
Abstract
Critical-size bone defects after compound fractures, infection, or tumor resection are challenging to treat. The same is true for fractures in patients with impaired bone healing due to metabolic diseases and cancer. Despite considerable progress over the last decade in surgical techniques, material design, and dedicated imaging approaches, these scenarios represent unsolved clinical problems. The high socioeconomic burden of such conditions justifies increasing interest in novel osteoinductive drugs for adjuvant therapeutic approaches. There is an increasing body of experimental and clinical literature on potentially promising effects of growth factors, anti-resorptive, and anabolic agents. The true clinical efficacy of these, however, is discussed controversially. Therefore, we aimed to critically examine the hypothesis that targeted adjuvant therapies have the potential to enhance bone regeneration in critical-size bone defects and under systemic conditions that impair bone healing. This first approach to the topic deals with small molecule drugs and compounds that influence the immune response and inflammatory processes. In particular, literature reporting on selective cyclooxygenase-2 inhibitors has been reviewed with respect to their local and systemic mode of action and to stimulate further research on bone healing under critical conditions.
Keywords
List of abbreviations
arachidonic acid
adenylate cyclase
adenosine/guanosine triphosphate
bone morphogenetic protein
calmodulin
cyclic adenosine monophosphate
calcineurin
COX-inhibiting nitric oxide donator
cyclooxygenase
cAMP response element-binding protein
5,5-dimethyl-3-3(3fluorophenyl)-4-(4methylsulfonal)phenyl-2 (5H)-furanone
Dickkopf-related protein
extracellular matrix
epidermal growth factor receptor
ETS domain-containing protein Elk
endothelial nitric oxide synthase
prostaglandin receptors
extracellular signal-regulated kinase
Food and Drug Administration
fibroblast/vascular endothelial growth factor
guanosine diphosphate
G-protein coupled receptor
glycogen synthase kinase-3ß
hypoxia-inducible factor
inhibitor of kappa B
inhibitor of kappa B kinase
interleukin
mitogen-activated protein kinase
mitogen-activated protein kinase kinase
matrix metalloprotease
mesenchymal stem cells
nuclear factor of activated T-cells
nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells
non-steroidal anti-inflammatory drug
osteoprotegerin
prostaglandin
phosphatidylinositol-3-kinase
protein kinase A
protein kinase C
poly-lactic-co-glycolic acid
phospholipase C
parathyroid hormone
rapidly accelerated fibrosarcoma
receptor activator of nuclear factor κB ligand
rat sarcoma
recombinant human
runt-related transcription factor 2
contraction of Sma and Mad (small mothers against decapentaplegic homolog)
sarcoma oncogene cellular homolog / proto-oncogene tyrosine-protein kinase
transgene
transforming growth factor-beta
tumor necrosis factor alpha
Wingless-related integration site
Introduction
Critical-size bone defects, meaning bone gaps that will not heal without therapeutic intervention (Fig. 1), after compound fractures, infection, or tumor resection still represent unsolved clinical problems [1]. The same applies for fractures in patients with impaired bone healing due to metabolic or inflammatory diseases and cancer, both with a high socioeconomic burden [2].
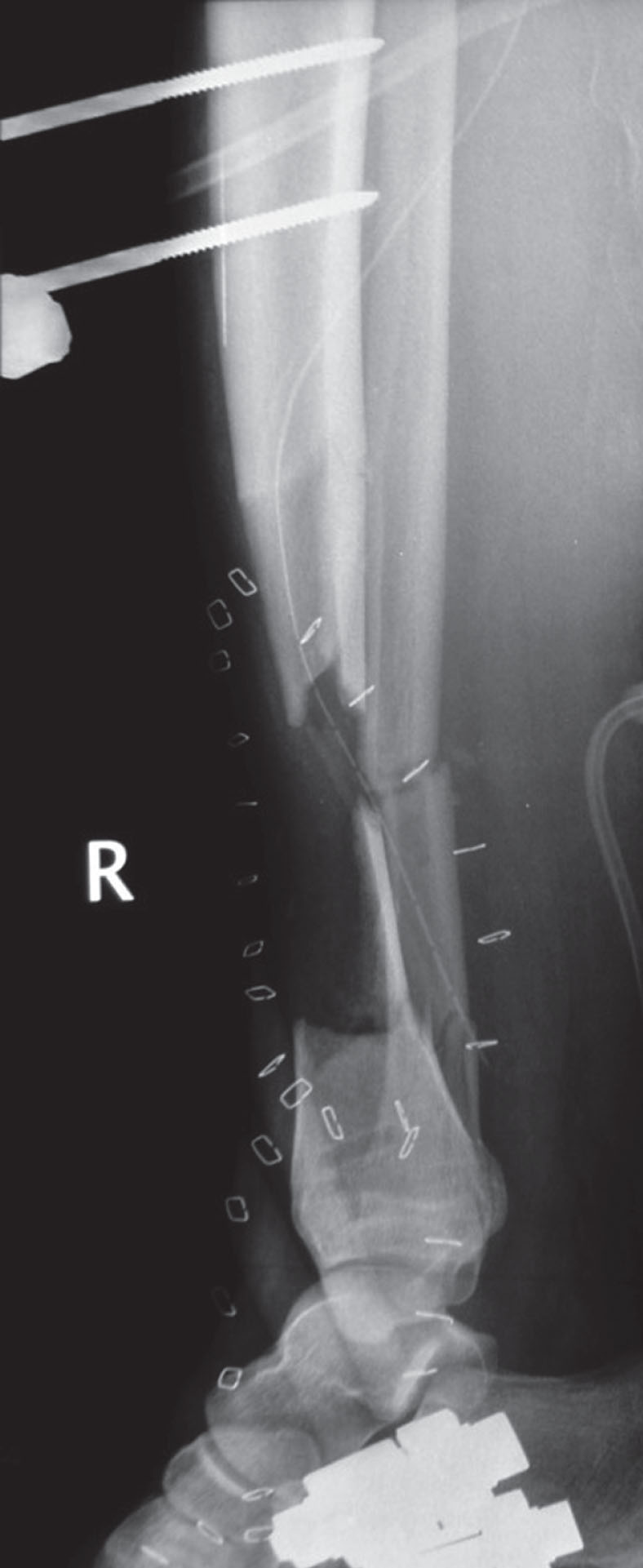
Lateral radiograph of an open lower leg fracture with a critical-size (7 cm) bone defect in the tibial metaphysis. The tibia is provisionally stabilized with an external fixator. The metallic clips outline the corresponding soft tissue defect that is covered with a collagen-based artificial skin graft.
