
Abstract
BACKGROUND:
Myocardial inflammation mediated by toll-like receptor 4 (TLR4) plays an active role in myocardial ischemia/reperfusion (I/R) injury. Studies show that heat shock protein 90 (HSP90) is involved in ischemic postconditioning (IPostC) cardioprotection. This study investigates the roles of TLR4 and HSP90 in IPostC.
METHODS:
Rats were subjected to 30 min ischemia, then 2 h reperfusion. IPostC was applied by three cycles of 30 s reperfusion, then 30 s reocclusion at reperfusion onset. Sixty rats were randomly divided into four groups: sham, I/R, IPostC, and geldanamycin (GA, HSP90 inhibitor, 1 mg/kg) plus IPostC (IPostC + GA).
RESULTS:
IPostC significantly reduced I/R-induced infarct size (40.2±2.1% versus 28.4±2.4%; P < 0.05); the release of cardiac Troponin T, creatine kinase-MB, and lactate dehydrogenase (191.5±3.1 versus 140.6±3.3 pg/ml, 3394.6±132.7 versus 2880.7±125.5 pg/ml, 2686.2±98.6 versus 1848.8±90.1 pg/ml, respectively; P < 0.05); and cardiomyocyte apoptosis (40.3±2.2% versus 27.0±1.6%; P < 0.05). Further, local and circulating IL-1β, IL-6, TNF-α, and ICAM-1 levels decreased; TLR4 expression and nuclear factor-KB (NF-κB) signaling decreased; and cardiac HSP90 expression increased. Blocking HSP90 function with GA inhibited IPostC protection and anti-inflammation, suggesting that IPostC has a HSP90-dependent anti-inflammatory effect.
CONCLUSION:
HSP90 may play a role in IPostC-mediated cardioprotection by inhibiting TLR4 activation, local and systemic inflammation, and NF-kB signaling.
Keywords
Introduction
Acute myocardial infarction is one of the leading causes of death and disability worldwide. Coronary reperfusion—the prompt opening of occluded blood vessels and the rapid recovery of coronary flow—is at present the definitive treatment for salvaging myocardium following ischemic injury. However, this sudden restoration of coronary flow has deleterious consequences and can lead to lethal ischemia/reperfusion (I/R) injury. Currently, I/R injury and its treatment are topics of intense investigation. Although the precise mechanism responsible for I/R injury remains unclear, it is widely accepted that the innate immune and inflammatory responses mediated by toll-like receptor 4 (TLR4) signaling are crucial to the development of heart damage [1–3].
TLR4 is an important class of protein molecules involved in the natural immune response and is mainly responsible for regulating endogenous or exogenous inflammation [4]. Apart from immune cells, TLR4 can also be expressed in cardiomyocytes and vascular endothelial cells. Once activated, TLR4 triggers a cascade of cellular signals that culminates in the activation of nuclear factor-KB (NF-kB), which leads to the production of related proinflammatory cytokines—such as tumor necrosis factor α (TNF-α), interleukin (IL)-1, IL-6, and intercellular cell adhesion molecule 1 (ICAM-1) [5]. The production of these cytokines could act as positive feedback to further activate NF-kB. Due to this induction of proinflammatory cytokines and the infiltration of chemokines, TLR4 is vital in the regulation of the immune response and the activation of the inflammatory response during myocardial I/R injury [6]. As such, TLR4/NF-kB signaling shows promise as a therapy for I/R injury.
Ischemic postconditioning (IPostC)—short and repeated ischemia/reperfusion treatment before reperfusion—can significantly reduce myocardial injury [7]. To date, IPostC cardioprotection has been demonstrated from animal models to clinical trials, though the mechanisms involved have not yet been elucidated [8–11]. Increasing evidence shows that reducing the inflammatory response during reperfusion is closely related to the cardioprotective effect of IPostC. However, its precise mechanisms that underly anti-inflammation are not yet fully understood, and few studies have examined the role of TLR4/NF-κB interaction in myocardial IPostC.
Heat shock protein 90 (HSP90), one of the most abundant and conserved molecular chaperones, is activated by cellular stress and is important for protein folding and maintaining stability. Previous studies have shown that HSP90 plays an important role in ischemic preconditioning and post conditioning to protect the myocardium against I/R injury [12, 13]. However, the potential mechanisms involved in HSP90 cardio protection during IPostC remain elusive, and its effect on TLR4-mediated inflammation during myocardial reperfusion is not well known.
