
Abstract
Establishing an endothelial cell (EC) monolayer on top of the blood contacting surface of grafts is considered to be a promising approach for creating a hemocompatible surface. Here we utilized the high affinity interactions between the EC plasma membrane expressed enzyme called endothelin converting enzyme-1 (ECE-1) and its corresponding substrate big Endothelin-1 (bigET-1) to engineer an EC-specific binding surface. Since enzymatic cleavage of substrates require physical interaction between the enzyme and its corresponding substrate, it was hypothesized that a surface with chemically immobilized synthetic bigET-1 will preferentially attract ECs over other types of cells found in vascular system such as vascular smooth muscle cells (VSMCs). First, the expression of ECE-1 was significantly higher in ECs, and ECs processed synthetic bigET-1 to produce ET-1 in a cell number-dependent manner. Such interaction between ECs and synthetic bigET-1 was also detectible in blood. Next, vinyl-terminated self-assembled monolayers (SAMs) were established, oxidized and activated on a glass substrate as a model to immobilize synthetic bigET-1 via amide bonds. The ECs cultured on the synthetic bigET-1-immobilized surface processed larger amount of synthetic bigET-1 to produce ET-1 compared to VSMCs (102.9±5.13 vs. 9.75±0.74 pg/ml). The number of ECs bound to the synthetic bigET-1-immobilized surface during 1 h of shearing (5dyne/cm2) was approximately 3-fold higher than that of VSMCs (46.25±12.61 vs. 15.25±3.69 cells/100×HPF). EC-specific binding of synthetic bigET-1-immobilized surface over a surface modified with collagen, a common substance for cell adhesion, was also observed. The present study demonstrated that using the substrate-enzyme affinity (SEA) of cell type-specific enzyme and its corresponding substrate can be an effective method to engineer a surface preferentially binds specific type of cells. This novel strategy might open a new route toward rapid endothelialization under dynamic conditions supporting the long-term patency of cardiovascular implants.
Introduction
Cardiovascular disease is one of the leading causes of death worldwide [1, 2], and severe occlusive vascular disease demands a vascular intervention or a replacement surgery [3]. Auto-grafts, commonly taken from the left internal thoracic artery (LITA) or from the saphenous vein, are the gold standards today for coronary artery bypass grafting (CABG) [4]. However, no suitable veins/arteries are available due to previous use or diseased vein wall in 5–30% of patients [5], and such limited supply of auto-grafts creates a great need for synthetic material-based small diameter vascular prostheses. Unfortunately, synthetic materials in vivo often did not support rapid endothelialization and resulted in graft failure, especially in small diameter vascular prostheses (Ø≤4∼6 mm) due to thrombosis and the formation of pseudo-intimal hyperplasia followed by restenosis [6, 7].
Formation of a functionally stable endothelial cell (EC) layer on the luminal surface of prosthesis can solve the problems of thrombogenicity and pseudo-intima formation [8–13]. Deutsch et al. as well as Dohmen et al. reported that vascular prostheses pre-seeded with human venous EC showed much longer patency compared to non-seeded ones, strongly emphasizing the importance of establishing a stable EC layer [14, 15]. Nevertheless, laborious preparation steps such as sampling of appropriate cells from patients and pre-seeding of prostheses in vitro call for the development of a cell-free, off the shelf vascular prostheses, which can be immediately used for the implantation [16] and recruit EC/EC-precursors from the neighboring tissues and/or from the blood after the implantation. Since the vascular wall and the blood contain heterogeneous cell populations that also contact a polymer surface in viv o, finding a way to give a surface EC specificity might be a key element to develop fully functional vascular prostheses. Although establishment of a strongly attached EC layer on top of an artificial surface is a task rather difficult to achieve [17, 18], it is an important requirement for the development of a fully functional small diameter vascular prostheses.
A number of different approaches to engineer hemocompatible surface have been tried, and one of them was to modify the surface in a way to mimic the extracellular matrix (ECM) [19–21]. For example, the tripeptide Arg-Gly-Asp (RGD) sequence is a frequently studied ECM constituent peptide sequence promoting cell adhesion via ligand-receptor interaction [22]. The RGD sequence is shared by many different ECM proteins such as fibronectin and collagen, as well as laminin [23, 24]. Other examples of integrin binding peptides to facilitate EC binding are the fibronectin-derived Arg-Glu-Asp-Val (REDV) motif and the osteopontin-derived Ser-Val-Val-Tyr-Gly-Leu-Arg (SVVYGLR) motif [25, 26]. Although all of these peptides demonstrated that they can promote EC adhesion, they did not show sufficient EC-specificity or preferential EC binding over other types of cells. To overcome such problem of insufficient EC-specificity, we introduce a new approach based on substrate-enzyme affinity (SEA) that might provide EC specificity in the present study.
One of the plasma membrane enzymes expressed in EC is endothelin converting enzyme-1 (ECE-1) which is responsible for producing endothelin-1 (ET-1) by cleaving its corresponding substrate big endothelin-1 (bigET-1). According to the induced fit model, for enzymes to process corresponding substrates, they form a transition state complex where they specifically recognize and bind to each other [27]. The present study exploits such specific interaction between enzymes and corresponding substrates to create an EC specific surface. In other words, it was hypothesized that a surface with chemically immobilized synthetic bigET-1 will preferentially attract ECs over the other types of cells found in vascular system.