Although numerous resorbable and non-resorbable bone substitute materials have been developed, the golden standard for treatment of critical-size defects is still autologous bone grafting combined with stable internal fixation. Recent developments in enhancing bone remodeling and regeneration include modification with organic and inorganic extracellular matrix (ECM) components as well as the addition of cells, cytokines, or recombinant growth factors [3–6].
Bone regeneration after injury is a well-orchestrated process that evolves in three stages. Any fracture is immediately followed by an initial inflammatory stage (Fig. 2) [7–10]. Damage of blood vessels due to the injury results in local hypoxia. The fracture hematoma contains active blood products such as platelets, fibrin, growth factors, and cytokines. Thereby, it serves as a matrix for immune cells, mesenchymal stem cells (MSCs) and endothelial cells. Together with leucocytes and neutrophils, macrophages enter fracture site. Macrophages phagocytose necrotic tissue and release signaling molecules inducing inflammation, angiogenesis and osteogenesis. Mediators at defect area include interleukins (ILs), transforming growth factor-beta (TGF-ß), tumor necrosis factor alpha (TNF-α), bone morphogenetic proteins (BMPs), vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and members of angiopoietin or ephrin family influencing cell populations like endothelial cells [11–13]. In response to these signaling molecules endothelial cells aggregate to form new blood vessels growing into the defect site. Furthermore, fibroblasts settle and MSCs differentiate into chondrocytes and osteoblasts [14, 15]. In the second phase of fracture healing, the reparative phase, damage site is vascularized and hematoma is resorbed and replaced with callus or cartilage tissue requiring appropriate oxygen and nutrient supply. Primary goal during this healing phase is revascularization of defect area since nutrients, oxygen, systemically circulating mediators and inflammatory cells can only enter defect site via newly formed capillaries [16–18]. When the defect is bridged by mineralized tissue, last healing phase, the remodeling phase, takes place. Again, growth factors and cytokines such as TGF, BMPs, and TNF-α stimulate pre-osteoclasts to differentiate to osteoclasts and to resorb newly formed bone. Then, osteoblasts synthesize new osteoid and secrete calcium phosphate for mineralization. During remodeling, number of cells and organic bone portion decrease while bone density and stability increase. Newly formed bone lamellae follow mechanical load and, finally, trabecular bone replaces undirected woven bone [19].
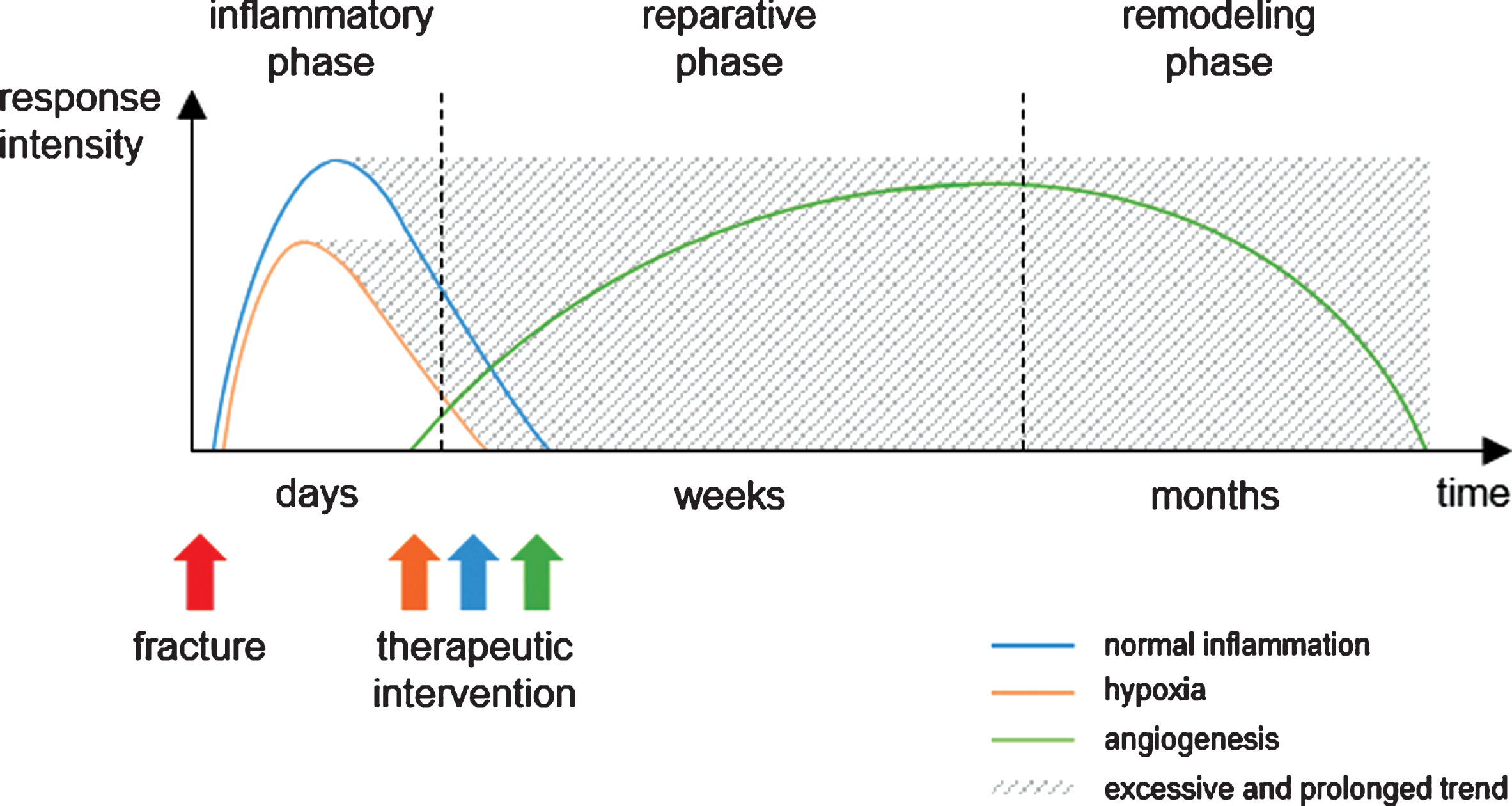
Hypothetical timeline of the essential processes during fracture healing. In the course of bone healing after fracture, inflammation and hypoxia immediately occur within days, whereas angiogenesis starts at a later time point. If hypoxia and inflammation become excessive, prolonged and chronic, deviating from the normal course, therapeutic intervention with anti-inflammatory, pro-angiogenic, and other agents to regulate these processes is necessary to ensure optimal healing (modified according to Casanova and coworkers as well as Namas and coworkers [191, 192]).