In the present study, the possible involvement of HSP90 in the inhibition of TLR4 activation, inflammation, and NF-kB signaling during IPostC was tested. A rat model of myocardial I/R was established to examine the effects of IPostC on infarct size, cardiomyocyte apoptosis, and the release of serum cardiac Troponin T (cTnT), creatine kinase-MB (CK-MB), lactate dehydrogenase (LDH), and inflammatory cytokines. The mRNA and protein levels of HSP90, TLR4, NF-kB, TNF-α, IL-1β, IL-6, and ICAM-1 were examined. The effects of the HSP90 inhibitor geldanamycin (GA) on IPostC were also investigated.
Materials and methods
Animals
Male Sprague-Dawley rats (weighing 260 to 300 g, n = 60) were obtained from Guangxi Medical University (Nanning, China). The rats were housed at 25±2°C, with luminosity cycles of 12 h light/12 h dark, and allowed free access to food and water. The study protocol was performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and was approved by the institutional ethics committee (2017 KY-E-089).
Myocardial ischemia/reperfusion model
The rats were anesthetized with pentobarbital sodium (50 mg/kg, intraperitoneal) and mechanically ventilated with oxygen-enriched room air using a rodent ventilator. A cannula was inserted into the left carotid artery for monitoring arterial pressure, and electrocardiogram leads were placed to measure heart rate. The thorax was opened at the fifth left intercostal space. A 6–0 polyproline ligature, along with a snare of a small plastic tube occluder, was placed under the left coronary artery (LCA). Myocardial ischemia was achieved by tightening the ligature around the plastic tube and confirmed by cyanosis of the myocardium and ST-segment elevation. After 30 min of ischemia, the ligature was untied to allow reperfusion for 2 h. At the end of reperfusion, the rats were euthanized. Blood samples and the anterior wall of the left ventricular myocardium were obtained for further analysis.
Experimental protocol
The rats were randomized into four groups (n = 15 for each group): (1) the sham group, in which the ligature was placed under the LCA without occlusion for 150 min; (2) the I/R group, on which ischemia was conducted for 30 min and reperfusion for 2 h; (3) the IPostC group, where three cycles of 30 s of reperfusion were alternated with 30 s of ischemia immediately at the onset of reperfusion; and (4) the IPostC + GA group, on which the HSP90 inhibitor GA (Sigma-Aldrich, St. Louis, MO, 10 mg/ml in dimethyl sulfoxide, further diluted to 500 mg/ml with 0.9% saline, 1 mg/kg, intraperitoneal) was administered 10 min before IPostC.
Determination of myocardial infarction size
Separate experiments were performed on the four groups of five animals each. After reperfusion, the LCA was religated and Evans blue dye (3%) was injected into the inferior vena cava to identify the area at risk. The stained heart was sliced into 2 mm slices, and incubated in 1 percent 2,3,5-triphenyltetrazolium chloride (TTC, Sigma-Aldrich) at 37°C for 15 min to delineate the size of the infarction. The infarct size (as a percentage of the area at risk) and the area at risk (as a percentage of the left ventricle) were quantified using digital imaging software (Image-Pro Plus version 6.0; Media Cybernetics, Bethesda, MD).
Activities of cTnT, CK-MB, and LDH in serum
At the end of reperfusion, 5 ml blood samples were collected and centrifuged at 2000 rpm for 10 min. The supernatant was stored in liquid nitrogen. The levels of cTnT, CK-MB, and LDH were determined with commercial kits (Roche Diagnostics, Mannheim, Germany) and an automatic analyzer 7600 (Hitachi, Tokyo, Japan).
Activities of inflammatory factors in serum
Serum levels of IL-1β, IL-6, TNF-α, and ICAM-1 were determined using an enzyme-linked immunosorbent assay kit (CUSABIO, Wuhan, China) according to the manufacturer’s instruction. Results were analyzed spectrophotometrically at 450 nm of absorbance and expressed as μg/ml of serum.
