
Abstract
Hexokinase 2 (HK2) is a metabolic sensor that couples glycolysis and oxidative phosphorylation of mitochondria by binding to the outer mitochondrial membrane (OMM), and it also has been implicated in induction of apoptotic process by regulating the integrity of OMM. When HK2 detaches from the mitochondria, it triggers permeability increase of the OMM and subsequently facilitates the cytosolic release of cytochrome c, a major apoptosis-inducing factor. According to previous studies, a harsh microenvironment created by ischemic heart disease such as low tissue oxygen and nutrients, and increased reactive oxygen species (ROS) can cause cardiomyocyte apoptosis. Under these conditions, the expression of HK2 in heart significantly decrease and such down-regulation of HK2 was correlated to the increased apoptosis of cardiomyocytes. Therefore, prevention of HK2 down-regulation may salvage cardiomyocytes from apoptosis. MicroRNAs are short, non-coding RNAs that either inhibit transcription of target mRNAs or degrade the targeted mRNAs via complementary binding to the 3’UTR (untranslated region) of the targeted mRNAs. Since miRNAs are known to be involved in virtually every biological processes, it is reasonable to assume that the expression of HK2 is also regulated by miRNAs. Currently, to my best knowledge, there is no previous study examined the miRNA-mediated regulation of HK2 in cardiomyocytes. Thus, in the present study, miRNA-mediated modulation of HK2 during ROS (H2O2)-induced cardiomyocyte apoptosis was investigated. First, the expression of HK2 in cardiomyocytes exposed to H2O2 was evaluated. H2O2 (500 μM) induced cardiomyocyte apoptosis and it also decreased the mitochondrial expression of HK2. Based on miRNA-target prediction databases and empirical data, miR-181a was identified as a HK2-targeting miRNA. To further examine the effect of negative regulation of the selected HK2-targeting miRNA on cardiomyocyte apoptosis, anti-miR-181a, which neutralizes endogenous miR-181a, was utilized. Delivery of anti-miR-181a significantly abrogated the H2O2-induced suppression of HK2 expression and subsequent disruption of mitochondrial membrane potential, improving the survival of cardiomyocytes exposed to H2O2. These findings suggest that miR-181a-mediated down-regulation of HK2 contributes to the apoptosis of cardiomyocytes exposed to ROS. Neutralizing miR-181a can be a viable and effective means to prevent cardiomyocyte from apoptosis in ischemic heart disease.
1. Introduction
Ischemic heart disease is one of the leading causes of death worldwide, including both Western world and industrialized developing countries [1]. In dire situations such as ischemia, where supply of nutrients and oxygen are shut down, cardiomyocytes are subjected to a life-or-death decision, and various cell death mechanisms (i.e., apoptosis) are activated as a result [2, 3]. Resultant loss of cardiomyocytes in the myocardium further promotes the development of various heart diseases that cause the functional demise of the myocardium, ultimately resulting in heart failure. One of the major death-inducing agents of ischemic heart is reactive oxygen species (ROS) [4, 5]. ROS are a family of highly unstable and reactive oxygen containing molecules derived from O2 due to O2 metabolism [6]. The type of ROS include oxygen-derived free radicals, such as superoxide anion (O2•-) and hydroxyl radical (OH•), and non-radical molecules like hydrogen peroxide (H2O2) [7].
Ischemia (or impaired blood flow) can cause tissue injury and organ dysfunction, and the duration and severity of ischemia determines the reversibility of the injury [8, 9]. Since prolonged tissue hypoxia and the consequent depletion of cellular ATP inflict ischemic tissue injury, timely restoration of blood flow (reperfusion), that replenishes cellular ATP and restores ionic balance within the cell, can minimize the magnitude of the hypoxic insult, and even result in full restoration of heart function [10–12]. However, a sudden resupply of oxygen to ischemic tissue results in a paradoxical injury to the ischemic tissue, called a reperfusion injury, that is not incurred during the period of ischemia [13]. Based on the observation that such injury was dependent on the reintroduction of molecular oxygen and accumulated empirical evidence indicating an imbalance between the rate of generation of ROS and the tissue’s capacity to detoxify these harmful reactive species during reperfusion injury, the notion that highly reactive and unstable ROS mediate the reperfusion injury has been a prevailing dogma [14, 15].
In a molecular mechanistic point of view, excessive ROS induce oxidative damage to wide range of biological macromolecules and disrupt the integrity and function of cellular membranes including the outer mitochondrial membrane (OMM) [7, 16]. Since increased OMM permeability or mitochondrial outer membrane permeabilization (MOMP) can lead to the release of pro-apoptotic contents of mitochondria intermembrane space (IMS) such as cytochrome c initiating apoptotic signaling pathways [17], inhibition of MOMP may help to prevent the apoptotic cell death incurred by ROS. Another molecule plays an important role in the maintenance of OMM permeability is hexokinase 2 (HK2) [18]. Four different isotypes of HK (HK 1–4, or I to VI) comprise the HK family, and they are originally known to initiate the conversion of glucose to glucose-6-phosphate during glycolysis [19]. Particularly, mitochondria bound HK2 acts as a metabolic sensor that couples glycolysis to oxidative phosphorylation [20].