Due to increasing body of knowledge on early and late events of bone healing there is an increasing interest in novel osteoinductive drugs for adjuvant therapeutic approaches. Experimental and clinical data suggest promising effects of growth factors as well as anti-resorptive and anabolic agents [8, 20–22]. In order to guarantee osteoinductive properties of delivered drugs or cells, biocompatible bone substitute materials have to support regeneration and restore physiological function. For this purpose, scaffolds and grafts can be functionalized with growth factors or cytokines by chemical modification, physical adsorption or encapsulation. Most frequently utilized bioactive molecules in bone regeneration are antibiotics and growth factors such as pro-angiogenic VEGF, immune-modulatory TGF-β, and osteogenic BMPs [23–26]. Instead of merely immobilizing bone regenerative factors, it appears attractive to incorporate mediator-secreting cells in bone grafts. Especially MSCs are of interest due to their regenerative potential. Endres and coworkers as well as Harada and coworkers implanted PLGA (poly-lactic-co-glycolic acid) scaffolds seeded with MSCs in critical-size femoral defects in rats or rabbits, respectively [27, 28]. This results in bridging of the defect by biomechanically stable mineralized bone suggesting osteogenic benefits from MSCs at the defect site, an effect mainly attributable to the release of paracrine factors by MSCs [29]. However, stem cell therapy is associated with ethical concerns and strict requirements for medical approval. Recent approaches focused on conditioned cell culture medium obtained from in vitro cultivation of MSCs. MSC culture supernatant contained angiogenic and osteogenic growth factors as well as cytokines and served as a mediator source for functionalization of tricalcium phosphate scaffolds. Implanted in rabbit calvarial defects, these grafts treated with conditioned medium enhanced new bone formation especially in early healing stage [30]. Additionally, anti-inflammatory, pro-angiogenic, anti-apoptotic, and cell-recruiting effects have been shown for implants coated with conditioned medium [31, 32].
Hyaluronic acid is a glycosaminoglycan with remarkable tunable mechanical properties and a highly tolerated structure, the latter due to its presence in ECM of all vertebrates. Hyaluronan has been reported as both a potential agent for bone regeneration and a drug delivery system in bone healing. In vitro application of hyaluronic acid resulted in enhanced expression of osteogenic markers in MSCs suggesting beneficial effects on bone formation [33]. This was confirmed by an in vivo study utilizing hyaluronan-coated implants in critical-size defects in rat femora [34]. A drug delivery system composed of hyaluronan hydrogels functionalized with growth factors induced formation of woven and lamellar bone in bone defects in rabbits [35]. So far, drug delivery systems lack ability to migrate to site of injury due to insufficient guiding processes. Furthermore, application of mediators in primary inflammatory healing stage results in either non-physiologically high concentrations of the applied factor or in fast degradation by immune cells like macrophages and neutrophils [36]. Therefore, new delivery strategies and systems are required enabling temporal specification as well as synergistic effects of growth factors. Covalent binding or crosslinking of biomolecules may result in protein denaturation, loss of bioactivity, or toxic residues leading to immune reactions and delayed healing [37]. To avoid these drawbacks, researchers focus on small molecules, which, due to their size, do not induce immune reactions but show stable bioactivity [38]. One of these small molecules with bone-related activity is FTY720, a sphingosine-1-phosphate analogue, with osteogenic and angiogenic activity. In critical-size tibial defects, FTY720-functionalized allografts enhanced bone density of newly formed bone and increased vessel formation [39]. The angiogenic potential of FTY720 is discussed in more detail elsewhere (see section ‘Agonistic targeting of lipid mediators’) [40]. Another small molecule with impact on bone healing is N-acetyl cysteine. This amino acid, immobilized on a collagen sponge, affected bone regeneration by inducing osteogenic differentiation of cells of the osteoblast lineage at defect site [41].
There is growing evidence that biological signaling molecules can be used to improve healing of critical-size bone defects, particularly if therapeutic agents such as growth factors, lipid mediators, and other promising molecules with therapeutic potential, are released in a targeted and controlled manner in different healing phases (Scheme 1). The following review systematically examines the hypothesis that targeted adjuvant therapies using immune- and inflammation-modulatory agents have the potential to enhance bone regeneration in critical-size bone defects and under systemic conditions that impair bone healing. Approaches focusing on pro-angiogenic agents and metabolism-modulatory compounds are discussed in detail elsewhere (Part II – Modulation of angiogenesis [40]; Part III – Further strategies for local and systemic modulation [42]).
A PubMed database search was performed in November 2018 using key words and phrases ‘agonists’, ‘anabolic’, ‘anti-resorptive’ ‘angiogenesis’, ‘antagonists’, ‘drugs’, ‘inflammation’, ‘inhibitors’, ‘local’, ‘small molecule compounds’, ‘systemic’ linked to the key words ‘critical bone defect’, ‘fracture’, and ‘healing’ by AND/OR as Boolean function.
Anabolic biological agents
The entire wound healing process requires interplay of many regulated aligned molecular mechanisms. Among others, cytokines and growth factors control these diverse processes including proliferation and differentiation [43]. Several strategies exist to enhance bone repair based on either anabolic treatment by inducing bone formation via osteoblasts or anti-resorptive approaches blocking bone resorption by osteoclasts [44, 45]. Currently, several anabolic biological agents, especially parathyroid hormone (PTH) analogs and BMPs are in common clinical use to improve fracture healing [20].
Parathyroid hormone (PTH)
PTH is a polypeptide hormone consisting of 84 amino acids and a potent natural anabolic agent regarding bone remodeling [21]. In general, PTH exerts both anabolic and catabolic effects on fracture healing depending on dose and administration. The anabolic influence on bone metabolism is mediated through several growth-promoting signaling pathways, whereas bone resorption and osteoclastogenesis occur after stimulation of RANKL (receptor activator of nuclear factor κB ligand) and inhibition of osteoprotegerin (OPG) expression. RANKL-mediated activation of IKK (inhibitor of kappa B kinase) – NFκB (nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells) signaling or PLC (phospholipase C) – NFAT (nuclear factor of activated T-cells) signaling leads to the transcription of proliferation-enhancing genes. It is known that continuous elevation of PTH levels provoke catabolic effects such as bone loss, while intermittent administration increases bone mass by activating bone formation [46–50]. The enhanced bone formation in the course of intermittent PTH application may be due to advanced osteoblast function and extended osteoblastic half-life based on suppressed apoptosis [51]. The main anabolic signaling, transmitted via G-protein coupled receptors (GPCRs), is subsequently associated with activation of adenylate cyclase (AC), protein kinase A (PKA), PLC and protein kinase C (PKC). This leads to phosphorylation of transcription factors like cAMP response element-binding protein (CREB) or further proliferation-enhancing target genes. Additional anabolic downstream signaling pathways of PTH include mitogen-activated protein kinase (MAPK) cascade, Wnt (Wingless-related integration site) signaling and ß-arrestin mediated extracellular signal-regulated kinase (ERK) signaling [21, 52–55]. Zhou and coworkers revealed a direct influence of PTH on osteoblast proliferation and differentiation via PKA – CREB signaling and Smad1/5/8 (small mothers against decapentaplegic homolog) – Runx2 (runt-related transcription factor 2) pathway leading to increased fracture healing [56]. The various capabilities to trigger diverse signaling pathways, all of which result in anabolic effects, demonstrate a potential benefit when being transferred to clinical use for the treatment of critical-size bone defects.