TUNEL (Terminal deoxynucleotidyl transferase-mediated dUTP-X nick end labeling) staining
Separate experiments were performed to determine cardiomyocyte apoptosis (n = 5 for each group). Paraformaldehyde-fixed heart tissue blocks were incubated with proteinase K, embedded in paraffin, and sectioned at 5μm using a microtome. Apoptotic cells were identified with a TUNEL detection kit (Roche Diagnostics) according to the manufacturer’s instruction. Staining was observed using a fluorescent microscope (excitation wavelength was 450∼500 nm and emission wavelength was 515∼565 nm), and 5 areas randomly selected from at least 100 cells were scored. Nuclei with brown staining indicated TUNEL-positive cells. The apoptotic index was determined as the percentage of TUNEL-positive cells versus the total number of myocytes. Image J software was used for quantitative analysis of apoptotic cells.
RNA extraction and quantitative reverse transcriptase-polymerase chain reaction (qPCR)
Total RNA from the heart samples were extracted and purified using the Trizol reagent kit (Invitrogen, CA, USA). Reverse transcription reactions were performed using a PrimeScript™ RT reagent kit (Takara Bionic, Otsu, Japan). qPCR was performed using SYBR Standard qPCR Mix (TaKaRa) on an ABI Prism 7500 system. The reaction conditions were 95°C for 30 s, then 40 cycles of 95°C for 5 s and 60°C for 30 s. GAPDH was used as the control. The mRNA levels were analyzed using the 2∧(–ΔΔCT) method. The following primers were used for detection:
Western blotting
Freshly frozen myocardial tissue samples were homogenized in a RIPA buffer (Beyotime, Shanghai, China). Protein concentration was determined using a BCA assay. Equal amounts of proteins were separated using sodium dodecyl sulfate-polyacrylamide gel (12%) and transferred to nitrocellulose membranes blocked with non-fat milk (5%) in Tris-buffered saline-Tween 20 (150 mM NaCl, 20 mM TriseHCl, 0.1% Tween 20, pH 7.4). The PVDF membranes were then exposed to HSP90, TLR4, NF-kB, IL-1β, IL-6, TNF-α and ICAM-1 rabbit monoclonal antibodies (all at a dilution of 1 : 1000, Abcam, Cambridge, UK). They were then subsequently incubated with peroxidase-conjugated secondary antibody (IRDye800CW goat anti-rabbit IgG (H + L) antibody, dilution 1 : 3000, LICOR, USA) and chemiluminescence substrates before being exposed to radiographic film. Results were analyzed quantitatively using ImageJ software (NIH, Bethesda, MD).
Statistical analysis
The data were expressed as the mean±standard deviation. Multiple group means were analyzed by a one-way analysis of variance, and a Student-Newman-Keuls (SNK) q-test was used for selected groups. P values less than 0.05 were considered statistically significant. All statistical analyses were performed using SPSS 23.0 (SPSS Inc, Chicago, IL, USA).
Results
Sixty rats were included initially in the study, of which two were later excluded: one in the I/R group because of ventricular fibrillation, and the other in the IPostC + GA group because of cardiogenic shock during reperfusion. The results presented are for the remaining 58 rats.
Expression of HSP90 in myocardium with IPostC
To determine whether HSP90 is involved in IPostC, alterations in the mRNA and the protein expression of HSP90 in myocardial tissue were identified. As shown in Fig. 1, IPostC significantly increased the mRNA and protein levels of HSP90 compared with the I/R group. Inhibiting HSP90 activity with the selective HSP90 inhibitor GA negated the increased expression of HSP90.
