
Abstract
Thrombotic events result from different pathologies and are the underlying causes of severe diseases like stroke or myocardial infarction. Recent basic research now revealed a link between food uptake, food conversion and gut metabolism. Gut microbial production of trimethylamine N-oxide (TMAO) from dietary nutrients like choline, lecithin and L-carnitine was associated with the development of cardiovascular diseases. Within this review we give a systematic overview about the influence of TMAO on blood components like platelets and endothelial cells which both are involved as key players in thrombotic processes. In summary, a mechanistic correlation between the gut microbiome, TMAO and cardiovascular diseases becomes obvious and emphasizes to the significance of the intestinal microbiome.
Background
Thromboses can be caused by several factors like genetic disorders (defects known to predispose for thrombosis include the hemostatic system like APC-resistance, a point mutation in the prothrombin gene (g20210A) and deficiency of anticoagulant protein, protein S or antithrombinIII), tumors or external influences (e.g. immobilization, surgical procedures, heart failure, polyglobulia, compression, pregnancy, infectious diseases, obesity, cytostatic drugs, radiotherapy, contraceptives, etc.) which can show different symptoms [1, 2]. In many cases, however, a clear cause for the thrombosis cannot be determined.
Recent findings from basic research point to a new link according to which both food intake and its metabolism in the intestinal tract can have an influence on cardiovascular risk factors and the development of thrombosis [3–6]. The gut microbiome includes the entire intestinal habitat, including resident microorganisms (the microbiota) and their metabolic byproducts. The mechanisms by which the microbiome may play a role in the development of thromboses are however still poorly understood [7, 8]. Independently from the well-known cardiovascular risk factors like advancing age, hypertension, diabetes mellitus, dyslipidemia, obesity or lifestyle choices (i.e. smoking, physical activity, diet, etc.) the gut microbiota has recently also been shown to contribute to major cardiovascular events [9].
By eating fatty foods, we ingest larger amounts of the lipid phosphatidylcholine (PC) and L-carnitine, which - as soon as they reach the intestines - are metabolized to choline (C) by microbiota. Intestinal bacteria use the bacterial enzyme mTMA-lyase to metabolize it into gaseous trimethylamine (TMA).
TMA is absorbed in turn and travels via the portal circulation to the liver, where it is oxidized by flavin containing monoxygenases (FMOs), primarily FMO3 [10], to Trimethylamin-N-oxide (TMAO) [10–12]. After 3 weeks of a choline provided diet in atherosclerosis-prone LDLR–/– mice a substantial increase in circulating TMAO was observed [13].
In this review we summarize the status of studies on the influence of TMAO on platelets and endothelial cells.
Platelets
Animal or cell culture studies seem to hint to an influence of TMAO on different pathophysiological processes which are involved in the development of atherosclerotic diseases. Among those processes platelet activation and aggregation is an essential step in the genesis and propagation of atherothrombotic complications [14–17]. Some groups hypothesized that the deleterious effects of TMAO might be due to its promotion of platelet aggregation [18–22]. Zhu et al. examined the effect of physiologically relevant levels of exogenous TMAO (100μM vs. vehicle) on platelet activation by ADP or thrombin in platelet-rich plasma isolated from healthy volunteers with low levels of TMAO (<2.4μM) [22]. The study revealed that direct exposure of platelets to TMAO enhanced submaximal stimulus-dependent platelet activation from multiple agonists through augmented Ca2+ release from intracellular stores [22].
In addition, they raised plasma TMAO levels in mice using intraperitoneal TMAO injection, and quantified the in vivo thrombosis potential in a carotid artery injury model using fluorescently tagged platelets and intravital microscopy (100μM vs. normal saline). At the time of the carotid artery injury (2–h following TMAO i.p. injection), a more than 10-fold increase in plasma TMAO was observed. The rate of thrombus formation increased by TMAO and the occlusion time was significantly shorter in TMAO injected mice (p < 0.001).
Also, in an in vivo study, Zhu et al. proved whether dietary nutrients enhance TMAO generation and subsequently modulate platelet function and thrombosis potential in vivo [22]. To explore this possibility, mice were placed on a chemically defined diet comparable to normal chow (0.08% total choline) versus the same chemically defined diet supplemented with either TMAO (0.12%) or choline (1%) over six weeks. Plasma TMAO increased in mice provided dietary supplementation with either choline or TMAO. In parallel, ex vivo platelet aggregation induced by ADP stimulation was significantly increased in these groups (p = 0.0002). Consistently, in vivo thrombosis potential, as monitored by time to blood flow cessation in the carotid artery following FeCl3 injury, was shortened - indicating a more prothrombotic phenotype - in mice with heightened TMAO levels.