The ingrowth of ECs from the existing vasculature at the site of anastomosis is one of the major sources of ECs that facilitate endothelialization of vascular grafts [28]. However, the ingrowth of vascular smooth muscle cells (VSMCs), known as anastomotic intimal hyperplasia, can also occur along with the ingrowth of ECs causing prosthetic graft failure [29]. Therefore, it is important that the luminal surface of a synthetic vascular prosthesis has a preferential binding ability towards EC, especially over VSMCs. As a consequence, in the present study, the ECE-1 expression and the synthetic bigET-1 processing ability of EC was examined in comparison to those of vascular smooth muscle cell (VSMC), another major cell type of vasculature. Furthermore, using synthetic bigET-1-immobilized glass substrates and a rheology testing device, binding of ECs under hemodynamic shear stress was examined and compared to that of VSMCs.
Material and methods
Cell culture
Human umbilical vein endothelial cells (HUVEC) were purchased from Lonza (Allendale, NJ, USA), and human coronary artery smooth muscle cells (HCASMC) were purchased from ECACC (European collection of cell cultures, UK). HUVECs were maintained in endothelial growth medium (EGM2, Lonza) and HCASMCs were maintained in smooth muscle cell growth medium (SmGM2, Lonza). All cells were maintained at 37°C in a 5% v/v CO2 incubation chamber.
Detection of the ECE-1 expression in vascular cells
For the detection of ECE-1 expression in HCASMCs and HUVECs, cells (103cells/well) grown in a 4 well chamber slides (30104, SPL Life Sciences Col, Ltd, Pocheon City, Korea) were subjected to immunostaining using polyclonal goat IgG human ECE-1 primary antibodies (AF1784, R&S Systems, Minneapolis, MN, USA) and FITC-conjugated anti-goat secondary antibodies (705-096-147, Jackson ImmunoResearch Europe Ltd., Suffolk, UK). Briefly, the cells were washed with PBS and fixed in 4% w/v paraformaldehyde for 10 minutes at room temperature. After PBS wash, the cells were permeabilized in 0.1% v/v Triton X-100 for 5 minutes and then phosphate buffered saline (PBS) washed. After 30 minutes of blocking in 1% w/v bovine serum albumin in PBS, the cells were incubated with the primary antibodies (1 : 100, 2% w/v bovine serum albumin (BSA) in PBS) overnight at 4°C. The cells were washed in PBS 3 times and successively incubated with the secondary antibodies (1 : 200, 2% w/v BSA in PBS) for additional 1 hour at room temperature. For visualizing the cytoskeletal structure, F-actin was stained with Texas Red-X phalloidin (Invitrogen, Waltham, MA, USA) and the nuclei were stained with DAPI. Immunofluorescence was detected by confocal microscopy (LSM710; Carl Zeiss Microscopy GmbH, Jena, Germany).
Reverse transcription polymerase chain reaction (RT-PCR)
Briefly, total RNAs from HCASMCs and HUVECs were extracted using TRIzol reagent (Ambion, Waltham, MA, USA) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized from 500 ng of RNA by AMV reverse transcriptase (Promega, Fitchburg, WI, USA). The primer sequences used were as follows: human ECE-1 (458 bp): Forward: 5′-TGT CTT CCT CGC TGG AAG TT-3′, Reverse: 5′-GCC GGA AAC ACA ATC TCA TT-3′; human β-actin (352bp): Forward: 5′-AAA CTG GAA CGG TGA AGG TG-3′, Reverse: 5′-CTC AAG TTG GGG GAC AAA AA-3.
Enzymatic processing of bigET-1 by ECs in suspensions
Synthetic bigET-1 (25 ng, 023-10, Phoenix Pharmaceuticals Inc., Burlingame, CA, USA) was incubated with various concentrations of suspended ECs (0.25∼8×104 cells/ml) in endothelial basal medium (EBM, Lonza) at 37°C, and 100μl of the supernatants were collected at 1 and 2 hours. The collected supernatants were centrifuged at 3,000 g for 5 minutes to precipitate the cells and the supernatants were used to detect the amount of ET-1 produced by enzymatic cleavage using Endothelin-1 Quantikine ELISA Kit (DET100, R&D systems, Minneapolis, MN, USA) according to the manufacturer recommended protocol. For the detection of bigET-1 cleavage in blood, 900μl of pooled blood collected from male balb/c athymic nude mice (5 weeks old, Koatech, Pyeoungtaek, Korea) via orbital blood collection was mixed with 100μl of EGM2 (Lonza) containing bigET-1 (0.1μg/ml) and HUVECs (10∼100 cells/ml) for 2 hours. The amount of ET-1 produced was determined by using human Endothelin-1 QuantiGlo ELISA Kit (QET00B, R&D systems). All experimental procedures for animal studies were approved by the Committee for the Care and Use of Laboratory Animals of Catholic Kwandong University College of Medicine and were performed in accordance with the Committee’s Guidelines and Regulations for Animal Care (CKU 01-2017-008).