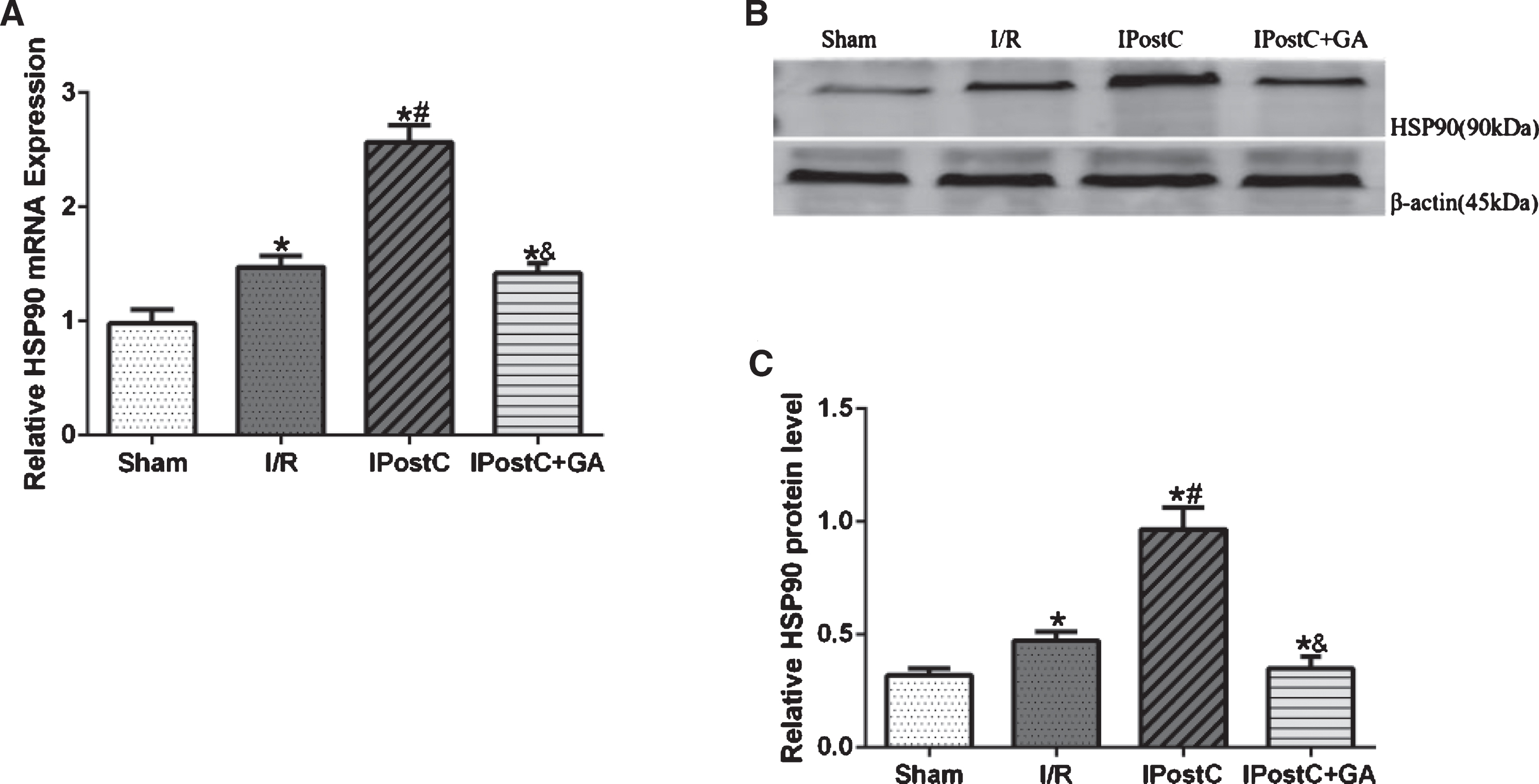
Effects of GA and IpostC on HSP90 mRNA and protein expression. (A) The mRNA levels of HSP90 were measured using qPCR in different groups. (B) Representative Western blots showing the expression of HSP90. (C) Graphic representation of HSP90. The data are presented as the mean±standard deviation. *P < 0.05 versus sham group; #P < 0.05 versus I/R group; n = 5 for each group.
As shown in Fig. 2, no myocardial infarction was observed in the sham group. The area of risk in the left ventricular was similar among all treatment groups. Infarct size in the I/R group averaged (40.2±2.1)%, and was significantly reduced in the IPostC group [(28.4±2.4)%, P < 0.05]. The administration of GA, a HSP90 inhibitor, significantly increased the infarct size to (36.7±2.4)% compared to the IPostC group (P < 0.05). These results indicate that GA offsets the reduction of infarct size observed with IPostC.