In various experimental and clinical studies, two amino terminal analogs of PTH (PTH 1 – 34 and PTH 1 – 84) have been extensively examined and showed improved bone formation as well as a shorter healing duration after intermittent application [8, 57–59]. Using a rodent model, Alkhiary and coworkers indicated a significantly accelerated bone healing in strength and stiffness after daily subcutaneous injection of 5 μg/kg to 30 μg/kg teriparatide (PTH 1 – 34) over three weeks [60]. Systemic administration of the active PTH fragment (PTH 1 – 34), which is approved by the FDA (Food and Drug Administration) for osteoporosis treatment, favored not only early callus formation but also exhibited positive effects on later stages of fracture healing [51]. Similar results have been demonstrated by Komatsubara and coworkers [61]. Consequently, PTH administration throughout all stages of fracture healing has a positive impact on bone repair outcome [8]. Enhancement of early fracture repair by PTH is based on the assumptions that PTH recruits and stimulates proliferation as well as differentiation of osteoprogenitor cells like chondrocytes and osteoblasts. This represents a critical step for initiating of fracture healing cascade [59]. Peichl and coworkers observed a reduced fracture healing time and an improved functional outcome in postmenopausal women suffering from osteoporosis after daily injection of 100 μg PTH 1–84 [62]. Intermittent application of PTH can elicit pleiotropic effects leading to an increase in number of osteoblasts or at least retardation of osteoblast apoptosis by stimulating transcription of pro-survival genes and inhibition of caspase activity [54, 63]. To shorten fracture healing time, it might be advantageous to combine systematically applied PTH with locally delivered agents such as BMPs due to possible synergistic effects [57].
Bone morphogenetic proteins (BMPs)
BMPs belong to the superfamily of TGF-β signaling molecules. These cytokines are able to stimulate proliferation, differentiation and chemotaxis of osteoprogenitor cells as well as angiogenesis resulting in enhanced bone formation [64]. Throughout all stages of fracture repair, BMP family members show different time-dependent expression rates [65]. BMP-2, for instance, displays high expression level within the first 24 hours after a bone defect and, therefore, seems to be essential in setting up bone repair process during early stages of regenerative fracture healing [66]. In a clinical context, BMP-2 should be applied therapeutically during primary inflammatory phase to achieve optimal healing process [8]. However, it is evident that BMP-2 is not only necessary for initiating of inflammation immediately after fracture but is also important of healing process at subsequent time points in order to enhance bone mineralization and stiffness [67]. Furthermore, BMP-3, -4, -7 and -8 exhibit distinct expression patterns in later stages of fracture healing linked to bone formation and remodeling. On the contrary, constitutive expression of BMP-5 and -6 during first three weeks of fracture repair indicates a regulatory role for these growth factors over the entire healing period [68]. Generally, BMPs perform their pleiotropic anabolic effects by inducing either Smad1/5/8-dependent canonical or Smad-independent signaling pathways such as MAPK cascade after binding to their receptors [69–72]. In addition, Baron and coworkers pointed out a complex cross-talk between BMP and Wnt signaling, which can be “synergistic or antagonistic, depending on the cellular context” [55]. Since several members of the BMP family exert individual functions during healing progression of critical-size bone defects, they represent important therapeutic targets.
In clinical trials, (recombinant) BMP-2 and -7 have been intensively examined. Accelerated fracture healing has been observed only in a part of the studies summarized by Virk and coworkers applying recombinant human (rh) BMP-2 (1.5 mg/ml rhBMP-2/absorbable collagen sponge) [21]. In addition, in some investigations an undesirably increased infection risk has been observed during BMP-2 therapy. Furthermore, Lee and coworkers examined the potential of BMP-2 in bone regeneration by inserting a hybrid scaffold, consisting of heparin sulfate and collagen loaded with low dose rhBMP-2 into rodent critical-size femoral defects [73]. BMP-2 loaded scaffolds increase bone formation. Several clinical studies cited in Cecchi and coworkers as well as in Virk and coworkers, depicted a shortened healing period and an enhanced fracture healing after BMP-7 therapy administrating about 3.5 mg BMP-7 once [21–69]. Taken together, BMP-7 seems to be more promising compared to BMP-2 concerning its safety profile and observed side effects. However, due to short half-life of the growth factors therapeutic effect is limited and requires both sustained and local delivery of BMPs [10]. Still controversial is, whether the FDA approved rhBMP-2 or -7 therapy improves bone healing in a greater extent than autologous bone grafting [8–74, 75]. Consequently, dual delivery of osteogenic and angiogenic agents like BMP and VEGF specifically released from carriers or scaffolds is another attempt in terms of enhanced fracture regeneration. Patel and coworkers showed improved bone repair upon simultaneous release of 2 μg BMP-2 and 12 μg VEGF in a rodent cranial critical-size defect model suggesting a synergistic effect [76]. Combination of BMP and VEGF enhanced differentiation of MSCs to osteoblasts by activating MAPK cascade or Wnt signaling pathway [77]. Moreover, Kempen and coworkers created a scaffold consisting of BMP-2-loaded microspheres surrounded by VEGF-loaded gelatin hydrogel [24]. In vivo, the hydrogel initially released a large burst of VEGF, whereas BMP-2 was released continuously. Dual compound release promoted bone formation in a rodent femoral defect model. However, increased fracture healing after combined treatment compared to single treatment with BMP-2 is still controversial. Possible issues like release system, loading dose as well as growth factor ratio and release kinetics of utilized cytokines should be taken into consideration [78]. Moreover, recombinant BMPs, even though showing preferable osteoinductive qualities, exhibit some disadvantages concerning clinical use. Applicability is limited with regard to a short half-life and requirement of high or multiple doses. In advance, BMP production is expensive and an optimal carrier matrix remains to be found [8, 21]. Difficulties in therapeutic application of common anabolic biological agents listed above accentuate necessity to develop new treatment approaches.
