations found to be increased in response to ROCK inhibition, strongly suggesting calcium dependency of the pathway. Phosphorylation of eNOS at Serine1177 position is also known to be associated with increased activity of the enzyme. In the present study 5μM Fasudil caused increased Ser1177 peNOS levels in RBC. This phosphorylation is related to enzymatic activities of kinases other than PI3K/Akt because: 1)AKT inhibition did not affect ROCK inhibition mediated eNOS activation and intracellular NO generation, 2)AKT activity (AKT phosphorylation at serine 473) did not changed by ROCK inhibition.
Small G-protein RhoA and its downstream effectors (ROCKs) was initially defined as enzymes that arrange cytoskeletal rearrangement and cytoskeleton related cellular functions such as cell migration and smooth muscle cell (SMC) contraction [14, 18]. In the context of NO bioavailability, RhoA-ROCK pathway is known to downregulate eNOS expression and inhibit eNOS enzyme activity [25, 26]. Active RhoA and ROCK pathway has also been demonstrated in unstimulated RBC and shown to negatively regulate RBC deformability under basal conditions [19]. In the present study, inhibition of RhoA/ROCK pathway, by using two different selective inhibitors, caused significantly increment in intracellular NO generation under basal conditions (Fig. 1a and b). This increment has shown to be blocked in the presence of a competitive NOS inhibitor, L-NAME, demonstrating the response is related to the enzymatic generation of NO (Fig. 2). These results confirm previous studies demonstrating ROCK mediated negative regulation of eNOS enzyme activity under basal conditions and disease states [24, 30].
eNOS is a constitutively expressed bi-domain enzyme comprised of an N-terminal oxidase domain and a reductase domain. It’s activity is regulated by both domains via direct protein-protein interaction or phosphorylation [31]. Calcium-calmodulin binds to calmodulin binding site on both domains resulting in activation of the enzyme via displacement of auto-inhibitory loops. Thus eNOS activity is proportional to intracellular calcium levels [31]. In the present study, ROCK inhibition via treating RBC with 5μM Fasudil resulted in intracellular free calcium increment (Fig. 4). Moreover, ROCK mediated enhancement of intracellular NO generation blunted in the presence of calcium-free media (Fig. 3). These results strongly demonstrate calcium dependency of eNOS activation upon ROCK inhibition. Studies evaluating the effect of ROCK mediated regulation of cell function, under basal states, also support our findings. These studies demosntrated ROCK inhibition cause enhanced intracellular calcium levels and agonist mediated calcium transients [32, 33]. Considering ROCK inhibition decreases calcium influx under disease states, ROCK inhibition induced calcium responses seem to be cell and/or condition specific.
In addition to calcium dependent mechanisms, eNOS activation is also regulated by phosphorylation from its tyrosine, threonine and serine residues by several enzymes. Although threonine 495, tyrosine 657 and serine 114 phosphorylation are shown to be related to inhibition of the enzyme, serine 1177 phosphorylation causes activation [31]. Whether Akt kinase is a well known enzyme related to serine 1177 phosphorylation, protein kinase A (PKA), AMP kinase (AMPK) and calcium-calmodulin dependent kinase (CAMKII) are other enzymes demonstrated to phosphorylate eNOS from the same region resulting in activation [31]. Because we and other laboratories have previously demonstrated Akt-kinase mediated serine 1177 phosphorylation and activation of eNOS in RBC, in the present study we sought to determine whether same pathway plays role on eNOS activation in response to ROCK inhibition [4, 34]. However demonstrating AKT inhibition does not affect neither intracellular NO generation, nor serine 1177 phosphorylation of eNOS, enzymes other than AKT seem to play role in ROCK inhibition mediated eNOS phosphorylation and eNOS activation in RBC (Figs. 5 and 6). Moreover, we found no change in serine 473 phosphorylation of AKT (Fig. 7), demonstrating increased activity of AKT, strongly suggesting AKT is not involved in ROCK inhibition mediated increment in eNOS activation in RBC, under basal conditions.
In summary, this study provides first evidences about ROCK mediated negative regulation of RBC-eNOS activity under basal conditions. Although stimulatory factors affecting RBC-eNOS activity have been well studied since its first demonstration, regulatory mechanisms of RBC-eNOS activity under basal states have not been fully clarified yet. Considering increased ROCK activity contributes to progression of several cardiovascular diseases and RBC are important regulators of blood pressure and blood flow regulation [8–13, 23], the pathway described herein might also be more important under pathological conditions.
Footnotes
Acknowledgments
This study supported by TUBITAK (project no: 116S271).