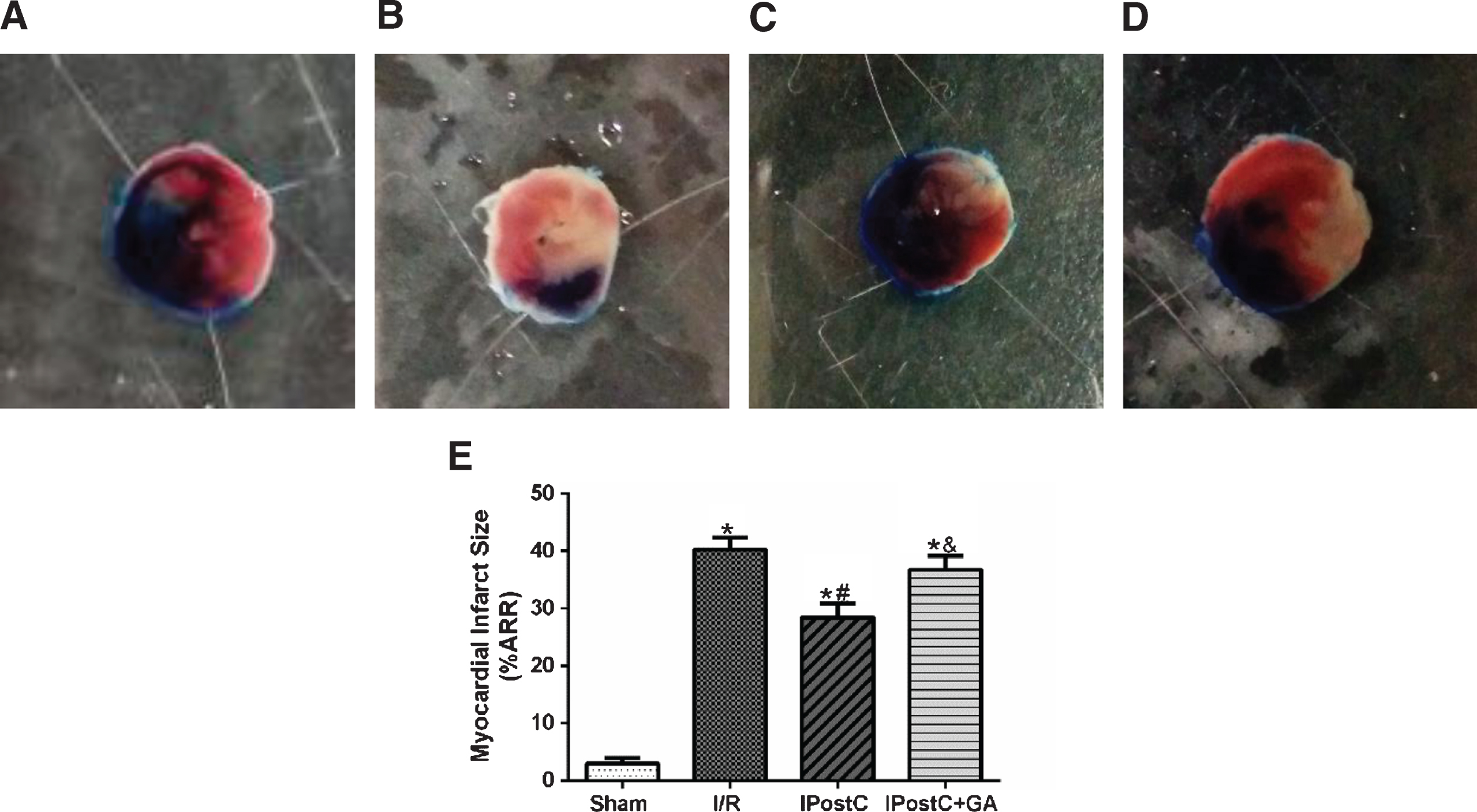
Effects of GA and IpostC on myocardial infarct size after cardiac I/R injury. (A) Sham group, (B) I/R group, (C) IPostC group, (D) IPostC + GA group, (E) The results presented in a bar graph are the mean±standard deviation. *P < 0.05 versus sham group; #P < 0.05 versus I/R group; &P < 0.05 versus IpostC group; n = 5 for each group.
Serum cardiac enzymes such as cTnT, CK-MB, and LDH, which were considered in the present protocol, are typical markers of myocardial injury. Their activities were significantly higher in the I/R group than in the sham group (Table.1). The IPostC group had significantly lower levels of cTnT, CK-MB, and LDH compared with the I/R group. The application of GA repressed IPostC-induced reductions in cTnT, CK-MB, and LDH release (see Table.1).
Role of HSP90 in cardiomyocyte apoptosis during IPostC
As shown in Fig. 3, the number of apoptotic cardiomyocytes identified by TUNEL staining was markedly lower in the IPostC group (27.0±1.6%) compared with the I/R group (40.3±2.2%). Treatment with GA significantly attenuated the anti-apoptosis effects of IPostC, suggesting that GA restrained the apoptotic- and cardiomyocyte-limiting effect of IPostC.