Adjuvant drug-assisted bone healing
Modulation of inflammation
Fracture healing is a multilayered procedure requiring tangled interactions on cellular and molecular levels between physical and chemical factors as well as diverse signaling pathways [79]. In order to achieve a successful fracture healing both inflammation and angiogenesis go along with essential stages of bone regeneration (Fig. 2) [40], with each bone healing phase being determined by defined key mediators [80–83]. Initially, local inflammatory reaction regulated in length and magnitude is essential for activating healing pathways, in turn being crucial for successful fracture repair [84, 85]. The proceedings during inflammation are essential for efficient and successful bone regeneration [86]. Acute inflammatory response occurs immediately after injury inducing important repair cascade and lasts naturally about one week in human, whereby the inflammatory reaction is resolved via feedback mechanisms [87–90]. For example, high expression level of inflammatory cytokines, such as IL-6, is measured within first four days post-fracture, whereas expression of cytokines shifted after day four in favor of bone remodeling [91]. However, IL-6 signaling is essential for fracture healing [92]. Initial phase of bone regeneration is characterized by low oxygen tension and recruitment of neutrophils to fracture site. This cell population is followed by macrophages having an essential function in the course of fracture healing via further secreted cytokine mediators [93, 94]. Additionally, other key events like proliferation and differentiation of MSCs to osteoprogenitor cells such as chondrocytes and osteoblasts take place [9, 95]. Based on above-mentioned cells and cytokines decisively involved in fracture healing, immunomodulation is an important and promising approach to improve bone repair [95]. At an early stage of osteogenesis, interaction or cross-talk between pro-inflammatory macrophages and other cells involved in healing process seems to initially accelerate bone formation due to cyclooxygenase–2 (COX-2)- and prostaglandin (PG)-mediated events [96]. If the brief essential acute inflammation, which is generally regulated and damped by reparative cytokines to establish a normal homeostatic state, becomes chronic and detrimental due to an unbalanced raised or prolonged inflammatory reaction it leads to increased bone resorption and decreased bone formation. In that case, fracture healing is at least delayed [84, 97–100]. Therefore, smart modulation of inflammatory processes provides a promising strategy to optimize treatment of critical-size bone defects [44, 82, 101]. Possible intervention may occur in terms of controlled inflammation mediators or adjustment of involved relevant enzymes.
Role of prostaglandins in bone healing
PGs being produced by enzymes called COX are the main lipid mediators in managing pain and inflammatory processes, especially during early phases of fracture healing [102, 103]. Starting from arachidonic acid (AA), PGs are formed in a three-step reaction [104]. However, rapid degradation of PGs by dehydrogenases may occur in vivo resulting in a short half-life of these mediators. Within first two weeks after fracture, PGs are substantially synthesized by osteoblasts [105]. Inhibition of PG production or function at least delays healing process [106]. PGs show an impact on bone formation as well as on bone resorption, while prostaglandin E2 (PGE2) seems to be the dominant eicosanoid regulating BMP und RANKL expression [105, 108]. Forced RANKL expression and associated reduction of antagonistic OPG in osteoblastic lineage mediates catabolic impact on bone. Thereby, activated osteoclasts accelerate bone resorption [103, 110]. Accordingly, activation of distinct signaling pathways leading to raised transcription of target genes favoring bone resorption further promotes bone loss. For instance, Kim and coworkers showed an enhanced matrix metalloprotease (MMP) expression as a result of PGE2 initiated cAMP (cyclic adenosine monophosphate) – PKA signaling [111]. Essential signaling of PGs for bone metabolism and remodeling proceeds stimulation of growth and differentiation via GPCR-mediated downstream signaling pathways (Fig. 3). After binding to one out of the four main receptor types, further signaling molecules like AC, PKA, and the transcription factor NFAT are stimulated by PG action [112, 113]. Especially the main GPCR types EP2R and EP4R (prostaglandin receptors) promote anabolic effects whereas EP4R is also known to be the most pro-angiogenic one [114]. Other frequently used signaling pathways by various receptor types include activation of phosphatidylinositol-3-kinase (PI3K), proteinkinase B (Akt) and MAPK cascade resulting in enhanced proliferation [115, 116]. Moreover, Wnt signaling is also a present PG-stimulated signaling pathway. Binding of a complex composed of Axin and glycogen synthase kinase-3ß (GSK-3ß) to the GPCR leads to its subsequent inactivation. Therefore, ß-catenin is relieved of the protein complex and after cytosolic accumulating initiates transcription of proliferation-enhancing target genes (Fig. 3) [117, 118]. The other two prostaglandin receptors EP1R and EP3R are thought to mobilize intracellular calcium and reduce second messenger cAMP [109].
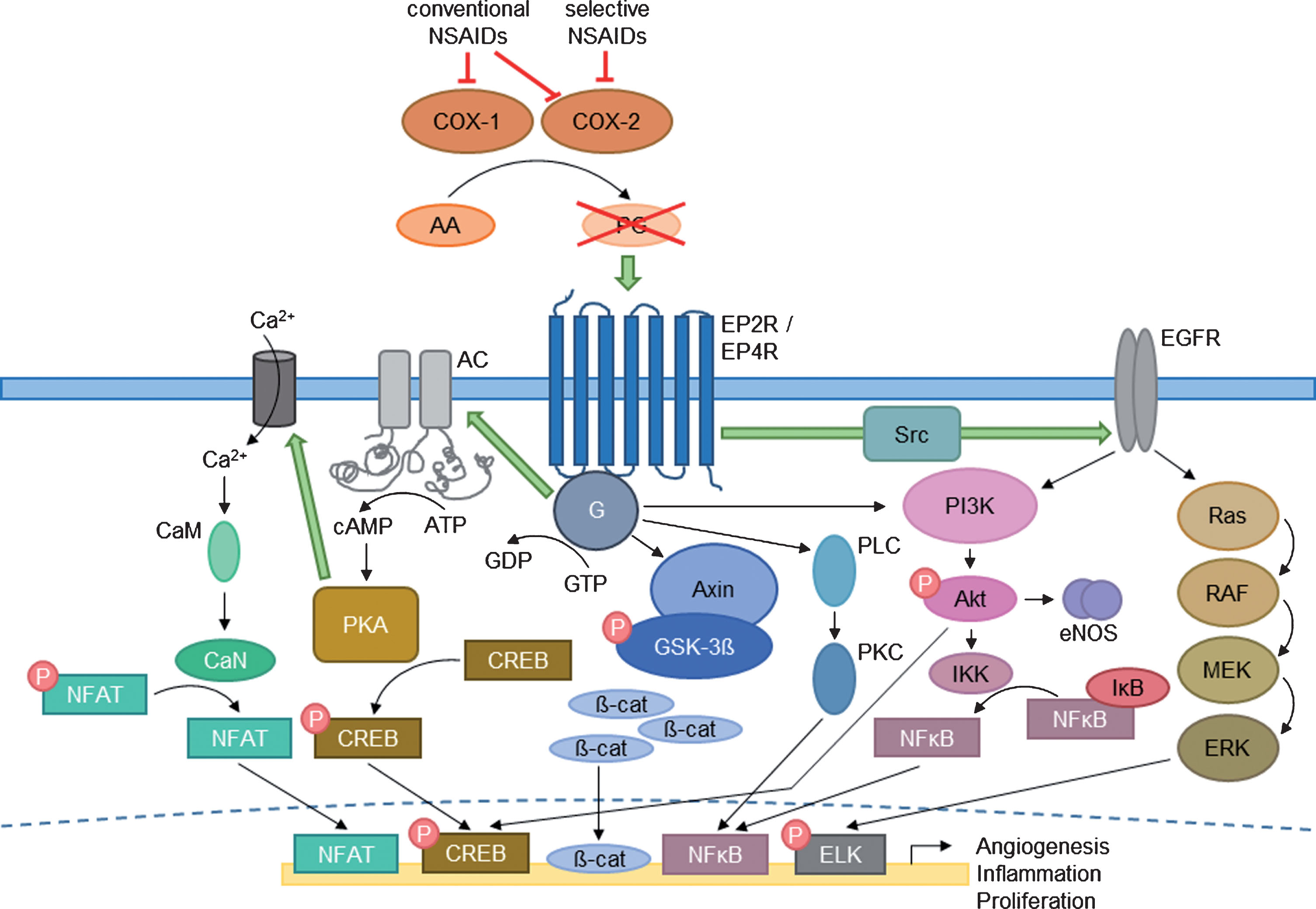
