
Abstract
Lipoxins and ATL appear to be the first recognized members of a new class of endogenous mediator that are anti-inflammatory or serve for the “pro-resolution” of inflammation. PGE2 can and may display anti-inflammatory properties in certain settings, but in most cases, it enhances inflammation in vivo. This is likely the result of numerous receptor isoforms and differential coupled mechanisms for PGE2 and its diverse role in human physiology. Since the integrated response of the host is essential to health and disease, it is important to achieve a more complete understanding of the molecular and cellular events governing the formation and actions of endogenous mediators of resolution that appear to control the magnitude and duration of inflammation. In view of the present body of evidence, it is not surprising that a protective action for inhibition of COX-2 was found in cardiovascular disease. Characterizing useful experimental systems with clinically relevant endpoints will also take a multidisciplinary approach and require a shift in our current thinking about inflammation and the role of lipid mediators.
Introduction
Inflammation is a physiologic series of responses generated by the host in response to infection or other insults. Inflammation can have rapid onset and last a short period of time (acute inflammation), or it can persist due to a continuous stimulus or injury (chronic inflammation). The Initial events of inflammation are derived from vascular reactions at the site of injury. Vascular changes are important for the induction of the response and are characterized by redness, heat, and swelling, usually accompanied by pain and loss of function collectively represent the “cardinal signs” of inflammation. These signs of inflammation are the result of vasodilatation and increased vascular permeability, leading to exudation of fluid and plasma proteins and recruitment of leukocytes to the site of injury [1].
Inflammation has the net effect of confining the injury or insult to an isolated area and serves as the first step in the initiation of the immune response, through which the infection is eliminated and the injury is repaired. In general, once the insult is eliminated, the inflammatory response resolves and subsequent immune reactions diminish. In the case of chronic inflammation, however, the persistence of the response leads to host tissue destruction and may result in irreversible pathological changes. Periodontal diseases are important chronic infections leading to chronic inflammation that result in a host-mediated