However, another recent study did not confirm these results [23]. Haissman et al. reported no evidence of TMAO-induced platelet hyperreactivity in 50 treated and 50 untreated HIV infected individuals [23] –a disease associated with platelet alterations and endothelial dysfunction [24]. The authors hypothesized that the lacking of association between TMAO and platelet function could be, that other factors might have a stronger effect on platelet function in HIV-infected individuals than TMAO. It is well known that in HIV-infected populations both immune activation, viral replication and specific cART drug classes have a strong influence on platelet activation and dysfunction and so might dilute the direct effect of TMAO on platelet function. [25–28].
Due to the few studies available so far, it is very difficult to arrive at a reliable assessment. Well-controlled acute and prolonged studies in humans are indispensable to expand our understanding of the influence of TMAO on quiescent or hyperaggregable platelets.
Endothelial cells
The situation regarding the influence of TMAO on endothelial function seems to be more consistent than for platelets, although there are only a few studies available. An in vitro study with human venous endothelial cells revealed that TMAO significantly triggered oxidative stress inducing the release of interleukin-1β and IL-18 in a dose- and time-dependent manner [29] which may lead to loss of function and even cell death [30, 31]. In addition, TMAO induced the production of reactive oxygen species (ROS), inhibited the mRNA and protein expression of endothelial nitric-oxide synthase (eNOS) and down-regulated the production of nitric oxide (NO). However, it is unknown, whether TMAO is involved in an inflammatory response through specific membrane receptor mediated signaling [29]. Sun's results are in line with a study in rats. Here, circulating TMAO levels correlated with reduced eNOS-derived NO production and increased vascular oxidative stress and inflammation thereby contributing to endothelial dysfunction [32].
Another study using human arterial endothelial cells revealed that the treatment with TMAO resulted in phosphorylation of the p38 mitogen-activated protein kinase/extracellular signal–related kinase/NF-κB pathway, enhanced p65 NF-κB nuclear localization, and induced inflammatory transcripts (cyclooxygenase 2, interleukin 6, E-selectin, and ICAM-1) [13].
TMAO was also shown to promote atherosclerosis through foam cell formation and interference with reverse cholesterol transport from the atherosclerotic plaque [33–35]. Studies in mice, supplemented with TMAO led to an enhancement of macrophage cholesterol accumulation and atherogenic plaque formation, indicating a causal relationship [11, 36].
Concluding, cell culture studies to date indicate that choline-derived metabolite TMAO can induce oxidative stress resulting in the release of inflammatory cytokines and endothelial dysfunction. Nevertheless, clinical studies in humans under different nutritional protocols leading to increased TMAO values should be performed to demonstrate the association between elevated TMAO levels and endothelial dysfunction.
Clinical studies
Gut microbiota and liver metabolism has raised much interest in the cardio-metabolic field of research [37]. The mechanisms through which TMAO alter cardio-metabolic disease risks are just beginning to be understood. During the last decade several studies have shown a noticeable association between TMAO plasma levels and cardiovascular disease (CVD) risk factors [1–11, 38]; this according to the extent of coronary atherosclerotic plaque burden [38–40].
Especially the group of Hazen provided strong support that high levels of TMAO predict an increased risk of cardiovascular diseases [9, 38–44]. TMAO showed a dose-dependent association with increased cumulative burden of incident thrombotic events as illustrated by Kaplan-Meier survival analyses in a cohort of subjects (n = 4007) during a follow-up of 3 years [38]. Stubbs et al. also found elevated TMAO concentrations to independently predict coronary atherosclerosis and mortality in patients with chronic kidney disease [45].
However, although TMAO levels were found to be increased in hyperglycemic subjects and patients with impaired kidney function in other studies, they were not associated with all-cause mortality, cardiovascular death, or hospitalizations [46] or history of myocardial infarction, the angiographically assessed presence of coronary artery disease or incident cardiovascular events during eight years of follow-up [47].