Anabolic signaling pathways of prostaglandins. Depicted are common downstream signaling pathways of PGs after conversion from AA via COX. Signaling includes G-protein (G) coupled prostaglandin receptors (EP2R/EP4R) and further activated adaptors such as AC – PKA – Calmodulin (CaM) – Calcineurin (CaN), PLC – PKC, PI3K – Akt – eNOS and IKK. Downstream signaling also activates MAPK cascade including Ras, rapidly accelerated fibrosarcoma (RAF), mitogen-activated protein kinase kinase (MEK), and ERK as well as a large part of Wnt signaling (Axin – GSK-3β). The several mediators enable transcription factors like NFAT, CREB, ß-catenin, and NFκB to trigger transcriptional processes of certain target genes resulting in enhanced angiogenesis, proliferation, and inflammation. The impact of several steroid types on the rate-limiting COX enzymes are illustrated in red (modified according to Blackwell and coworkers [109]).
Generally, healing of critical-size bone defects may benefit from anabolic properties of PGs (Table 1). Animal studies have proven both anabolic and catabolic effects on bone metabolism after systemic application of PGE2. Differences become evident regarding the way of administration. Continuously applied PGE2 causes loss of bone mass partly due to enhanced matrix degradation by activated MMPs, whereas intermittent application favors bone formation recognizable on the basis of promoted chondrogenesis and osteoblast activity [103, 109]. Nevertheless, clinical application is still controversial due to short half-life and biphasic effects of PGs on osteoblasts and osteoclasts. The last is dependent on applied animal model, dosage as well as administration type and frequency to achieve the optimal initiated signaling cascade [119]. In order to reduce undesired systemic side effects of PG therapy, including diarrhea and lethargy, selective EP2R and EP4R agonists mimicking favorable effects of PGE2 came into focus of research for locally enhanced fracture healing [103, 120–123]. Li and coworkers, Kyllönen and coworkers as well as Graham and coworkers reviewed several EP2R and EP4R agonists [22, 125]. A selective EP2R agonist, named CP-533,536 (3-pyridyl sulfonamide derivative [126]), displayed enhanced bone formation and strength in various animal models after multiple administrations. Moreover, described positive effect on fracture healing has been also shown by applying the EP4R agonists ONO-4819 [120] (heptanoic acid derivative) and CP-734,432 [127] (lactam analog of PGE2 [128]), both being able to activate already mentioned common signaling pathways such as PI3K and MAPK cascade [114]. However, results of animal studies imply that EP4R agonists are more potent in inducing bone formation [110]. Further, Kamolratanakul and coworkers investigated a new strategy to further improve bone regeneration by dual delivery of 100 μg selective EP4 agonist ONO-AE1-437 (butanoic acid derivative) and 0.5 μg BMP-2 being released gradually and locally from a hydrogel in a murine critical-size calvarial defect model [121]. The authors demonstrated significantly induced bone formation after applying drug combination once resulting from therapeutically beneficial synergistic interplay of the two agents. This highlights a new hydrogel-based approach for treatment of critical-size bone defects. Furthermore, recent studies indicate that novel EP1R antagonists may favor fracture healing, which could also be a promising approach in the future [129].
Summary of in vitro and predominantly in vivo studies regarding the effect of promising inflammation-modulatory drugs on bone metabolism
COX-1 and -2 are globular enzymes accelerating the rate-limiting conversion of AA to PGs at the beginning of the PG synthesis [112]. Generally, COX-1 being constitutively expressed by osteoblasts and osteoclasts maintains physiological homeostasis as a so-called “housekeeping enzyme” [105, 130]. However, COX-1 activity does not seem to exert critical functions during the complete healing process but synthesized PGs still cooperate in a way during fracture healing [86, 132]. On the contrary, COX-2 expression is rapidly induced and regulated locally and thereby coordinates pain management and inflammation processes [104, 133]. In osteoblasts, PTH, among other factors, probably induces expression of COX-2 via cAMP – PKA – NFAT signaling [134]. A bone fracture activates COX-2 immediately and COX-2 is the dominating and essential COX isotype regarding bone repair, especially in early stages of the healing process. Inhibition of COX-2 activity is detrimental on osteogenesis and angiogenesis resulting in impaired fracture healing [9, 135]. Involvement of COX-2 in early osteogenesis stimulation, more precisely in osteoblast differentiation and maturation, leads to positive impact on fracture healing due to resultant pro-angiogenic PG production [112, 137]. In later stages of bone formation, COX-2 modulates osteoblast differentiation and maturation via adjusted gene expression of genes like Runx2 and osterix [107, 138–141]. Gerstenfeld and coworkers demonstrated a raised COX-2 expression throughout the first two weeks of healing process [142]. An impairment of the COX-2 activity in that specific period has resulted in restricted or at least delayed fracture repair. After three weeks, expression rate has weakened and returned to a basal activity. Importance of COX-2 function in initial healing of critical-size bone defects caused scientific research concerning enhancement of COX-2 activity [86]. Rundle and coworkers increased COX-2 concentration at local fracture site by overexpressing COX-2 using a retroviral vector system [143]. Thereby, they detected both an accelerated fracture healing and a faster bone formation confirming the essential role of COX-2 in bone repair. Nevertheless, if natural resolving of initial inflammation is incomplete, clinical intervention with appropriate agents to dampen inflammatory response in order to prevent a hyperergic or chronical inflammatory reaction and ensure an optimal healing process is required.