destruction of the supporting tissues of the dentition. The pathogenesis of periodontal diseases has been the subject of intense study over the past two decades. The periodontal diseases are infectious diseases caused by predominantly Gram-negative bacteria. However, as our understanding of the pathogenesis of the periodontal diseases grows, it is becoming clear that most of the tissue damage that characterizes periodontal disease is caused by the host response to infection and not by the infectious agent directly. Investigation into the mechanism of action of host-mediated tissue injury has revealed that the neutrophil plays an important role in destruction of host tissues. The biochemical pathways and molecular mediators are responsible for regulation of the inflammatory response in diseases such as periodontitis along with lipid mediators of inflammation. Pro-inflammatory mediators, such as prostaglandins and leukotrienes, are balanced by counter-regulatory signals provided by a class of molecules called lipoxins. Figure 1 depicts the role of lipid mediators in inflammation.
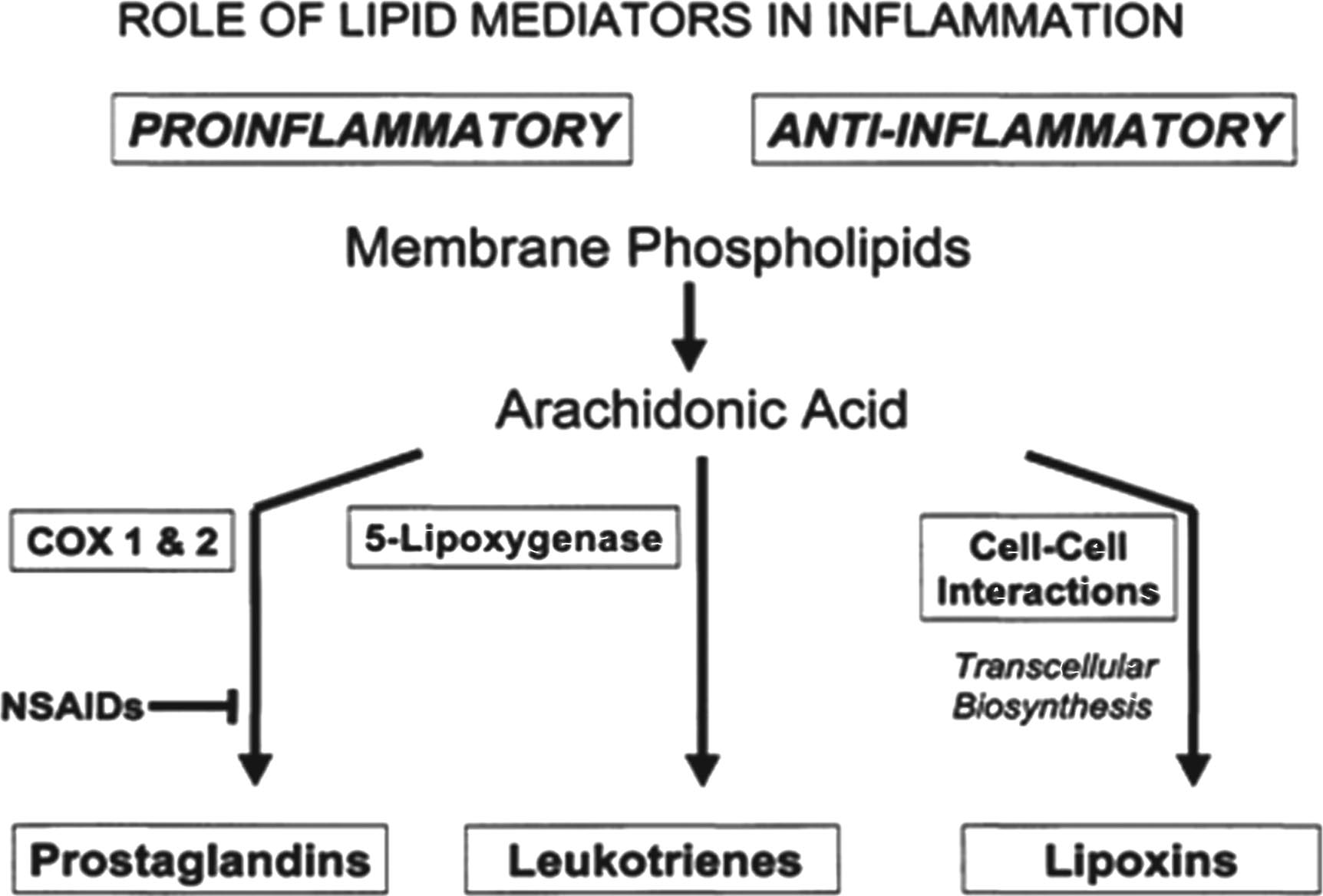
Role of lipid mediators in inflammation.
Thornton JM, Yin K (2021) studied the specialized Pro-resolving Mediators (SPMs) are bioactive fatty acids that are shown to be highly effective in promoting resolution of infectious inflammation and survival in several models of infection [2]. SPMs do not have direct anti-inflammatory effects by inhibiting or directly blocking this process but can actively reduce neutrophil infiltration into inflamed tissues, enhance efferocytosis and bacterial phagocytosis by monocytes and macrophages and simultaneously inhibit inflammatory cytokine production [3]. One such SPM is Lipoxin (LX) which will be discussed in this review in depth.
Lipoxins (LXs) are derivates obtained from omega 6 arachidonic acid. Lipoxins are the first Specialized Pro-resolving Mediators (SPMs) which have been proposed to resolve inflammation including periodontitis and peri-implantitis [4].
Once inflammation is initiated, the cascade of inflammatory events is an amplified loop until the infection is contained via immune reactions, or injury is confined. These early actions of the host response are later replaced by more specific mechanisms and eventually become redundant. Thus, it is important for the inflammatory response to be limited and resolved. Many molecules play a counter regulatory role in this resolution stage of inflammatory response to control the magnitude and duration of the inflammatory response [5].