In addition to this catabolic enzymatic activity, HKs also have non-enzymatic activities involving the maintenance of the OMM permeability, inhibiting apoptosis [21]. Such anti-apoptotic function of HK2 is achieved by interacting with the voltage-dependent anion channel (VDAC) that facilitates the trafficking of small metabolites across the OMM [22, 23]. While HK2 bound VDAC interacts with anti-apoptotic Bcl-2 family members, preventing the accumulation of Bax/Bak proteins at the OMM [24], detachment of HK2 from VDAC causes Bax-induced cytochrome c release and subsequent apoptosis [25]. This HK2-mediated regulation of apoptosis was also reported in heart disease. According to a previous study, the expression of HK2, as well as the binding to mitochondria, significantly decreased following ischemia/reperfusion (I/R) injury compromising the heart function [26]. Therefore, preventing the down-regulation of HK2 in heart disease may improve the survival of cardiomyocytes by protecting cells from ROS-mediated apoptosis.
Regarding the negative regulation of HK2 in ROS-mediated cardiomyocyte apoptosis, noncoding RNAs such as microRNAs (miRNAs) are highly suspected to play a crucial role [27]. MiRNAs are approximately 22-nucleotide-long RNAs that negatively regulate gene expressions of corresponding target genes by either inhibiting messenger RNA (mRNAs) translation or degrading mRNAs [28]. In fact, miRNA-mediated down regulation of HK2 has been frequently demonstrated [29–31]. Based on these reports, it was hypothesized that HK2-targeting miRNAs facilitate the ROS-induced apoptosis of cardiomyocytes. Therefore, in the present study, the possibility of miRNA-mediated HK2 regulation during ROS-induced death of cardiomyocytes was investigated in vitro.
Materials and methods
Culture of rat cardiomyocytes and H2O2 treatment
The embryonic cardiomyocyte cell line H9c2 is commonly used to study heart disease including I/R injury [32]. H9c2 cell line (American Type Culture Collection) was cultured in high glucose-DMEM (GIBCO, Waltham, MA, USA) containing 10% FBS (Atlas Biologicals, Fort Collins, CO, USA) and 1% antibiotics (GIBCO). The cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2. For H2O2 treatment, 9M of stock H2O2 was added to the culture media at a final concentration of 100 μM to 600 μM.
Cell viability assay
To measure cell viability, CCK reagent (cell counting kit-8, Dojindo) was added to each well for a final concentration of 0.5 mg/mL and the cells were incubated at 37°C for 2 hours. The absorbance of the medium was measured at 450 nm using a microplate reader.
PI/Annexin V double staining for apoptosis
To determine the apoptosis of cells, the annexin V-FITC/propidium iodide (PI) apoptosis detection kit (BD Biosciences) was used. Briefly, cells were collected and re-suspended in 200 ml of buffered medium. Annexin V solution (10 ml) was added to the cell suspension, which was then incubated for 15 min in the dark at RT. Then, 5 μl of PI was added, and the cells were analyzed by flow cytometry (BD ACCURI C6 cytometer, BD Biosciences). Annexin V/PI double negative cells represented viable cells, annexin V positive/PI negative cells represented early apoptotic cells, and annexin V/PI double positive cells represented late apoptotic cells.
Tetra methyl rhodamine methyl ester (TMRM) staining
Mitochondrial membrane potential was determined by using a tetramethylrhodamine methyl esters (TMRM) fluorescent dye (Invitrogen) [33]. In healthy cells with functioning mitochondria, TMRM accumulates in active mitochondria with intact membrane potentials. Loss of the mitochondrial membrane potential decreases TMRM signal. To evaluate mitochondrial membrane permeability, the cells were loaded with 200 nM of TMRM for 30 minutes at 37°C in medium. TMRM signal was detected by a laser confocal microscopy with absorbance peak at 548 nm and emission peak at 574 nm.
Immunocytochemistry
The cells were cultured in 4-well slide chambers, washed twice with PBS, and fixed in 1% para-formaldehyde solution for 10 min. The cells were then washed twice with PBS before permeabilization using 0.1% Triton X-100 for 10 min. Next, the cells were blocked for 1 h in blocking solution (2% bovine serum albumin and 10% horse serum in PBS) and incubated with Cytochrome C antibody (Santa Cruz Biotechnology). FITC-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) were then used. Immunofluorescence was detected by a laser confocal microscopy (LSM710; Carl Zeiss).
Transfection of miRNAs and anti-miRNAs
Transfections of miRNA mimics and anti-miRNAs were performed using siLentFect™ Lipid reagent (Life Science Research). Mature specific miRNAs (Genolution Pharmaceuticals, Inc., Korea) were used at a final concentration of 100 nM. Anti-miRNAs was used at a final concentration of 50 nM. After 4 hours of incubation in a CO2 incubator at 37°C, the medium was changed to 10% FBS con taining DMEM.
Western blot
The cells were washed once in PBS and lysed in lysis buffer (Cell Signaling Technology) with protease and phosphatase inhibitor cocktail. Protein concentrations were determined using the BCA protein assay kit (Thermo Science). Proteins were separated in a SDS–polyacrylamide gel and transferred to the PVDF membrane (Millipore). After blocking the membrane with 0.1% Tris-buffered saline–Tween 20 (TBS-T, 0.1% Tween 20) containing 10% skim milk for 1 hr at room temperature, the membrane was washed twice with TBS and incubated with primary antibodies for overnight at 4 °C. The membrane was washed three times with 0.1% TBS-T for 5 min and then incubated for 1 hr at room temperature with horseradish peroxidase (HRP)-conjugated secondary antibodies. After extensive washing, the bands were detected by enhanced chemiluminescence reagent (ECL, Santa Cruz Biotechnology). The band intensities were quantified using NIH Image J version 1.34e software. The primary antibodies for HKII and β-actin were from Cell Signaling (28675) and Sigma (A1978), respectively.