Immobilization of bigET-1
Plain cover glass for the rheology testing chamber with diameter of 2.5 cm were cleaned with piranha solution (3/1, v/v, conc. H2SO4/30% v/v H2O2.) at 75°C for 1 hour and rinsed thoroughly with MilliQ water. These substrates were immersed into a 1 wt.% toluene solution of 5-hexenyltrichlorosilane for 1 hour to obtain substrates having vinyl-terminated self-assembled monolayers (SAMs) [30]. The vinyl-functional groups were then oxidized to – COOH groups under Lemieux condition (0.5 mM of KMnO4, 14.7 mM of NaIO4 and 3 mM of K2CO3 for 24 h), followed by washing with MilliQ water [31]. The formed carboxylic acid groups were further activated by using 45 mM 1-(3-(dimethylamino) propyl)-3-ethylcarbodiimide hydrochloride (EDC) and 15 mM N-hydroxysuccinimide (NHS) for 3 hours at room temperature [32]. The glass substrates were then thoroughly rinsed with MilliQ water (Supplementary Figure 1A). To form amide bond between the activated carboxylic acid group of the glass substrates and the amine group of proteins, known concentration of protein solution was applied to the activated surface for up to 24 hours. Unbound proteins were washed off under running water for 5 minutes. Protein binding was verified by staining with Bradford solution after protein immobilization (Supplementary Figure 1B).
Enzymatic processing of surface-immobilized bigET-1 by vascular cells under static condition
Cover glass with activated carboxylic acid group was immersed and incubated in bigET-1 solution (10 ng/ml, 2 ml) overnight. The synthetic bigET-1-immobilized cover glass was placed in a 35 mm culture dish, and either HUVECs (4×104 cells/ml, 2 ml) or HCASMCs (8×104 cells/ml, 2 ml) were seeded on top of the cover glass. The cells were cultured in a CO2 incubation chamber for 24 hours under static condition. The culture media were collected and the amount of ET-1 produced was determined by using Endothelin-1 Quantikine ELISA Kit (R&D systems).
Vascular cell binding to surface-immobilized bigET-1 under shear stress
The test of cell binding under shear stress was performed using the method described previously with minor modifications [18]. On the day of the experiment, synthetic bigET-1-immobilized sample glass disks (immersed in 5 ml of 100 ng/ml bigET-1 solution for 3 hours) were thoroughly washed with MilliQ water and then placed in the sample chambers of the rheology testing device (Rheobasic®, Engtech GmbH, Ulm, Germany). Suspensions of HUVECs or HCASMCs (2×105 cells in 700μl of the respective culture media) were applied to the respective sample chambers. Immediately, the cells were exposed to a shear stress of 5 dyne/cm2 for 1 hour. After 1 hour of shearing, the sample disks were washed by 3 changes of culture medium, and microphotographs were taken from 8 different spots from each sample disk. To evaluate the cell type specific binding of the surface, the number of cells was counted from each high powered field (100×HPF) microphotograph.
Comparison of bigET-1 to other adhesion proteins for EC binding under shear stress
To verify the EC-specificity of the synthetic bigET-1-immobilized surface, the adhesion of HUVECs and HCASMCs to the BSA-immobilized surface and the human collagen type IV-immobilized surface under shear stress was examined. For BSA-immobilized surface, activated sample glass disks were immersed in a BSA solution (10 mg/ml, 5 ml) for 3 hours. For collagen-immobilized surface, the sample glass disks were immersed in a collagen solution (0.1 mg/ml, 5 ml) for 3 hours. Suspensions of HUVECs or HCASMCs (2×105 cells in 700μl of the respective culture media) were applied to the respective sample chambers, and the cells were exposed to a shear stress of 5 dyne/cm2 for 30 minutes.
Statistical analysis
For all quantification analyses, at least three independent experiments were performed. The quantitative data were expressed as the arithmetic means±S.D. (standard deviation) of at least 3 independent experiments. For two sample comparisons, a two-sided t-test was used. P values less than 0.05 were considered to be significant.
Results
ECE-1 expressions in vascular cells
To examine the expression of ECE-1 in major vascular cells, HUVECs and HCASMCs were subjected to immunocytochemical staining using anti-human specific ECE-1 antibodies. The expression of ECE-1 was apparently pronounced in HUVECs compared to that of HCASMCs (Fig. 1A). The mRNA expressions of ECE-1 in HUVECs and HCASMCs were examined by RT-PCR, and the ECE-1 mRNA expression in HUVEC was significantly higher compared to that of HCASMC (2.35±0.31 vs. 0.3 9±0.02, arbitrary unit, p < 0.001) (Fig. 1B).