Inflammation-modulatory agents
Non-specific non-steroidal anti-inflammatory drugs
Generally, non-specific non-steroidal anti-inflammatory drugs (NSAIDs) are drugs commonly used in pain management after any injury or disease [86, 136]. NSAIDs include diverse compounds such as aspirin, ibuprofen, paracetamol and further ones. These drugs inhibit both COX isoforms equally by competing with AA substrate for binding [104, 109]. Inhibition of inducible COX-2 elicits anti-inflammatory effects due to reduced PG production. Effects of NSAIDs on bone metabolism depend on initiation and treatment duration. Nevertheless, observed impact of these drugs is mainly negative because of impaired bone formation based on decreased PG secretion being crucial, especially in the first phase of healing. However, application of NSAIDs at the late stage of fracture healing might lead to an increased risk of nonunion [144]. Therefore, only a specific timeframe for NSAID treatment during fracture healing seems to be available. On the contrary, repression of constitutive COX-1 leads to adverse side effects in kidneys and gastrointestinal tract based on altered physiological homeostasis [131, 146]. Several NSAIDs are reviewed by Chen and coworkers regarding bone and soft tissue healing [147]. According to Boursinos and coworkers, indomethacine is the most frequently investigated non-specific NSAID [131]. The authors summarized a number of animal studies (Table 1) applying about 1–2 mg/kg/day and 2–10 mg/kg/day indomethacine to mice and rats or rabbits. All experiments resulted in impaired and delayed bone repair, reduced mechanical properties due to a decrease in bone strength, and interfered blood flow, regardless of whether administration was short- or long-term, even when applied locally. It also became evident from these studies that the earlier the drug was administered post-operatively, the greater the resultant negative inhibitory effect was. On the contrary, investigations of therapeutic impact of ibuprofen provided controversial results. A study applying 30 mg/kg/day ibuprofen to mice did not determine any negative impact on mechanical properties during bone repair, whereas other groups showed an inhibitory effect due to a reduction in callus mass. Kidd and coworkers continuously treated rats with 30 mg/kg/day ibuprofen and pointed out a negative impact on bone resorption as well as formation during remodeling phase [148]. In addition, O’Conner and coworkers indicated a delay but no inhibition of fracture healing due to periodic COX-2 activity, when multiple administrations of short-acting NSAIDs are arranged with interruptions of about 4 h because intermediate COX-2 activity limits negative effects, which compares favorably with long-acting drugs [149]. Varying degrees of no impact to inhibitory effects of ibuprofen application are likely to be dose- and administration-dependent with the majority of studies showing delayed bone healing [108].
Selective COX-2-inhibitors – COXIBs
Selective COX-2 inhibitors, also called COXIBs, such as drugs like celecoxib, diclofenac, or meloxicam are a new generation of NSAIDs with improved efficacy and safety regarding reduced adverse impact on gastrointestinal tract [104, 136]. Basic structure of most synthetic COXIBs consists of a diaryl-substituted heterocycle comprising a mono- or bicyclic core structure [150–155]. Similar to non-specific NSAIDs, outcome of COXIBs with regard to impaired bone healing varies dose- and duration-dependent from minor to no impact on fracture repair (Table 1) [138]. For instance, both short- and long-term treatment with diclofenac delayed fracture healing in rodents, having a tibial defect [156]. Certainly, selective COXIBs elicit adverse effects on a smaller extent compared to non-selective NSAIDs implying that the more selective the inhibitors are the less negative outcome occur during the healing process [105–141]. Therefore, COXIBs seem to be more suitable for clinical administration. De Vasconcelos Gurgel and coworkers indicated a decreased fracture healing rate after continuous administration of 3 mg/kg selective COX-2 inhibitor meloxicam in a rodent model [139]. Impairment is potentially based on interference of PG production blocking their anabolic and angiogenic effects or at any rate altering local signal transduction [142]. As a consequence, essential proliferation- and differentiation-promoting signaling pathways are inhibited such as the important canonical Wnt – ß-catenin signaling demonstrated by Nagano and coworkers [117]. The authors detected an impaired osteoblastogenesis in vitro after celecoxib addition based on degradation of a transcription factor and inhibited expression of Wnt signaling target genes. Suppression of Wnt signaling and resultant osteoblast growth leads to attenuated fracture healing. Further, Endo and coworkers showed that short-term administration of selective NSAIDs causes at least a delay of the healing process, in particular when applied during early healing phases [157]. This has also been shown by Simon and coworkers [158]. The authors applied celecoxib in a dose range of 2 mg/kg to 4 mg/kg in a rodent model and detected an impaired fracture healing if administration took place during the first two weeks of bone repair. Beyond that, celecoxib administration before fracture or after the critical initial healing phase has caused no delayed or impeded fracture healing. Specifically, short-term application of high selective COXIBs in low therapeutical doses in a later stage results in minor and reversible inhibitory effects [131–141]. Short-term treatments did not influence outcome of the healing process significantly due to a rapid recovery of PG levels after canceled application reversing the inhibition [43, 159]. In addition to inhibition of osteoblastic proliferation and differentiation through impaired PG production, Moreno-Rubio and coworkers demonstrated an effect on osteoclasts upon long-term treatment with the selective COX-2 inhibitor celecoxib [160]. One possible reason for this could be inhibition of local PGE2 accumulation, which further suppresses RANKL expression, and, concomitantly, stimulation of osteoclasts finally impeding resorptive signals. Janssen and coworkers investigated influence of COXIBs on bone repair from another point of view [161]. The authors determined not only an impaired osteogenesis but also a reduced chondrogenic differentiation. This leads to attenuated fracture repair based on alterations in the growth plate. Moreover, Reuben and coworkers as well as Wickerts and coworkers illustrated not only the negative impact on bone repair but also adverse effects on cardiovascular system such as myocardial infarction and stroke based on inhibited prostacyclin activity [109, 162–164].
Comprising, several reviews summarized a wide range of animal and clinical studies revealing time- and duration-dependent impact of NSAIDs on bone healing being mainly negative especially during early fracture healing [43, 165–167]. Impairment is based on inhibited proliferation and differentiation of osteoblasts probably based on attenuated PG synthesis [168]. These effects depend on drug specificity in a dose- and duration-dependent manner, whereas a greater delay in fracture healing was observed due to treatment with non-selective NSAIDs compared to COXIBs [142]. The adverse effects became clear through reduced mechanical strength, stiffness, bone density, and blood flow [108, 169]. Indeed, discontinued NSAID administration resumes normal fracture healing processes reversing the above-mentioned negative impact. However, fundamental delay of fracture healing by use of COX-2 inhibitors should be taken into consideration. Treatment should be avoided in the early inflammatory phase and in patients with preshaped delay in healing processes such as occurrence of trauma-related sepsis or comorbidities, among others [107, 171]. In addition, an alternative pain management should be introduced if delayed bone repair is an acute issue [106].