Luciferase assay using the 3’UTR of HK2
The 3’UTR sequences of HK2 was amplified using primers with XbaI (forward) and EcoRI (reverse) endonuclease sites. The 3’UTR fragment was then cloned into the pmirGLO vector. HeLa cells were plated at a density of 1×105 cell/well in a 12 well plate, and then transfected with either pmirGLO control vector or pmirGLO vector with HK2 3’UTR using Lipofectamine LTX. After 48 hours, relative luciferase activity was measured by using Dual Luciferase assay kit (Promega) according to the manufacturer’s instructions. The Renilla luciferase was used for normalization.
Rat I/R injury model
All experimental procedures for animal studies were approved by the Committee for the Care and Use of Laboratory Animals, and performed in accordance with the Committee’s Guidelines and Regulations for Animal Care (CKU 01-2019-008). I/R-injury was produced in male Sprague-Dawley rats (200±50 g) by surgical occlusion of the left anterior descending coronary artery. Briefly, after induction of anesthesia with zoletil (0.8 ml/kg) and rompun (0.2 ml/kg), the rats were intubated, for ventilation (62 strokes/min, tidal volume 8–10 ml/kg). After intubation, the third and fourth ribs were cut to open the chest, and the heart was exteriorized through the intercostal space. The left coronary artery was then ligated 2–3 mm from its origin with a 6-0 prolene suture (Ethicon, Somerville, NJ, USA). Reperfusion was conducted after 1 hour of ischemia. For anti-miR181 treatment, microRNA (5 μg/head) and reagent mixture were prepared in 60 μl and injected from the injured region to the border using a Hamilton syringe (Hamilton Co., Reno, NV, USA) with a 30 gauge needle. Throughout the operation, animals were ventilated with 95% O2 and 5% CO2 using a Harvard ventilator (Holliston, MA, USA). Animals were sacrificed 2 weeks after the surgery for analysis.
Statistical analysis
Quantitative data were expressed as the means±S.E.M (standard error of measurement) of at least 3 independent experiments. For statistical analysis, one-way ANOVA with Bonferroni correction was performed using the OriginPro 8 SR4 software (ver. 8.0951, OriginLab Corporation, Northampton, MA, USA). A p value of less than 0.05 was considered to be statistically significant.
Results
H2O2 increased apoptosis by disrupting mitochondrial membrane potential
It has been reported that H2O2 induces apoptosis of cardiomyocytes [34]. In the present study, H2O2 was used to simulate the oxidative stress. According to the data, H2O2 decreased cell viability of cardiomyocytes in a concentration-dependent manner. To be specific, 300 μM or higher concentra-tions of H2O2 significantly decreased cell viability of cardiomyocytes after 6 hours of treatment (Figure 1A). Since 6 hours of treatment with 500 μM of H2O2 induced more than 20% of cell death of cardiomyocytes, this particular condition was use for further experiments.
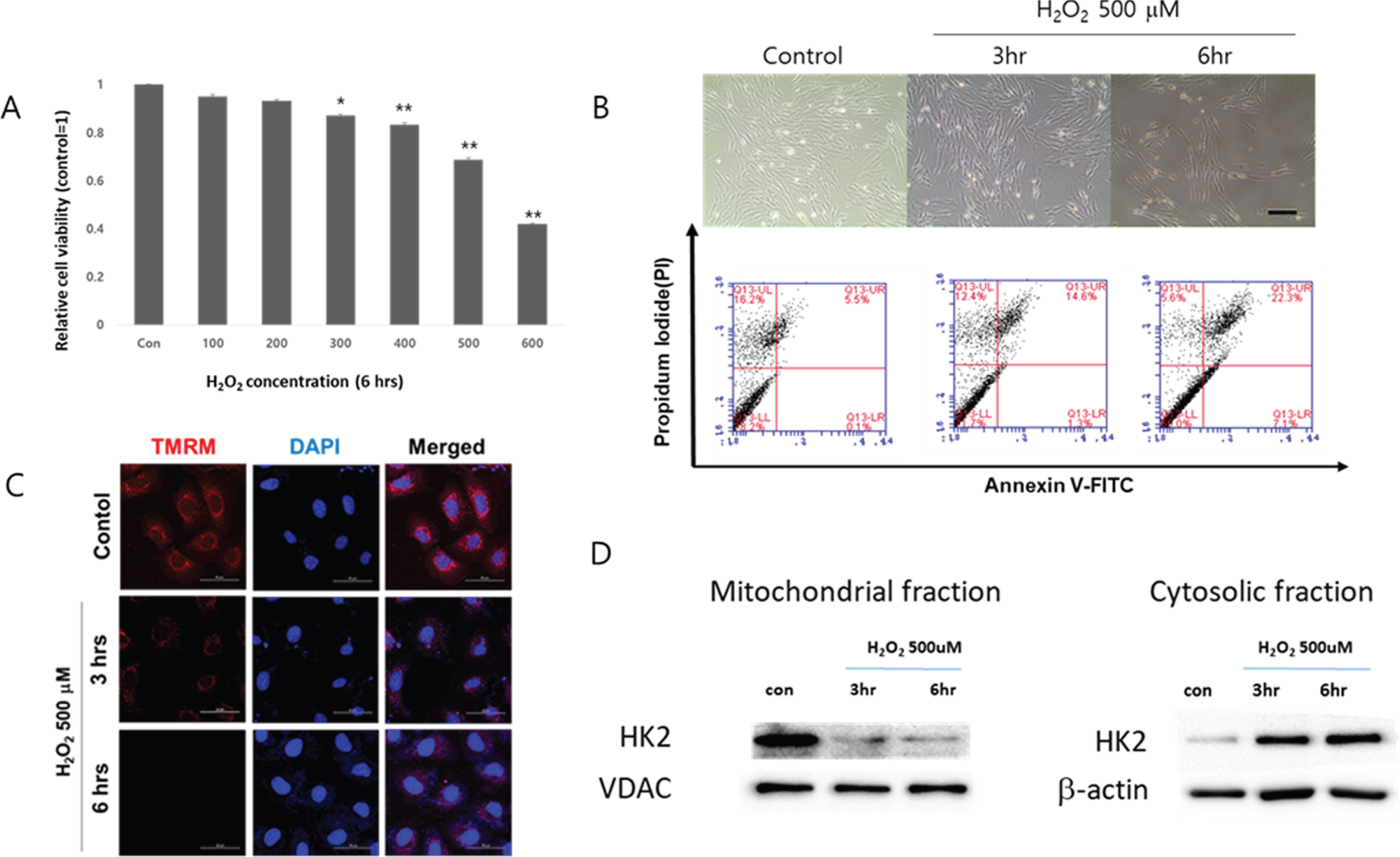