Endogenous lipid-derived mediators have been demonstrated to moderate the host response and coordinate the resolution of inflammation. Recently, several novel lipid mediators have been described as potential anti-inflammatory molecules, illustrating the importance of endogenous generation of lipid mediators with anti-inflammatory properties. An important example of lipid mediators with inflammation-resolving properties is the lipoxins (LX). Lipoxins contain a trihydroxytetraene group and are members of the eicosanoid family that are produced within the vascular lumen, primarily via platelet-leukocyte transcellular biosynthesis. Lipoxins can be generated by several different pathways. In general, cell-cell interactions result in the generation of lipoxins, while single cells also can produce lipoxins (Fig. 2).

Synthesis of anti- inflammatory lipoxins.
Lipoxin generation is a very rapid process that is activated by inflammation, atherosclerosis, and thrombosis [6]. Cell-cell interactions, that occur during these events and lead to generation of lipoxins, can also induce transcellular biosynthetic routes that lead to amplification signals such as leukotrienes and prostaglandins or to braking signals that involve novel compounds [7]. Therefore, lipoxin production is an important stage of the inflammatory response.
Biosynthesis of lipoxin was first demonstrated in 1984. It was shown that insertion of molecular oxygen into the 15-carbon (C15) position of arachidonic acid is essential for lipoxin production. In addition, it was also reported that lipoxin generation through this pathway required the action of 15-LO. Once oxygenated at the C15 position, arachidonic acid is converted into 15-hydroperoxyeicosatetraenoic acid (15-HPETE), which is a substrate for 5-LO in leukocytes. This molecule is rapidly converted by hydrolases to either 5S, 6R, 15S-trihydroxy-7, 9, 13-trans-1l-cis-eicosatetraenoicacid (lipoxin A4; LXA4), and/or, via lipoxin B4 hydrolase, to 5S, 14R, 15S-trihydroxy-6,10,12-trans-8-cis-eicosatetraenoic acid (lipoxin B4; LXB4) [8]. LXA4 and LXB4 are vasoactive molecules, primarily vasodilatory in vivo, and regulate leukocyte functions. While the 15-LO-initiated route synthesizes lipoxin, 5-LO activation blocks leukotriene synthesis. Thus, these series of events can be viewed as an inverse relationship between leukotriene and lipoxin synthesis. When neutrophils produce lipoxins by carrying the alcohol group in 15-HETE to either the R or S configuration, leukotriene formation is dramatically reduced.
The role of the neutrophil in lipoxin generation is crucial. It has been shown that primed neutrophils are another source of LX biosynthesis. This mechanism is mediated by the esterification of 15-HETE in inositol-containing phospholipids within the cell membrane. Cells rapidly convert 15-HETEs into their inositol-containing lipids. When stimulated by various agonists, neutrophils release 15-HETE, which is further transformed into lipoxin. This pathway suggests that precursors of lipoxin synthesis can be stored within membranes of inflammatory cells and then released by stimuli activating PLA2. Furthermore, membrane priming of neutrophils generates bioactive lipid mediators that have implications for second messengers, such as 15-hydroxy-phosphatidyl inositol diphosphate and diacylglycerol, which contain 15-HETE that may alter their intracellular signaling activities. As is the case with 15S-HETE, 15R-HETE is also rapidly esterified into membrane phospholipids and could probably serve as a reservoir for epimeric lipoxin formation [8].
5-Lipoxygenase-initiated pathway
The second route for LX biosynthesis occurs with the interaction of human neutrophils with platelets in the blood vessels. In this model, cell-to-cell interaction involves 5-LO in neutrophils and 12-LO in platelets for the insertion of molecular oxygen into arachidonic acid. Under resting conditions, unstimulated neutrophils do not generate considerable amounts of lipoxin, and the majority of neutrophil-generated LTA4 by the 5-LO pathway is released into the extracellular environment. When platelets adhere to neutrophils, however, they convert LTA4 into a cation (carbonium) by 12-LO. As a result, platelet 12-LO reduces hydrogen at the C13 position and inserts oxygen into the CIS position of LTA4, converting LTA4 either to LXB4 at the C14 position or LXA4 at the C6 position. Both LXB4 and LXA4 generation is exclusively mediated by 12-LO. Thus, 12-LO functions as a “lipoxin synthase” in platelets [9]. These observations with isolated intact platelets from human peripheral blood were also confirmed with recombinant 12-LO.