Effects of GA and IpostC on apoptosis after myocardial I/R. (A) Sham group, (B) I/R group, (C) IPostC group, (D) IpostC + GA group. Apoptotic cardiomyocyte nuclei appear brown stained, whereas TUNEL-negative nuclei appear blue. Mean apoptotic index was counted in each group (A–D), results presented in a bar graph (E) are the mean±standard deviation. Arrow indicates TUNEL-positive cells. *P < 0.05 versus sham group; #P < 0.05 versus I/R group; &P < 0.05 versus IpostC group; n = 5 for each group (×200).
Compared to the IPostC group, the I/R group expressed higher serum levels of IL-1β, IL-6, TNF-α, and ICAM-1 (see Table 2), suggesting that the I/R-related inflammatory response was significantly suppressed by IPostC. The IPostC group treated with GA did not differ from the I/R group, indicating that HSP90 assisted the anti-inflammatory effect of IPostC.
Levels of CK-MB, cTnT and LDH in serum
Levels of CK-MB, cTnT and LDH in serum
Data are presented as the mean±standard deviation. *P < 0.05 versus sham group; #P < 0.05 versus I/R group; &P < 0.05 versus IpostC group; n = 8 for each group.
Levels of IL-1β, IL-6, TNF-α and ICAM-1 in serum
Data are presented as the mean±standard deviation. *P < 0.05 versus sham group; #P < 0.05 versus I/R group; &P < 0.05 versus IpostC group; n = 6 for each group.
Similar to the results obtained for the serums, both the protein and mRNA levels of IL-1β, IL-6, TNF-α, and ICAM-1 were significantly lower in the IPostC group than in the I/R group. Treatment with GA before IPostC increased IL-1β, IL-6, TNF-α, and ICAM-1 expression at both the mRNA and protein levels (see Fig. 4). This suggests that IPostC could induce anti-inflammatory effects via HSP90 activation.

Effects of GA and IpostC on TNF-α, IL-1β, IL-6, and ICAM-1 mRNA and protein expression. (A) The mRNA levels of TNF-α, IL-1β, IL-6, and ICAM-1 were measured using qPCR in different groups. (B) Representative Western blots showing the expression of TNF-α, IL-1β, IL-6, and ICAM-1. (C) Graphic representation of TNF-α, IL-1β, IL-6, and ICAM-1 expression. The data are presented as the mean±standard deviation. *P < 0.05 versus sham group; #P < 0.05 versus I/R group; &P < 0.05 versus IpostC group; n = 5 for each group.
TLR4 signaling plays a pivotal role in the progression of myocardial I/R by mediating the expression of proinflammatory cytokines [14]. To confirm whether the TLR4 signaling pathway was modulated by IPostC, the mRNA and protein expressions of the TLR4 and NF-kB pathways were examined. As shown in Fig. 5, the expression levels of these two factors were significantly lower in the IPostC group than in the I/R group, and the effects were counteracted by GA. These results indicate that IPostC protects the heart from I/R-induced inflammatory injury by restraining the TLR4 and NF-kB signaling pathways, and these effects relate closely to HSP90.