H2O2 increased apoptosis by disrupting mitochondrial membrane potential. (A) Cell viability of cardiomyocytes exposed to increasing concentrations of H2O2 for 6 hours. *p < 0.01, **p < 0.001 compared to untreated control. The quantitative data were expressed as the means±SEM of at least 3 independent experiments. Cell viability was determined by using CCK. (B) Upper panel: Morphological examination of H2O2 treated cardiomyocytes. The cells were exposed to 500 μM of H2O2 for up to 6 hours. Scale bar = 200 μm. Bottom panel: The percentage of cells undergoing apoptosis were determined by flow cytometry using PI/Annexin-V double-staining. (C) Mitochondrial membrane potential was determined by tetramethylrhodamine methyl esters (TMRM) staining. The cells cultured in a chamber slide were treated with 500 μM of H2O2 for up to 6 hours and then loaded with 200 nM of TMRM for 30 minutes at 37°C in medium. The fluorescent signal of TMRM was detected using a confocal microscopy. DAPI was used to stain nuclei. Scale bar = 50 μm. (D) Time dependent changes of mitochondrial and cytosolic expressions of HK2 in H2O2-treated cardiomyocytes. VDAC: voltage dependent anion channel, VDAC and β-actin are used as loading controls for mitochondrial and cytosolic fraction, respectively.
When the cardiomyocytes were treated with 500 μM of H2O2, cell detachment was obvious from 6 hours (Figure 1B, upper panel) and the percentage of the cells positively stained for both PI and anne-xin V, which indicates late apoptosis [35], increased suggesting the type of cell death inflicted by H2O2 includes apoptosis (Figure 1B, bottom panel). Intact mitochondrial membrane effectively accumulates TMRM [36]. TMRM staining indicated that H2O2 disrupted mitochondrial membrane potential in a time-dependent manner (Figure 1C), and the amount of HK2 in mitochondrial fraction time-dependently decreased, suggesting H2O2 increased detachment of HK2 from mitochondrial outer membrane (Figure 1D).
To identify miRNAs targeting HK2, 3 different miRNA-target protein prediction databases (Target-Scan, miRWalk, and miRNA.org) were used. According to these databases, 4 miRNAs were simultaneously predicted to target rat HK2 (miR-9, miR-139, miR-425, and miR-181) (Figure 2A). When cardiomyocytes were transfected with those 4 miRNAs, only miR-181 significantly decreased the expression of HK2 (Figure 2B). Therefore, miR-181 was selected as a primary candidate miRNA that may facilitate the expression of HK2 under oxidative stress. To further verify whether miR-181 directly regulates the expression of HK2, a luciferase assay using pmirGLO vector containing the 3’UTR of rat HK2 was conducted (Figure 2C). The luciferase assay are used to evaluate the effect of miRNA-dependent post-transcriptional regulation of target genes [37]. Transfection of 100 nM of miR-181 to cardiomyocytes significantly decreased the expression of luciferase gene linked to the 3’ untranslated region (3’UTR) of HK2 (pmirGLO-HK2) compared to the control (Figure 2D). This indicated that the expression of HK2 is directly regulated by miR-181.