Concomittant metabolic pathway: Caveat
A concomitant metabolic pathway which also influences platelets and endothelial cells has to be considered when the results of TMAO studies are evaluated. Endothelial cells metabolize arachidonic acid by cyclooxygenase (COX), lipoxygenase (LO), and cytochrome P450 (CYP) pathways [48–53]. Among these, CYPs produce oxygenated metabolites that cause hyperpolarization, vasodilation and contribute to the regulation of vascular tone [47, 53–55]. Preliminary studies in animal models have shown that the epoxymetabolites derived from eicosapentaenoic (EPA) and docosahexaenoic acid (DHA) have a vasodilating action that largely exceeds that of the arachidonic (AA)-derived epoxides [56, 57]. In addition, it could be shown, that 11,12-EET (EET: epoxyeicosatrienoic acids) after 60 minutes of preincubation significantly decreased the rate of ristocetin-induced platelet aggregation [57]. However, 17,18-EEQ (EEQ: epoxyeicosatetraenoic acids) and 19,20-EDP (EDP: epoxydocosapentaenoic acids) were effective already at 1μM, whereas 5-fold higher concentrations were required with 11,12-EET [57].
Figure 2 shows the metabolic pathway for AA, EPA or DHA. “Western diets” rich in n-6 PUFAs (PUFA: polyunsaturated acids) result in a predominance of arachidonic acid-derived metabolites, whereas marine foodstuffs rich in n-3 PUFAs shift the profile to eicosapentaenoic and docosahexaenoic acid-derived metabolites. Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) omega-3 polyunsaturated fatty acids may compete with AA for CYP450-dependent bioactive lipid mediator formation [58].
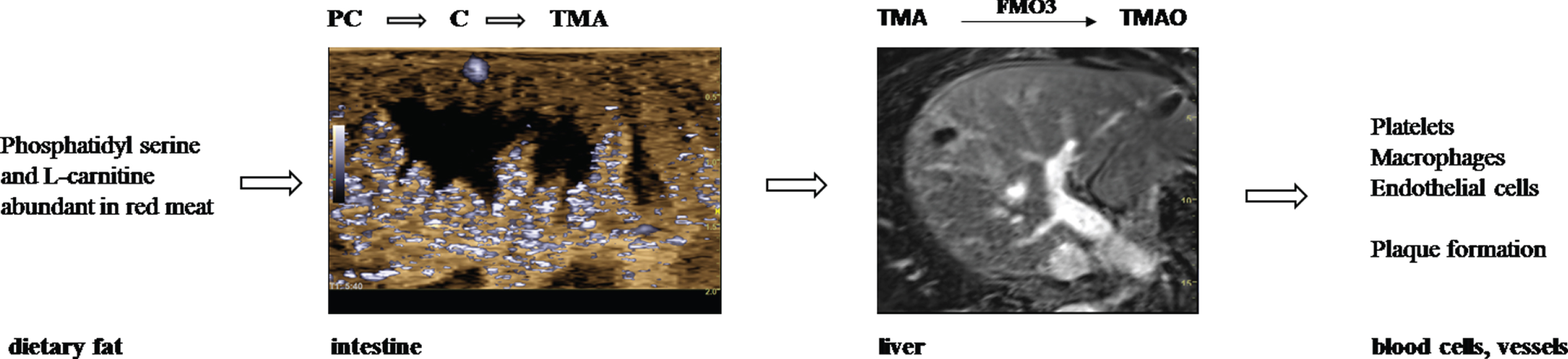
Pathway from dietary fat via intestine and liver to dysfunctional blood cells and atherosclerosis.

Cytochrome P450 epoxygenases accept arachidonic acid (AA), eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) as efficient alternative substrates. The products include isoform-specific sets of regioisomeric epoxyeicosatrienoic acids (EETs), epoxyeicosatetraenoic acids (EEQs) and epoxydocosapentaenoic acids (EDPs). (reprinted from [58], Copyright 2018, with permission from Pergamon Press).
In addition, Omega-3 PUFAs can exert a positive action by reverting the microbiota composition and increase the production of anti-inflammatory compounds [59]. Thus, the type of ingested food (rich in meat: then metabolizing 11,12-EET or rich in fish: then metabolizing 17,18-EEQ or 19,20-EDP) determines the extent of activation of thrombocytes and endothelial cells by eicosanoids, which can considerably counteract with the influence of TMAO on platelets and endothelial cells. This aspect must be taken into account in the conduct of clinical trials and should lead to a very controlled diet during studies (what can be done very stringently in animal experiments).
Taken together, it is quite likely that the gut microbiome can influence the balance of vascular homeostasis in critical features like platelet aggregation and endothelial dysfunction providing a novel mechanistic connection between microbes and thrombotic events. Variations in baseline and alterations within the microbiome should be considered as an influential factor. Therefore, Leslie & Annex suggested quite recently that studies of the microbiome within and outside cardiovascular medicine are likely to, and indeed should, increase [60].
Conflict of interest
The authors have no conflict of interest to report.