Corticosteroids
Corticosteroids such as cortisone, prednisone or dexamethasone inhibit phospholipases and thus hinder AA release from membrane resulting in further inhibited PG synthesis [172]. These steroids elicit anti-inflammatory as well as immunosuppressive effects. Among others, patients with rheumatoid arthritis, asthma, and organ transplantation receive systemic steroid therapy. However, adverse effects in the context of continuous corticosteroid administration are well known. These include for example osteoporosis and an enhanced fracture risk due to delayed growth factor production [137–173]. Pountos and coworkers summarized a variety of in vivo investigation regarding the impact of above mentioned corticosteroids on fracture healing [167]. Outcome varies from no inhibitory effect to retardation of bone healing depending on animal model, duration, and drug dose. Generally, steroids act detrimental on bone repair due to inhibition of osteoblastogenesis and induction of apoptosis via amplified reactive oxygen species production resulting in prolonged healing time, impaired bone strength, and reduced callus formation [131, 175]. Even administration of corticosteroids over a period longer than one week reduces bone healing [172]. Compared to systemic administration that inhibits healing process, administration of low doses of corticosteroids may promote wound healing and microcirculation [176, 177]. In therapeutic use, corticosteroids cause rapid bone loss within the first six months of applied therapy due to forced bone resorption. Afterwards, bone resorption rate remains at a lower but continuous level based on insufficient bone formation [173, 179].
A certain class of corticosteroids, the glucocorticoids, bind to glucocorticoid receptors and thereby reduce cell replication as well as differentiation of osteoprogenitor cells [175, 181]. Glucocorticoid receptors are nuclear receptors altering gene expression after ligand-binding induced dimerization and seem to take part during late fracture healing phase [182–184]. Glucocorticoid administration inhibits osteoclastogenesis as well as osteoblastogenesis, while for the latter, monomer activity is already sufficient as inhibition proceeds via suppressed cytokines [185]. Further, steroid administration promotes apoptosis of osteoblasts but not of osteoclasts. This leads to a decreased number of osteoblasts and maintained quantity of osteoclasts due to suggested prolonged life span of mature osteoclasts [178]. These persistent osteoclasts are probably responsible for early rapid bone resorption [186]. Prohibited osteoclast apoptosis potentially results from glucocorticoid-based increased RANKL expression in line with decreased expression of decoy receptor OPG. In long-term, these steroids further raise collagenase activity leading to matrix degradation and increasing bone loss [180]. However, application of short-acting glucocorticoids in low doses does not appear to have a significant adverse effect on bone metabolism [187].
By using the synthetic glucocorticoid dexamethasone, Wagner and coworkers showed an inhibition of transcription factor hypoxia-inducible factor-1 (HIF-1) being essential for fracture healing related processes [40–182]. Further, HIF-1 inhibition prevents downstream transcription of HIF-1 target genes like VEGF resulting in reduced bone formation. Therefore, long-term administration of glucocorticoids alters proliferation-enhancing signal transduction leading to a decrease in bone mineralization and strength. Yao and coworkers showed that these effects are due to impaired gene expression of target genes regulating the above-mentioned processes [188]. Beyond this, expression of certain genes such as Dickkopf-related protein and sclerostin inhibiting Wnt signaling is significantly increased diminishing bone formation [42]. The authors also demonstrated an improvement of bone mass when applying the anabolic PTH or a bisphosphonate coincidently. Thereby, PTH leads to a reduced expression of Wnt antagonists further promoting signaling to enhance bone formation while bisphosphonates support bone mineralization. In addition to growth factor- or Wnt-mediated signaling pathways MAP and PI3 kinases are also inhibited. Reduced bone formation and mineralization is observed even when glucocorticoids are not applied systemically [179–189]. Corticosteroids have been subject of animal studies many years ago. Waters and coworkers reviewed some investigations in which 10–25 mg/kg/day cortisone have been administered to rabbits resulting in decreased callus formation and retardation of healing process [190]. On the contrary, application of 8–20 mg/kg/day cortisone to rats did not significantly influence healing process but also had detrimental effects in some investigations. This gives rise to the assumption that impact of corticosteroids is dependent on dose, model species, and examined fracture type. Further, Waters and coworkers investigated the effect of 0.15 mg/kg prednisone applied systemically to rabbits [190]. The authors detected an impaired fracture repair due to decreased callus size and density.
Studies have shown that inhibition of COX-2 and consequent decreased PG production results in impaired bone repair, especially when applying corticosteroids and non-selective NSAIDs at early healing stages. In conclusion, we suggest that short-term administration of a high selective COX-2 inhibitor in low therapeutic dose is suitable to modulate inflammatory reactions applied after the first two weeks of fracture healing. Nevertheless, since above mentioned effects, regarding fracture healing of critical-size defects are mainly based on direct delivery of several agents without biomaterial support, it should be taken into consideration that different impacts may occur when drugs are released locally via biomaterials with the delivery being controlled in terms of optimal dosage and treatment duration.
Conclusion
The treatment of bone defects of critical-size is very difficult due to the complexity of cellular interactions and microenvironmental conditions, especially in presence of relevant comorbidities. Therefore, new therapeutic strategies to improve bone regeneration are being developed. In this regard, use of inflammation-modulating compounds is considered very critically. The innate immune response and inflammatory processes are an essential component of the healing process and poorly balanced interventions quickly lead to undesired effects. Each case, therefore, must be considered individually and type of medication, possible co-medications, dosage, start and duration of therapy have to be optimized. However, development of new active substances as well as discovery and characterization of new drug targets will significantly advance this therapy concept. Regarding selective inhibition of COX-2 promising approaches comprise, e.g., use of hybrid or bi-functional compounds like nitric oxide releasing COXIBs, which are discussed in detail elsewhere [40].
Author contributions
All authors have jointly conceived and written this review article. All authors read and approved the final manuscript.
Conflicts of interest
The authors declare that they have no conflict of interest. The founding sponsors had no influence on the conception of the article, the interpretation of literature data or the conclusions drawn, and in the decision to publish this review article.
Footnotes
Acknowledgments
We wish to apologize to those researchers whose works have not been mentioned due to restrictions in space. The authors are thankful to the Deutsche Forschungsgemeinschaft (DFG) for supporting this work within the Collaborative Research Center Transregio 67 “Functional Biomaterials for Controlling Healing Processes in Bone und Skin - From Material Science to Clinical Application“ (CRC/TRR 67/3). The authors also thank the Helmholtz Association for funding a part of this work through the Helmholtz Cross-Programme Initiative “Technology and Medicine – Adaptive Systems”.