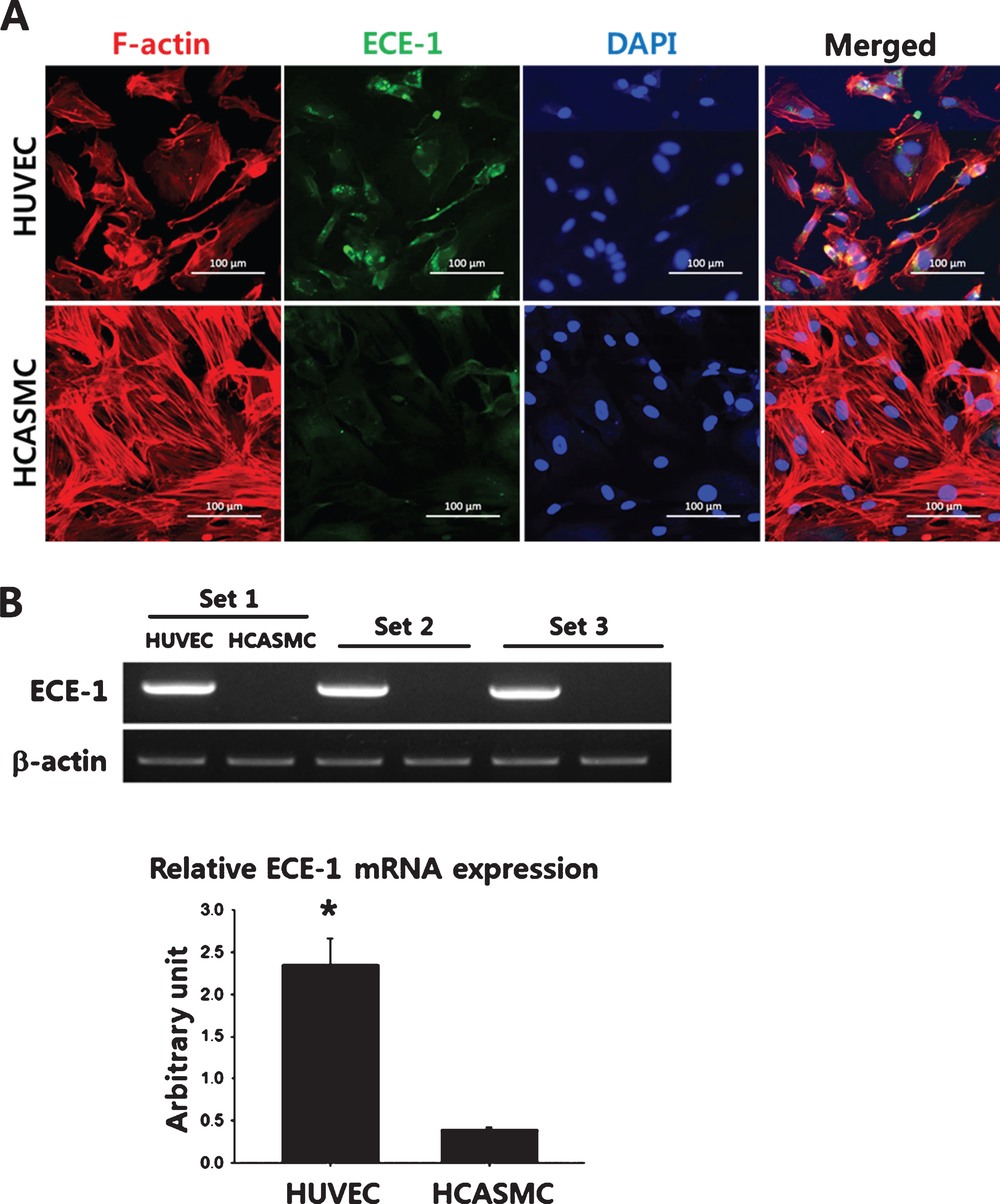
Expression of ECE-1 in ECs and VSMCs. (A) Representative immuno-cytochemical staining images of ECE-1 in HUVECS and HCASMCs. The cells were seeded in a 4 well chamber slide (103cells/well) and allowed to attach and spread for 24 hours. The expression of human ECE-1 was immunostained using human ECE-1-specific primary antibodies and FITC-conjugated secondary antibodies. Cytoskeletal organization and the nuclei were visualized by Texas Red-X phalloidin and DAPI staining, respectively. Scale bar = 100μm. (B) The mRNA expression of ECE-1 in HUVECs and HCASMCs was determined by RT-PCR. The quantitative data were expressed as the means±S.D. of at least 3 independent experiments. *p < 0.001.
To examine whether artificially synthesized bigET-1 could be processed by ECs, 25 ng of synthesized bigET-1 was incubated with increasing number of HUVECs (0–8×104 cells/ml) in culture media for up to 2 hours. As shown in the Fig. 2A, the amount of ET-1 produced significantly increased with EC numbers in samples collected at 1 hour and 2 hour (*p < 0.05, significance indications for 2 hour samples are not shown). Furthermore, the amount of ET-1 produced by 2 hours of incubation was significantly higher compared to that by 1 hour incubation at all the cell numbers tested (#p < 0.01). Even smaller number of HUVECs (80–100 cells/ml) suspended in mouse blood were able to significantly increase the amount of ET-1 produced by the enzymatic cleavage of synthetic bigET-1 (100 ng/ml) within 2 hours (Fig. 2B).