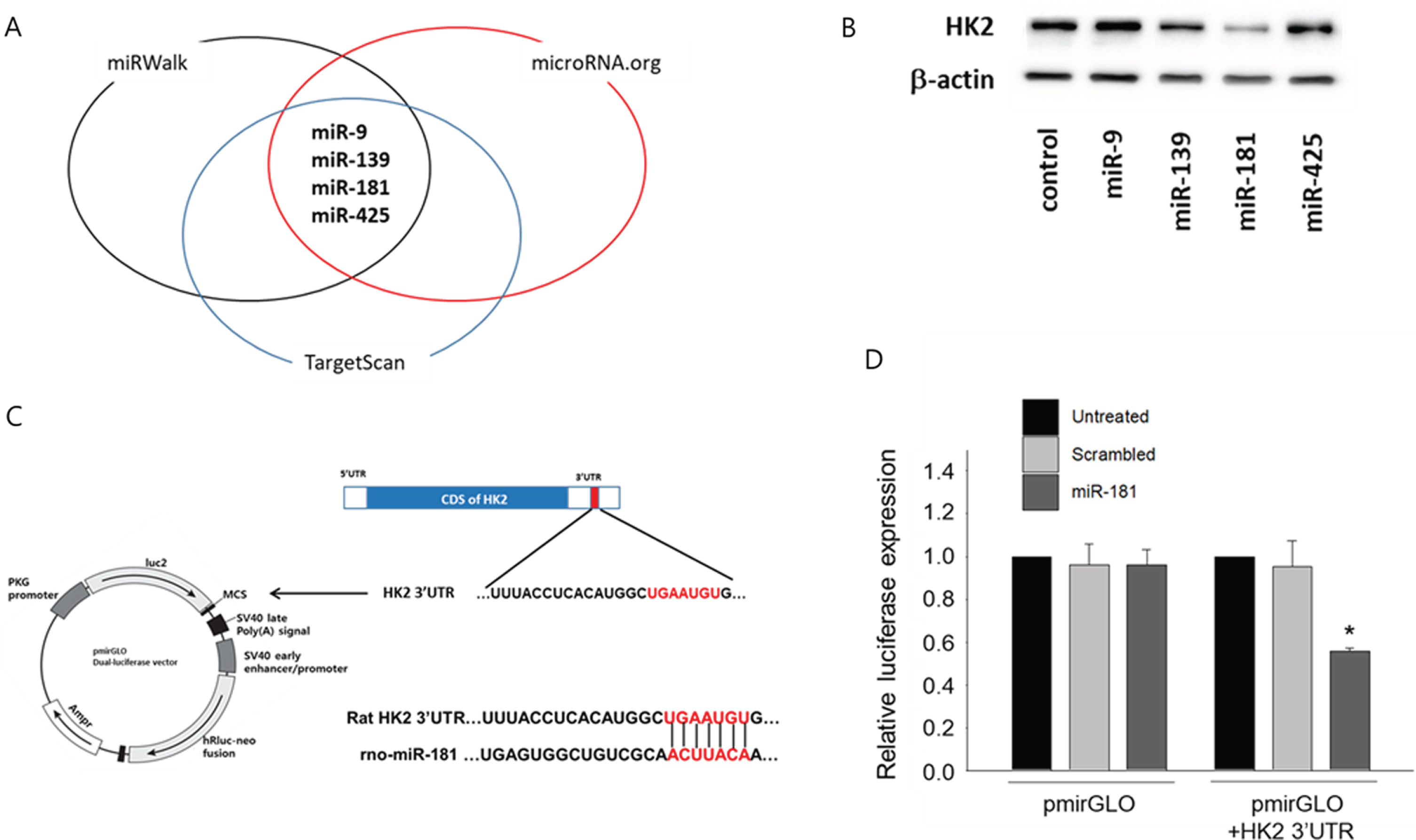
Identification of miR181 as miRNA targeting HK2. (A) Screening of candidate miRNAs targeting HK2. To identify miRNAs directly targeting HK2, in silico screening using 3 different miRNA-target gene prediction databases (miRWalk, TargetScan, and microRNA.org) were utilized. A total of 4 different miRNAs were predicted to target HK2. (B) Effect of candidate miRNAs on the expression of HK2. Cardiomyocytes were transfected with each candidate miRNAs (100 nM, each) and the expression of HK2 in total protein was evaluated by Western blot. (C) Construction of luciferase vector containing the 3’UTR sequences of HK2. The miR-181 binding sequence within the 3’UTR of HK2 was amplified using primers with XbaI (forward) and EcoRI (reverse) endonuclease sites. The 3’UTR fragment was then cloned into the pmirGLO vector. (D) MicroRNA-181 targets HK2. HeLa cells were plated at a density of 1×105 cell/well in a 12 well plate, and then transfected with either pmirGLO control vector or pmirGLO vector with HK2 3’UTR. To verify the effect of miR-181 on the expression of HK2, the cells were further treated with either scrambled miRNA or miR-181 (100 nM, each) for 48 hours. Relative luciferase activity was measured by using Dual Luciferase assay kit (Promega) according to the manufacturer’s instructions. *p < 0.05 compared to untreated control.
Since it was hypothesized that miR-181 contributed to the down-regulation of mitochondrial HK2 expression, endogenous miR-181 was neutralized using anti-miR-181 and its effect on mitochondrial HK2 was examined. When cardiomyocytes were exposed to H2O2 with or without miR-181 neutralization using anti-miR-181, mitochondrial HK2 expression was attenuated compared to H2O2 only treated group (Figure 3A). Upon morphological examination, anti-miR-181 apparently prevented cardiomyocytes from H2O2-induced detachment (Figure 3B) and H2O2-induced decrease of cardiomyocyte viability was significantly recovered by anti-miR-181 (Figure 3C), indicating suppression of miR-181 may be an effective way to enhance cell survival. Furthemore, neutralization of miR-181 using anti-miR-181 decreased the percentage of the cells positively stained for both PI and annexin V (Figure 3D), suggesting that the neutralization of miR-181 was effective in preventing H2O2-induced cardio-myocyte apoptosis.