Transcellular lipoxin synthesis requires cell adhesion. Therefore, cell adhesion characteristics play an important role in lipoxin metabolism. Indeed, a study on P-selectin-deficient knockout mice has showed that the adhesion is selectin-dependent, and when adhesion between the cells is blocked by specific antibody, transcellular lipoxin biosynthesis was also blocked [10].
LXA4 levels were restored and neutrophil infiltration was normalized when the P-selectin-deficient mice were infused with wild-type platelets. These observations suggest that platelet-neutrophil adherence and transcellular biosynthesis are important inflammatory events that regulate neutrophil recruitment by initiating the formation of lipid mediators that suppress pro-inflammatory responses. Thus, the role of platelets in lipoxin generation during platelet-neutrophil interaction within the blood vessels might be an important factor in regulating the extravasation of neutrophils. The vascular route of platelet-leukocyte interactions elevates the formation of lipoxin by transcellular conversion of LTA4. This appears to be a major route for lipoxin generation, particularly when the platelet COX-1 is inhibited by non-steroidal anti-inflammatory drugs (NSAIDs).
Additional pathways for lipoxin generation
Neutrophil-released LTA4 that is converted by the 15-LO in epithelial cells, particularly in tracheal epithelial cells, can also generate lipoxins by an LTA4-dependent mechanism. In this pathway, another biosynthetic route involves 5,6-dihydroxyeicosanoids, which are also substrates for conversion to lipoxin. These reactions can include both 15-LO or 12-LO conversion of 5,6-dihydroxyeicosanoid substrates to lipoxin, which are enhanced by cell-cell adhesion interactions. Lipoxin generation can also be mediated by single cells. In this model, primed neutrophils in inflammatory disorders such as asthma generate lipoxin entirely from endogenous sources of arachidonic acid from a single cell type.
Aspirin-triggered lipoxins
Aspirin may also play an important role in the generation of lipoxins. In this transcellular biosynthetic scheme, cyclooxygenase-2 (COX-2) switches its catalytic activity in the presence of aspirin, generating 15R-HETE instead of prostaglandins. Thus, aspirin inhibits prostaglandin biosynthesis by both COX-1 and COX-2 [11]. COX-2, when acetylated by aspirin in endothelial or epithelial cells, is enzymatically active and converts arachidonic acid to 15R-HETE, which is released and transformed through transcellular routes to form 15-epi-lipoxins by leukocytes. The activated and adherent leukocytes possess 5-LO and transform 15R-HETE to a 5(S)-epoxytetraene within leukocytes, which carries its C15 position alcohol in the R configuration. This intermediate leads to the formation of both 15-epi-LXA4 and 15-epi-LXB4 which carry the R configuration at C15. 15-epi-LXA4 is more potent than native LXA4 in inhibiting neutrophil adhesion, and 15-epi LXB4 inhibits cell proliferation. COX-2 is increased in inflammatory reactions and in disease states including colon cancer, and is normalized upon aspirin intake. Aspirin-triggered lipoxins (ATL) can serve as potential endogenous anti-inflammatory signals or mediators of some of aspirin’s beneficial actions. These beneficial actions of aspirin include prevention of myocardial infarction and protection from colorectal adenoma, as well as other forms of cancer.
Thus, in addition to serving as an inhibitor of eicosanoid biosynthesis, aspirin can also trigger the biosynthesis of various compounds, such as the endogenous generation of 15-epimeric lipoxins, via acetylation of COX-2 at sites of inflammation in vivo. Meanwhile, aspirin also affects COX-2-bearing vascular endothelial cells or epithelial cells and their co-activation with neutrophils. In this model, inflammatory cytokines induce COX-2 to generate 15R-HETE when aspirin is administered [12]. This intermediate carries a C15 alcohol in the R configuration that is rapidly converted by activated neutrophils to 15-epi-lipoxins rather than 15S native lipoxins, which result from LO interactions. These actions of LXA4 and ATL, their endogenous epimeric counterpart (15-epi-LX), were first found in experiments with isolated cell types in vitro and have been confirmed and demonstrated in several acute murine models of inflammation and second-organ reperfusion injury. Neutrophil infiltration in lung, skin, and sites of wound healing is dramatically inhibited by both intravenous and topical application of stable analogs of both LXA4 and aspirin-triggered 15-epi-LXA4(ATL). 15-epi-LXA4 and LXA4 analogs inhibit interleukin-lB (IL-IB), tumor necrosis factor-, and IL-8 expression while stimulating IL-4 release in vivo, and interact with a common receptor on human and murine leukocytes [13, 14].