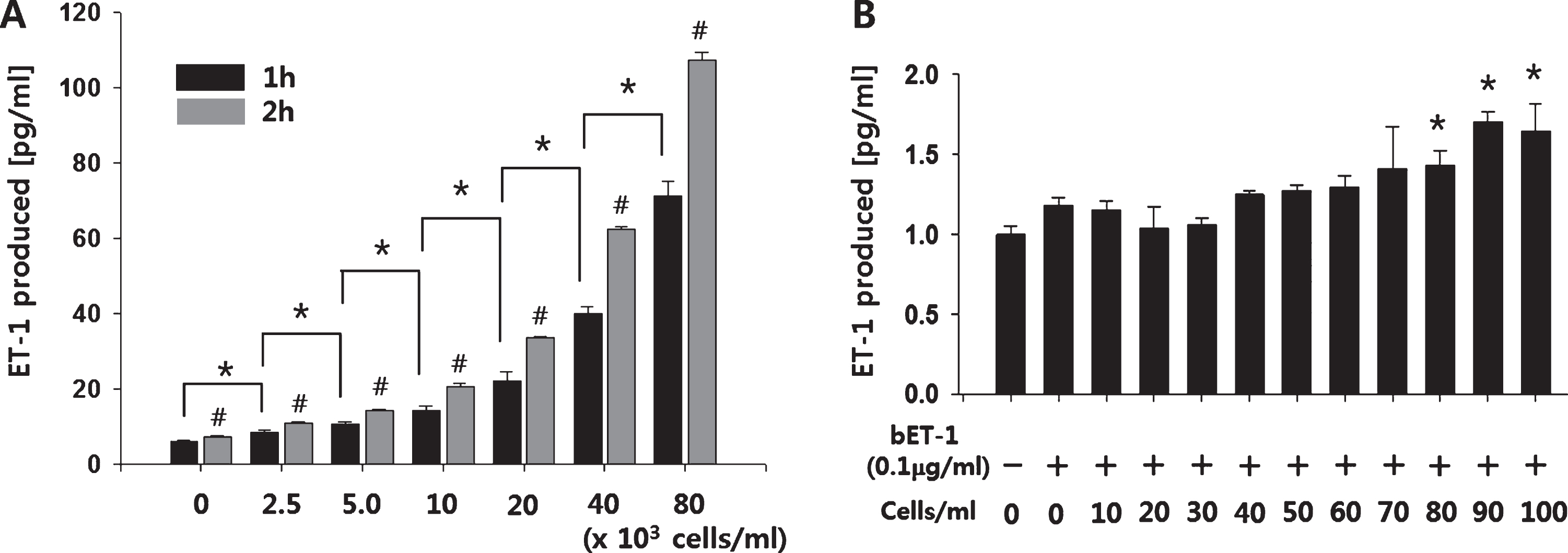
EC number-dependent cleavage of synthetic bigET-1. (A) Synthesized bigET-1 (25 ng) was incubated with increasing number of ECs at 37°C for up to 2 hours, and the amount of ET-1 produced by the cleavage of bigET-1 was measured using an ET-1 ELISA kit. *p < 0.05, significance indications for 2 hour samples are not shown. #p < 0.01 compared to matching 1 hour sample. (B) For the detection of synthetic bigET-1 cleavage in blood, 900μl of mouse blood was mixed with 100μl of endothelial growth medium containing synthetic bigET-1 (100 ng/ml) and HUVECs (0∼100 cells/ml) for 2 hours. The amount of ET-1 produced was determined by using an ET-1 ELISA kit. The quantitative data were expressed as the means±S.D. of at least 3 independent experiments. *p < 0.05.
To investigate any possible adverse effect of surface modification using synthetic bigET-1 on cell adhesion and also to verify that the surface-immobilized synthetic bigET-1 could be processed in an EC-specific manner, HUVECs (4×104 cells/ml) or HCASMCs (8×104 cells/ml) were seeded on top of the bigET-1-immobilized cover glass. Upon microscopic examination, no unusual abnormality in either initial cell adhesion (10 minutes after seeding) or subsequent cell spreading (24 hours after seeding) was observed. Furthermore, under static culture condition, there was no apparent EC-specific binding and spreading effect was observed (Fig. 3A). However, HUVECs processed significantly larger amount of the surface immobilized synthetic bigET-1 compared to HCASMCs so that the amount of ET-1 produced by HUVECs was approximately 10-times larger than that produced by HCASMCs (102.86±5.13 vs. 9.75±0.74 pg/ml, p < 0.001) (Fig. 3B). Considering the number of HCASMCs seeded was 2-times higher than that of HUVECs, actual fold difference between HUVEC and HCASMC could have been close to 20.

Enzymatic cleavage of immobilized bigET-1 by HUVEC and HCASMC. Either HUVECs (4×104cells/ml, 2 ml) or HCASMCs (8×104cells/ml, 2 ml) were cultured on bigET-1-immobilized glass cover slides for 24 hours. (A) Representative images of vascular cells adhere (10 minutes after seeding) and spread (24 hours after seeding) on the bigET-1-immobilized surface. (B) The amount of ET-1 produced by enzymatic cleavage of bigET-1 was measured at 24 hours using an ET-1 Elisa kit. EBM: endothelial basal medium, SmBM: smooth muscle basal medium. The quantitative data were expressed as the means±S.D. of at least 3 independent experiments. *p < 0.001 compared to HCASMC.