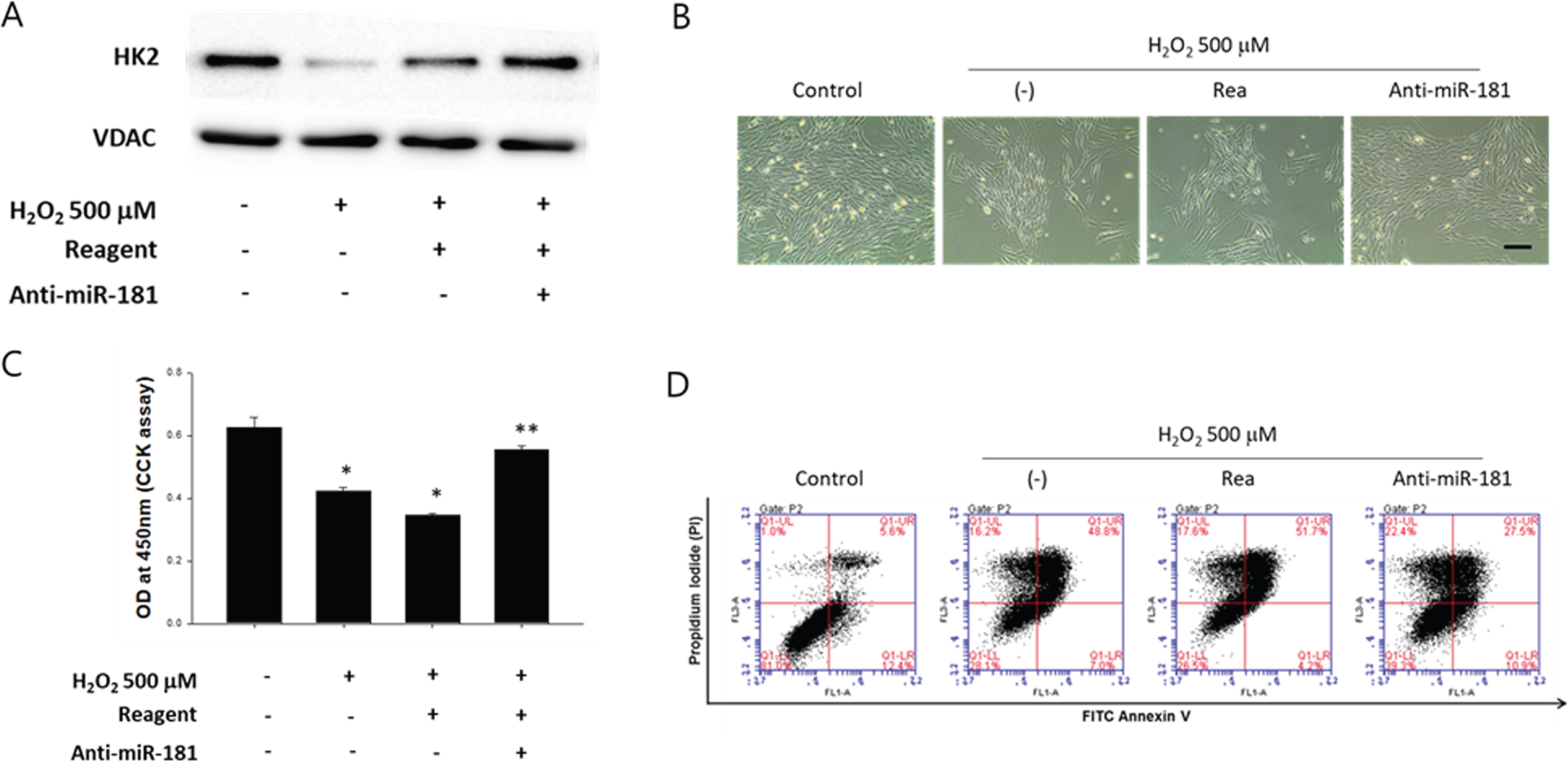
Effect of miR181 neutralization on H2O2-induced cardiomyocyte apoptosis. (A) Effect of anti-miR-181 on the expression of mitochondrial HK2. Cardiomyocytes were transfected with anti-miR-181 (100 nM) and exposed to 500 μM of H2O2 for 6 hours. The expression of HK2 in mitochondrial fraction was evaluated by Western blot. (B) Morphological examination of anti-miR-181 treated cardiomyocytes under oxidative stress. The cells with or without neutralization of miR-181 using anti-miR-181 (100 nM) were exposed to 500 μM of H2O2 for 6 hours. Scale bar = 200 μm. Rea: transfection reagent only. (C) Cell viability of cardiomyocytes exposed to 500 μM of H2O2 for 6 hours with or without miR-181 neutralization using anti-miR-181. *p < 0.05 compared to untreated control, **p < 0.05 compared to H2O2 treated group. The quantitative data were expressed as the means±SEM of at least 3 independent experiments. Cell viability was determined by using CCK. Rea: transfection reagent only. (D) The cells with or without miR-181 neutralization were treated with 500 μM of H2O2 for 6 hours and the percentage of cells undergoing apoptosis were determined by flow cytometry using PI/Annexin-V double-staining.
In a mechanistic point of view, cleaved form of caspases indicates their activation [38]. H2O2 induced cleavage of caspase-3 and -9 in cardiomyocytes but it was attenuated by anti-miR-181 (Figure 4A). Neutralization of miR-181 also attenuated H2O2-induced mitochondrial membrane potential disruption (Figure 4B) and ctyochrome C release (Figure 4D). Additionally, to investigate the effect of miR-181 neutralization in vivo, anti-miR-181 (5 μg/head) was directly delivered to the heart via a local injection immediately after I/R-injury. Masson’s trichrome staining is a common staining method for collagen [39]. Masson’s trichrome staining of 2 weeks post-injured heart indicated that anti-miR-181 attenuated cardiac fibrosis which commonly follows a massive death of cardiomyocytes (Figure 4D). Taken altogether, it was speculated that anti-miR-181 suppressed cardiac fibrosis by neutralizing endogenous miR-181 which mediates cardiomyocyte apoptosis following I/R-injury.