Bioactive ATL and LXA4 analogs compete with [3H]-LXA4 binding to LXA4 receptors. Thus, these inhibitory actions of LX and ATL analogs are likely to be mediated by specific lipoxin receptors present in rodent and human cells [15]. LXB4 is a positional isomer of LXA4, carrying alcohol groups at carbon 5S, 14R, and 15S positions, instead of the C5S, 6R, and 15S positions present in LXA4. Aspirin-triggered LXB4 carries a 15R alcohol, hence 15-epi-LXB4. Although LXA4 and LXB4 show similar activities in some biologic systems, in many others they have distinct actions (Fig. 3).
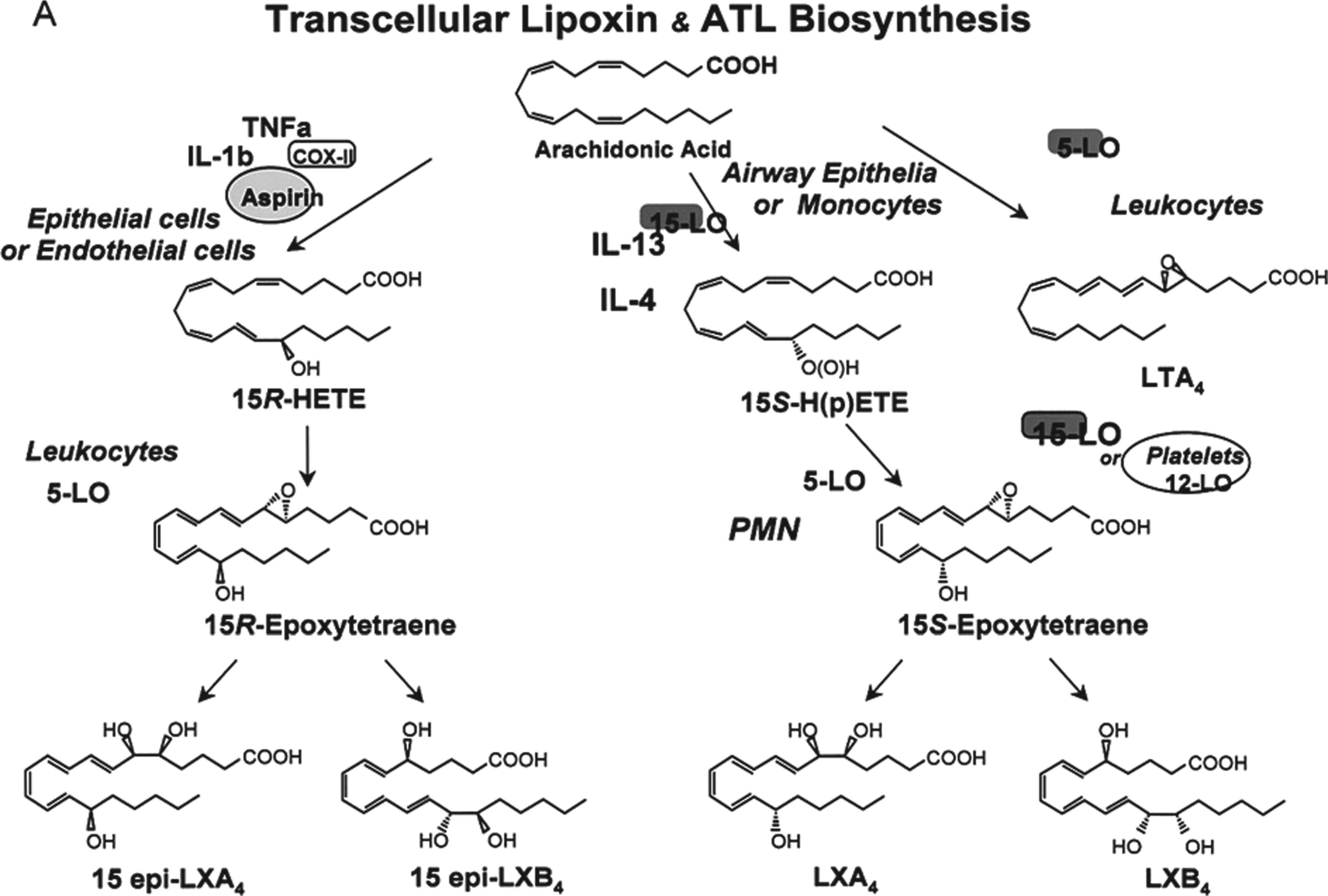
Depicts the transcelualar lipoxin and atl biosynthesis.