Once implanted, vascular prosthesis will be continuously exposed to a physiologic shear stress caused by blood flow. Therefore, an ideal blood contacting surface should be able to preferentially attract ECs under shear stress. To assess the EC-specific binding capacity of the synthetic bigET-1-immobilized surface, adherence of HUVECs and HCASMCs (2×105 cells in 700μl of the respective culture media) to the synthetic bigET-1-immobilized surface was evaluated under shear stress. The number of HUVECs adhered to the surface after 1 hour of shear stress (5 dyne/cm2) was significantly higher compared to that of HCASMCs (46.25±12.61 vs. 15.00±3.69 cells/100×HPF, p < 0.001) (Fig. 4A). Additionally, adherence of HUVECs and HCASMCs to BSA-immobilized or collagen type IV-immobilized surface under shear stress was also examined, and EC-specific affinity of the modified surface was only observed in the synthetic bigET-1-immobilized surfaces (Fig. 4B). Specifically, both the BSA + bigET-1-immobilized surface (28.75±12.45 vs. 0.50±0.87 cells/100×HPF, p < 0.005) and the bigET-1-immobilized surface (18.00±3.74 vs. 1.25±1.30 cells/100×HPF, p < 0.001) showed statistically significant EC-preferential binding over VSMCs. Although the difference between the number of HUVECs and the number of HCASMCs adhered to the BSA-immobilized surface was also statistically significant (3.50±1.00 vs. 0.00±0.00 cells/100×HPF, p < 0.001), the cell numbers adhered was too low to have scientific significance.
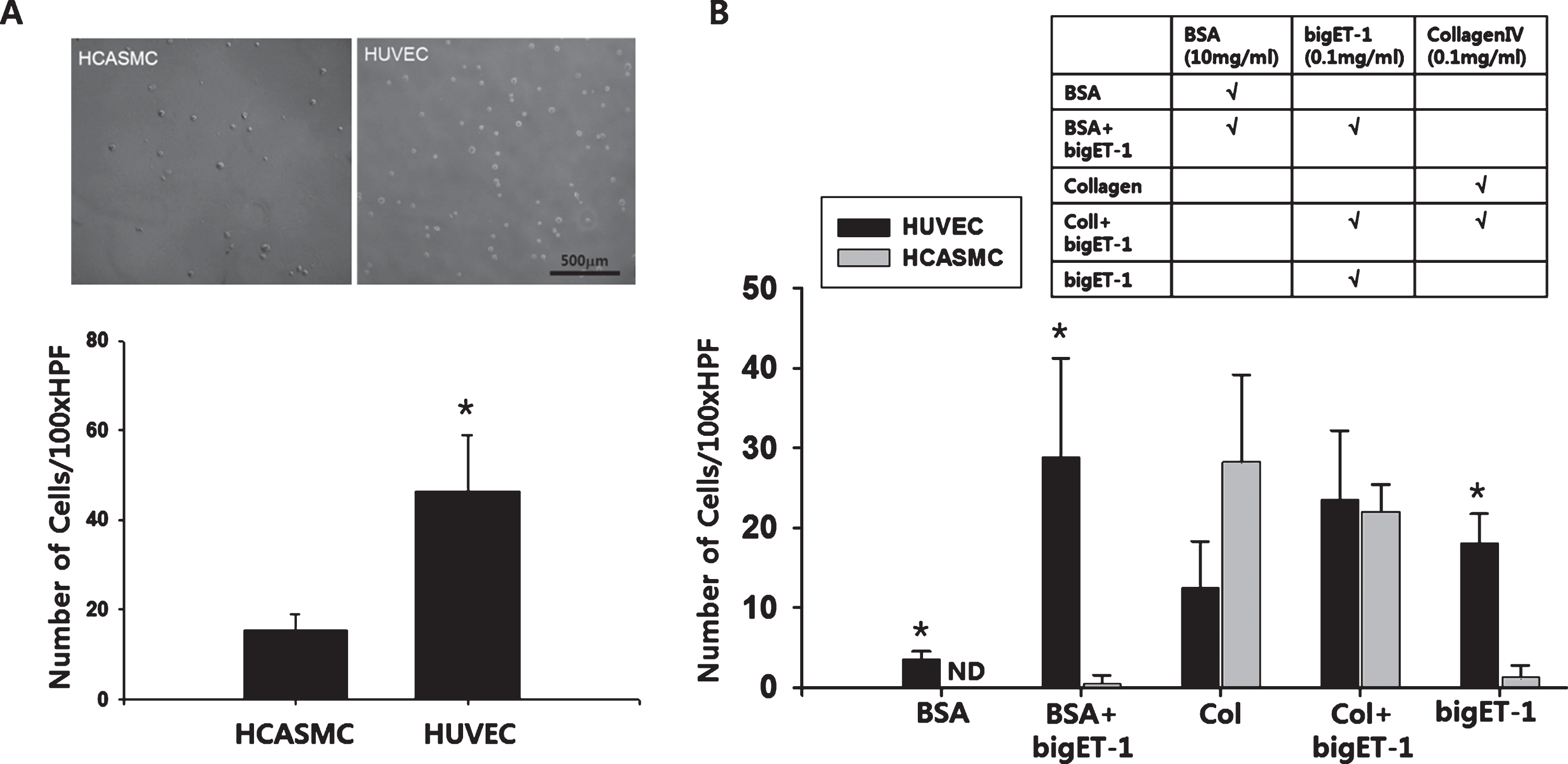
EC-specific binding of bigET-1-immobilized surface under shear stress. (A) Suspension of HUVECs or HCASMCs (2×105cells) were allowed to bind to the bigET-1-immobilized surface (immersed in 5 ml of 100 ng/ml bigET-1 solution for 3 hours) under shear stress (5dyne/cm2, 1 hour). After 1 hour of shearing, the samples were washed by 3 changes of culture medium, and high powered field microphotographs (100× HPF) were taken from 8 different spots from each sample disk. The number of cells was counted from each microphotograph. (B) Glass surface was modified with BSA, Collagen, bigET-1, or combinations of them as indicated. Proteins allowed to form amide bonds to the surface for 3 hours. HUVECs and HCASMCs (2×105cells) were allowed to bind under shear stress (5dyne/cm2) for 30minutes. The number of surface-bound cells was counted from each 100× HPF microphotograph. ND: not detected. The quantitative data were expressed as the means±S.D. of at least 3 independent experiments. Statistical significance compared to the matching HCASMC group is indicated.