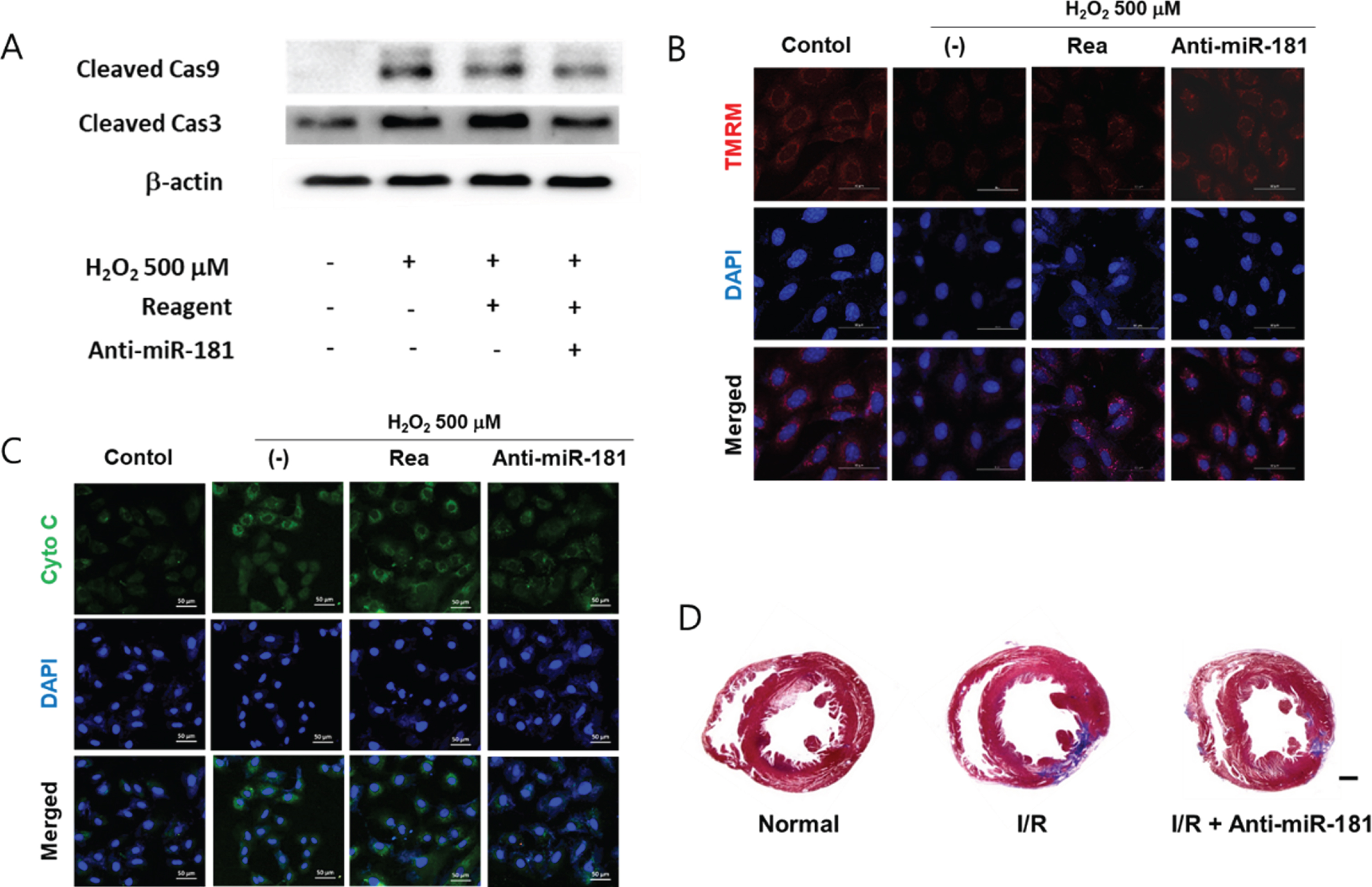
Anti-apoptotic effect of miR-181 neutralization. (A) Effect of anti-miR-181 on the activation of caspases. Cardiomyocytes were transfected with anti-miR-181 (100 nM) and exposed to 500 μM of H2O2 for 6 hours. The expression of cleaved caspase 9 and 3 was evaluated by Western blot. Rea: transfection reagent only. (B) Effect of miR-181 neutralization on the H2O2-induced mitochondrial membrane potential change. Mitochondrial membrane potential was determined by TMRM staining. The cells were treated with 500 μM of H2O2 for 6 hours with or without anti-miR-181 pretreatment. The fluorescent signal of TMRM was detected using a confocal microscopy. DAPI was used to stain nuclei. Scale bar = 50 μm. (C) Immunocytochemical detection of cytochrome C following miR-181 neutralization. The cells cultured in a chamber slide were treated with 500 μM of H2O2 for up to 6 hours with or without anti-miR-181 pretreatment. The fluorescent signal of cytochrome C was detected using a confocal microscopy. DAPI was used to stain nuclei. Scale bar = 50 μm. (D) Effect of anti-miR-181 on cardiac fibrosis. Representative images of Masson’s trichrome-stained sections of each group demonstrating fibrosis. The heart samples were harvested at 14 days after I/R-injury. Scale bar = 2 mm.