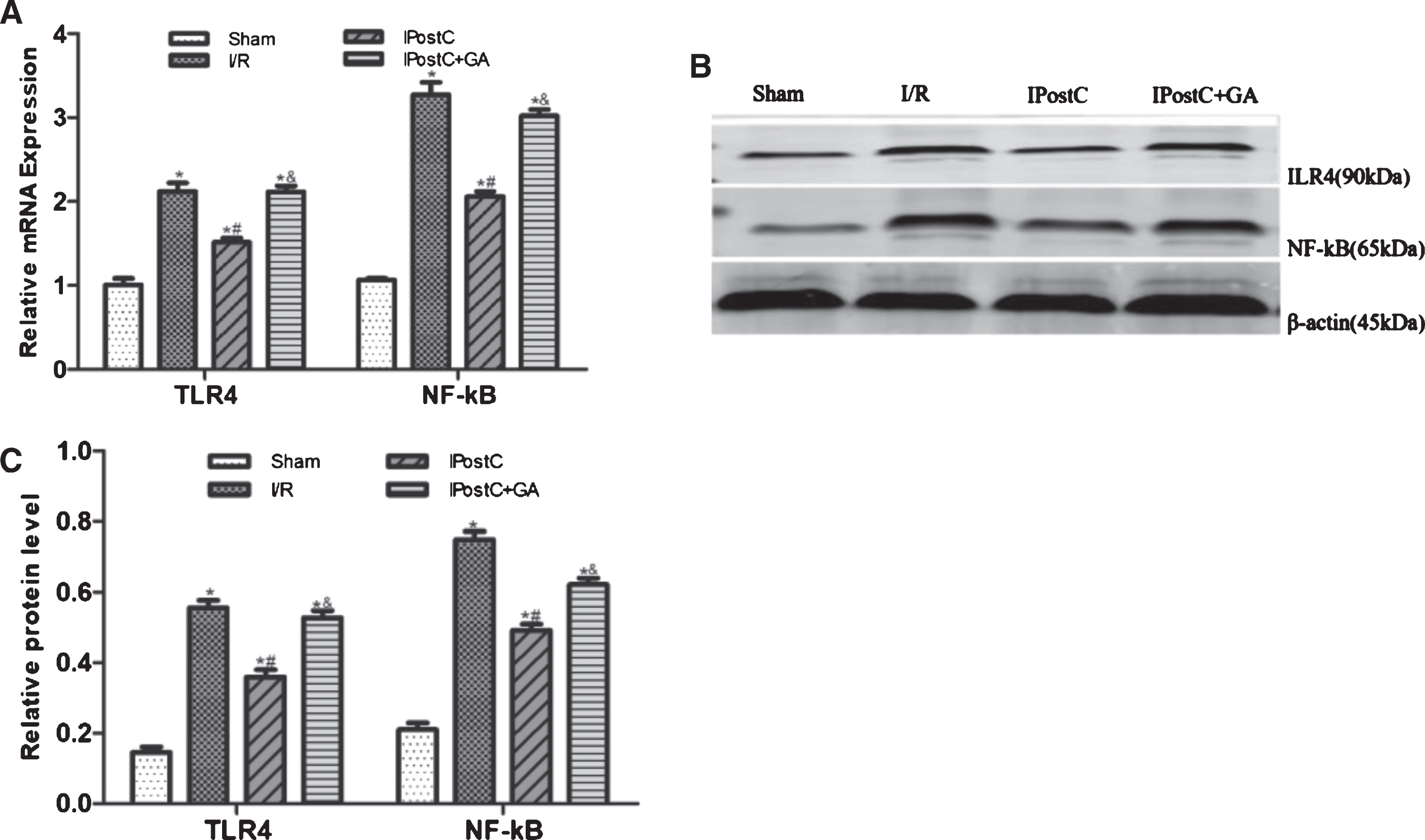
Effects of GA and IpostC on TLR4 and NF-kB mRNA and protein expression. (A) The mRNA levels of TLR4 and NF-kB were measured using qPCR in different groups. (B) Representative Western blots showing the expression of TLR4 and NF-kB. (C) Graphic representation of TLR4 and NF-kB expression. The data are presented as the mean±standard deviation. *P < 0.05 versus sham group; #P < 0.05 versus I/R group; &P < 0.05 versus IpostC group; n = 5 for each group.
The main finding of the present study was that IPostC inhibits I/R-induced TLR4 activation and inflammation in an HSP90-dependent manner. IPostC significantly increased the mRNA and protein levels of HSP90 and reduced I/R-induced TLR4 and NF-kB activation, local and systemic inflammation, infarct size, cardiomyocyte apoptosis, and the release of cTnT, CK-MB, and LDH. This study is the first to suggest that HSP90 is involved in IPostC-mediated cardioprotection against I/R-induced inflammatory injury, potentially by inhibiting TLR4 activation, local and systemic inflammation, and NF-kB signaling. The data reveal a novel mechanism involved in IPostC protection.
The accumulation of inflammatory cytokines affects the pathogenesis and process of myocardial I/R injury [15, 16]. Once stimulated, the inflammatory process tends to spread. The IL-1 and IL-6 families are proinflammatory cytokines released from necrotic cardiomyocytes or activated leukocytes, which can bind to receptors on the cell membrane and activate downstream signaling pathways to mediate ischemic myocardial damage [17, 18]. TNF-α can increase the expression of ICAM-1, which influences leukocyte trafficking in I/R injury [19]. Locally accumulated pro-inflammatory cytokines such as TNF-α, IL-1, and IL-6 can induce apoptosis or necrosis of reperfused myocardium. However, therapeutic strategies to improve I/R injury by inhibiting these cytokines have been shown to be effective in reducing cardiomyocytes apoptosis and infarct size [20–22]. In agreement with previous findings, this study’s results demonstrate that the number of apoptotic cardiomyocytes and the mRNA and protein levels of IL-1β, IL-6, TNF-α, and ICAM-1 are significantly elevated following myocardial I/R, but IPostC reverses this trend.