For cardiovascular implants, especially for small diameter vascular prostheses, a fully hemocompatible blood contacting surface is important in order to prevent current vascular implant-associated problems such as implant obstruction due to the formation of thrombus or intimal hyperplasia that can lead to the failure of the implant [33, 34]. Currently, there is no single surface modification strategy commonly accepted as the best approach to create a surface with EC-specific binding capability [35]. Therefore, testing various options to engineer hemocompatible surface is well justified. In the present study, a substrate-enzyme affinity (SEA)-based surface modification was tested as a strategy to give an artificial surface EC-specific binding capability. In our approach a cell type-specific enzyme shall react with the corresponding substrates immobilized on a surface, thus enabling a cell type-specific adhesion under shear stress to resist an detachment of EC by shear forces.
The major sources of ECs for the endothelialization of implanted vascular grafts include the ingrowth of ECs from the existing vasculature at the site of anastomosis [28]. However, existing VSMCs can also migrate into the implanted vascular grafts causing prosthetic graft failure [29]. Therefore, it was important to choose an EC plasma membrane enzyme that is preferentially expressed in ECs over VSMCs. The EC plasma membrane enzyme ECE-1 has been reported to be mostly expressed in ECs of blood vessels [36, 37], and our data also confirmed the pronounced expression of ECE-1 in ECs compared to VSMCs (Fig. 1). The major substrate of ECE-1 is bigET-1, which is enzymatically cleaved by ECE-1 to produce bioactive ET-1 [38]. Although big ET-1 has two additional isoforms, bigET-2 and bigET-3, the specificity of ECE-1 for bigET-1 is higher compared to the others [39]. Furthermore, a previous study reported that VSMCs could also process bigET-1, but the enzyme activity of VSMCs was negligible compared to that of ECs [40]. Consequently, bigET-1 was selected as a potential candidate molecule for the surface modification.
In the enzyme-substrate interaction, conformational stability of the participating proteins (i.e., both enzymes and substrates) is critical for enzymatic activity [41–43]. In other words, 3D structures of both the enzyme and the corresponding substrate may significantly affect the enzymatic activity. Unlike endogenous bigET-1 that has 2 disulfide bonds (Cys1-Cys15and Cys3-Cys11) known to significantly affect the enzyme activity of ECE-1 [44, 45], synthesized bigET-1 used in the present study was in a linear form without any disulfide bonds. Therefore, EC-dependent enzymatic processing of the synthetic bigET-1 was then examined prior to surface immobilization or shear stress test. As shown in the Fig. 2A, the amount of ET-1 produced was proportional to the number of ECs, indicating that the synthesized bigET-1 could be properly processed by ECs even without disulfide bonds.
Besides aforementioned migrating ECs at the site of anastomosis, blood circulating ECs and/or endothelial progenitor cells (EPCs) could be another major source of ECs for the endothelialization of implanted vascular prostheses. According to a previous study, the expression of ECE-1 in EPC-derived progenitor cells from both cord blood and G-CSF mobilized peripheral blood was higher than in HUVECs [46]. Furthermore, implanted vascular prostheses will be continuously exposed to blood flow. Therefore, it was critical to confirm that the enzyme-substrate interaction we designed works properly in the presence of blood. The data using mouse blood indicated that even smaller number of HUVECs successfully produced ET-1 from synthetic bigET-1 in blood (Fig. 2B). Speaking of properly function in blood, ECE-1 is a member of the M13 subfamily of zinc endopeptidases which also includes the Kell blood group protein, a blood-borne enzyme [47]. Since they share some sequence homology and structural similarities [48], there was a concern that the Kell blood group protein also interacts with bigET-1. However, literature search indicated that the Kell blood group protein preferentially processes bigET-3 not bigET-1 [49]. Therefore, it was assumed that other ECE-1 related enzymes such as the Kell blood group protein, or the cells expressing such enzymes, less likely interact with the bigET-1 until empirically proven otherwise in future studies. After taking into account all these considerations, ECE-1 and bigET-1 pair was finally selected as the model system to be tested in the present study.
For the present study, plain glass was used as the base-substrate because it can be easily functionalized with various chemical functional groups such as carboxylic acid, amine, and aldehyde. Additionally, transparent glass substrate enables the observation of adhered cells under the microscope without extra cell staining steps. The chemical modification starts with the formation of carboxylic group. The advantage of this modification is that the surface carboxylic groups can be easily activated and reacted with free amine groups of proteins to form an amide bond, which is facilitated in aqueous media by the coupling reagents EDC and NHS (Supplementary Fig. 1A). Using this strategy, one can chemically immobilize various biomolecules containing free amine groups such as amine-modified DNA, peptides, and proteins [50]. A Bradford-staining showed that the modified surface successfully bound common protein BSA (Supplementary Fig. 1B). Another important advantage of this strategy is that it is also easily applicable to various polymer films that could potentially serve as base material for vascular grafts [51].