In various cardiovascular diseases such as I/R injury, loss of functional cardiomyocytes can lead to cardiac fibrosis and eventual demise of heart function. Thus finding an effective strategy to enhance the survival of the cardiomyocytes under pathologic condition is clinically important. In the present study, we have investigated the miRNA-dependent regulation of HK2 in cardiomyocytes exposed to ROS and report that augmentation of HK2 expression improves the survival of cardiomyocytes exposed to ROS.
The N-terminus of HK2 have a hydrophobic mitochondrial binding domain [40, 41], which facilitates the bind of HK2 to the mitochondria embedded voltage-dependent anion channel (VDAC) [42]. This complex consisted of HK2 and VDAC forms the mitochondria permeability transition pore (PT-pore) [43], maintaining mitochondrial integrity and preventing bcl-2 homologous antagonist killer (BAK) and bcl-2 associated protein X (BAX) mediated mitochondrial cytochrome c release and subsequent apoptosis [44, 45]. Therefore, under physiologic condition, HK2 functions as an anti-apoptotic factor by regulating the mitochondria membrane permeability [41, 46]. Our data also demonstrated that ROS increased cardiomyocyte apoptosis (Figure 1B) and membrane potential change (Figure 1C), and this was coincide with the dissociation of HK2 from mitochondria (Figure 1D). Since miRNAs are involved in virtually every physiological and pathologic biological processes, it was hypothesized that the ROS-induced decrease of mitochondrial HK2 expression may be also regulated by miRNA.
To our best knowledge, miRNA-mediated regulation of HK2 expression is most frequently reported in cancer cells. MiR-143-mediated down-regulation of HK2 in colon cancer cell is a good example [47]. Since miR-143 is also reported to increase apoptosis of cancer cells [48, 49], while miR-155 that suppresses miR-143 expression increased HK2 expression and promoted breast cancer cell proliferation [50], it is reasonable to assume that down-regulation of HK2 contributes to cellular apoptosis. These studies involving the miRNA-mediated regulation of HK2 in cancer cells strongly indicated that modulation of HK2-targeting miRNAs to prevent apoptosis is a feasible approach. A recent study also demonstrated that miRNA-mediated regulation of key molecules involved in the reperfusion-induced apoptosis of cardiomyocyte reduced reperfusion injury [51], suggesting important role of miRNA-mediated regulation in cardiomyocyte apoptosis. Therefore, a number of candidate miRNAs that are predicted to target HK2 (Figure 2A) was examined, and miR-181 was selected as a primary candidate that regulates HK2. As the data demonstrated in the present study, miRNA-181 directly targets HK2 (Figure 2B and D). This agrees with a previous study reported that H2O2-induced up-regulation of miR-181 in rat cardiomyocytes, contributing to apoptotic death of cardiomyocytes [52]. Taken altogether, it could be concluded that miR-181 contributes to the ROS-induced apoptosis of cardiomyocytes by down-regulating HK2.
To further verify the legitimacy of the hypothesis, the effect of miR-181 neutralization using anti-miR-181 on cardiomyocyte apoptosis was examined. In in vitro studies, neutralization of miR-181 significantly attenuated the ROS-induced decrease of cell viability (Figure 3B and C) and prevented cardiomyocytes from apoptosis (Figure 3D). Delivery of anti-miR-181 suppressed cleavage of caspase 9 and 3 (Figure 4A) and ROS-induced disruption of membrane potential was also recovered by miR-181 neutralization (Figure 4B). Since miR-181 neutralization prevented ROS-induced mitochondrial membrane potential, as a consequence, it also prevented cytochrome release into cytosol (Figure 4C). These data further supported the hypothesis that ROS- and miRNA-induced loss of HK2 contributes to apoptosis of cardiomyocytes.
The clinical importance of cardiomyocyte death under pathologic condition is that it can eventually lead to cardiac fibrosis and ultimate demise of heart function [53, 54]. Upon massive cardiomyocyte death due to pathologic condition, various cytokines, such as transforming growth factor-beta (TGF-β) triggers the activation of fibroblasts or myofibroblast (myoFB) formation, which results in deposition of the fibrogenic extracellular matrix [55] and increased myocardial stiffness with reduced compliance [54]. Therefore, an ultimate purpose of preventing cardiomyocyte apoptosis is to prevent cardiac fibrosis following cardiac injury. Interestingly, neutralization of miR-181 using anti-miR-181 attenuated I/R-induced cardiac fibrosis in the present study (Figure 4D), indicating that neutralization of miR-181 could be a viable method to attenuates ROS-induced cardiomyocytes death and subsequent car-diac fibrosis.
Conclusions
In summary, the present study demonstrates that ROS down-regulates HK2 that functions as an anti-apoptotic regulator under physiologic condition and further shows that neutralization of miR-181 using anti-miR-181 attenuates cardiomyocytes death and subsequent cardiac fibrosis. Neutralization of miR-181 can be an effective method for enhancing the survival of cardiomyocytes during pathologic insults involving ROS production.
Footnotes
Acknowledgments
This study is supported by grants funded by the Korea Ministry of Science, ICT and Future Planning (NRF-2015M3A9E6029519), Ministry of Education (NRF-2016R1D1A1B03935124 and NRF-2020R1I1A2064710).
Conflict of interests
The authors declare no conflict of interests.