The regulatory mechanisms of inflammation in ischemic cardiomyocytes are complicated. Increasing evidence shows that pattern recognition receptors, especially TLR4, are the major initiators of the myocardial inflammation in myocardial I/R by activating NF-kB and producing inflammatory cytokines [23, 24]. Inhibiting TLR4 signaling pathways with the TLR4 antagonist suppresses NF-κB activation, attenuates proinflammatory cytokine expression, and reduces myocardial infarction [25]. There is also ample evidence that ischemic preconditioning attenuates myocardial I/R injury by inhibiting the activation of the TLR4/NF-kB signaling pathway [26, 27]. However, the exact role of the TLR4/NF-κB interaction in myocardial IPostC is unclear. The present study’s results demonstrate that IPostC simultaneously suppresses TLR4 activation and NF-κB signaling, supporting the notion that the inhibition of both TLR4 and NF-κB signaling may contribute to IPostC-induced cardioprotection. Still, the underlying mechanisms of these effects still need to be elucidated.
HSP90 is a highly conserved molecular chaperone that contributes to the correct folding and post-translational modification of proteins. The novel cellular function of this chaperone is that it can process a large number of client proteins, including TLR4 [28, 29]. Emerging studies suggest that the up-regulation of HSP90 is one of the main mechanisms for protecting the heart from I/R injury [30–32]. Although a large body of evidence suggests that HSP90 participates in IPostC cardioprotection, no study has addressed the link between HSP90 and TLR4 in IPostC. The present study assesses whether the chaperone HSP90 interacts with TLR4 in IPostC. The experimental results indicate that IPostC shows higher levels of HSP90 and lower levels of TLR4. However, the inhibition of HSP90’s function by GA prevents the inhibitory effect of IPostC-related TLR4, indicating that HSP90 is involved in the inhibition of TLR4 during IPostC. To the best of current knowledge, this is the first description of the interaction between HSP90 and TLR4 in IPostC, suggesting a possible mechanism for HSP90’s involvement in IPostC protection. Further in-depth study is required to provide more comprehensive information about the interaction between HSP90 and TLR4 in IPostC protection.
Geldanamycin is known as a potent small molecule inhibitor of the HSP90 function. GA binds to HSP90 N-terminal ATP binding site, inhibits HSP90 ATPase activity, affecting the dissociation of mature chaperone HSP90 complex and the degradation of HSP90 client proteins in proteasomes by E3 ligase. The mechanisms involved in proteasomal degradation of HSP90 client proteins and destabilization of HSP90 complexes are relatively well understood, but little is known about effects of GA on the modulation of HSP90 expression at the mRNA and protein level. Several studies indicated that GA could induce expression of many stress proteins, such as GRPs and HSP90, in a concentration-dependent and cell-specific manner [33, 34]. Several studies showed that 17AAG or 17DMAG (GA’derivatives) inhibited HSP90 gene expression in breast cancer and lung cancer [35, 36]. Smith et al. found that in the most sensitive cells (MEXF 276L, human melanoma cell lines) the expression of the target protein HSP90 was decreased upon treatment with either 17AAG or 17DMAG. The mode of depletion of HSP90 by 17AAG and 17DMAG in MEXF 276L cells was under investigation in their laboratories. It was, however, likely that reduction of HSP90 expression was related to an enhanced ubiquitinylation of HSP90 when bound to drug and thus its accelerated degradation in the ubiquitin-proteasome system [37]. While our experiments found GA reduced the expression of HSP90 in cardiomyocytes, the underlying mechanism is presently unresolved and will be subject to future investigations.
In conclusion, the present results indicate that HSP90 plays a central role in IPostC-mediated cardio protection by inhibiting TLR4 activation, local and systemic inflammation, and NF-κB signaling. The observations described in this study add to the knowledge system of IPostC-related cardioprotection against I/R injury and may be used to identify potential therapeutic targets.
Disclosure
The authors report no proprietary or commercial interest in any product mentioned or concept discussed in this article.
Footnotes
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (No. 81560068) and the Natural Science Foundation of Guangxi Province (No.2015GXNSFAA139198).