On the other hand, one of the possible disadvantages of this approach is that the orientation of binding cannot be controlled. The binding strategy used was based on the random covalent bonding between the activated carboxyl groups of the surface and the free amine groups of applied proteins. Usually, there are more-than-one free amine groups presented on proteins, and which of them participates in the amine bonds cannot be controlled. Furthermore, ECE-1 recognizes the sequence of bigET-1 from histone 27 to glycine 34 (His-Val-Val-Pro-Tyr-Gly-Leu-Gly) to physically interact [52]. Therefore, it could be possible that if the free amine groups of the histone 27 or the proline 30 were used up for the immobilization, ECE-1 might not be able to recognize the synthetic bigET-1 immobilized. To investigate such possibility, ECs and VSMCs were cultured on a synthetic bigET-1-immobilized surface for 24 hours under static condition. Another important purpose of the static culture on the synthetic bigET-1-immobilized surface was to examine whether the synthetic bigET-1-immobilized surface supports proper adhesion and subsequent spreading of the cells.
According to our data, both HUVECs and HCASMCs properly attached and spread on the synthetic bigET-1-immobilized surface (Fig. 3A), indicating that there was no significant adverse effect of surface modification on cell adhesion. Furthermore, more importantly, even less number of HUVECs produced significantly larger amount of ET-1 compared to HCASMCs (Fig. 3B), strongly suggesting that at least some of the synthetic bigET-1 immobilized to the surface maintained ECE-1 recognition motif available. Nevertheless, exactly how much of the immobilized synthetic bigET-1 maintained ECE-1 recognition motif available was not determined, and it remains one of the limitations of the present study. To eliminate the uncertainty of recognition motif availability after immobilization, different immobilization strategies that can give more control over the binding orientation such as the His-tag approach [53] will be considered in future studies.
Since the SEA-based surface modification strategy is intended to work with blood flow, EC-specific binding capacity of the synthetic bigET-1-immobilized surface under shear stress was examined. Compared to HCASMCs, significantly higher number of HUVECs adhered to the synthetic bigET-1-immobilized surface under shear stress (Fig. 4A). This result highlights an important aspect of the SEA-based surface modification strategy that the enzyme-substrate interaction was specific to ECs and strong enough to withstand the applied shear stress of 5 dyne/cm2, a shear stress that occurs in many vascular regions [54]. Furthermore, to exclude the possibility that the observed EC-specific binding under shear stress was due to general protein modification rather than due to the synthetic bigET-1 modification, additional rheology testing using BSA-immobilized and collagen-immobilized samples were conducted.
According to our data, EC-specific affinity of the modified surface was observed only when the synthetic bigET-1 was used (both alone and in combination with BSA) for the surface modification (Fig. 4B). Although the BSA-immobilized surface also showed significant difference between the number of HUVECs and the number of HCASMCs adhered, even the number of HUVECs adhered was insignificant. Taken together, these results confirmed that the modification with synthetic bigET-1 could give a surface EC-specific binding capacity under shear stress. In small diameter vascular prostheses, that are intended to replace or bypass the coronary arteries, the arterial wall shear stress ranges from 7 to 70 dynes/cm2 [55, 56]. Therefore, one can argue that withstanding a shear stress of 5 dyne/cm2 may not be clinically significant as it is. Nevertheless, as a proof of concept study, testing the feasibility of using SEA-based modification to engineer a surface selectively attracting ECs under shear stress has its own significance.
Another limitation of the present study is the lack of proven correlation between the amount of the protein immobilized to the surface and the number of cells adhered under shear stress. It would be necessary to know the protein density on the surface after the modification in order to optimize this SEA-based approach. In the future, a more thorough surface analysis following the surface modification such as infrared spectroscopy or Raman spectroscopy [57] and/or multicomponent protein patterning in combination with other proteins that support EC binding and proliferation [58] would be helpful to improve the surface modification strategy. Since enzymatically cleaved bigET-1 produces the most potent vasoconstrictor ET-1 [59], future studies should use bigET-1-derived peptides, not the original bigET-1 itself. Using such derivatives will enable us to achieve EC-specific binding without causing the problem of excessive ET-1 production.
In the present study, the SEA-based approach was explored with regard to its potential as a cell type-specific surface modification strategy. Our data indicated that this SEA-based approach worked properly to give a surface EC-specific binding capacity. Even the most recent EPC capturing study still utilized a more than decade-old strategy based on the interaction between EC expressed molecules and its corresponding antibodies [60, 61]. Therefore, the SEA-based strategy could be the next generation alternative to engineer a shear-resistant EC-specific binding surface for hemocompatible cardiovascular implants.
Footnotes
Acknowledgments
Authors thank the Helmholtz Association for funding of this work through program-oriented funding and through grant number SO-036 (Twinning grant: “Novel strategies of vascular regeneration after coronary interventions based on vascular healing drugs, improved stent hemocompatibility and predetermined drug release” from the BCRT). This work was also supported by the Korea Science and Engineering Foundation grant (NRF-2016R1D1A1B03935124).