Lipoxins, as other autacoids, are rapidly biosynthesized in response to stimuli, act locally and then are rapidly enzymatically inactivated. The major route of lipoxin inactivation is through dehydrogenation by monocytes that convert LXA4 to 15-oxo-LXA4, followed by specific reduction of the double bond adjacent to the ketone. 15-Hydroxy/oxo-eicosanoid oxidoreductase (15-PGDH) catalyzes the oxidation of LXA4 to 15-oxo-LXA4. This compound is biologically inactive and is further converted to 13,14-dihydro-15-oxo-LXA4 by the action of LXA4/PGE 13,14-reductase/LTB4 12-hydroxydehydrogenase (PGR/LTB4DH). Moreover, reduction of the 15-oxo group by 15-PGDH yields 13,14-dihydro-LXA4, revealing an additional catalytic activity for this enzyme. LXB4 can also be dehydrogenated by 15-PGDH at carbon 5 to produce 5-oxo-LXB4, therefore sharing a common route of inactivation. It has been shown that 15-oxo-LXA4 is also produced from LXA4 in mouse whole blood, suggesting that the mouse shares with humans a pathway for LXA4 inactivation (Fig. 4).

Depicts the mechanism of inactivation of lipoxins.
In view of the rapid transformation and inactivation of lipoxin by monocytes and, potentially, other cells in vivo, it was highly desirable to design lipoxin analogs that would resist this metabolism and maintain their structural integrity and potential beneficial biological actions. Lipoxin analogs were constructed with specific modifications of the native structures of LXA4 and LXB4, such as the addition of methyl groups to carbon 15 and carbon 5 of the LXA4 and LXB4 structures, respectively, to block dehydrogenation by 15-PGDH. 15R/S-methyl-LXA4 is a racemic stable analog of both LXA4 and 15-epi-LXA4. Additional analogs of LXA4 were synthesized with a phenoxy group attached to carbon 16 and replacing the ω-end of the molecule. This design permits 16-phenoxy-LXA4 to resist potential ω-oxidation and to be protected from dehydrogenation in vivo. Fluoride was added to the para-position of the phenoxy ring to yield 16-(para-fluoro)-phenoxy-LXA4 in order to hinder degradation of the phenoxy ring. The aspirin-triggered counterpart of 16-(para-fluoro)-phenoxy-LXA4, 15-epi-16-(para-fluoro)-phenoxy-LXA4, was also synthesized. These modifications not only prolong the half-life of the compounds in blood but also enhance their bioavailability and bioactivity.
The ATL are less effectively converted in vitro to their 15-oxo metabolite than LXA4. This indicates that the dehydrogenation step is highly stereospecific and suggests that, when ATL are generated in vivo, their biological half-life is increased by about two-fold compared to that of native LXA4, thereby enhancing their ability to evoke bioactions. Hence, biologically stable analogs of lipoxin and ATL can be engineered to enhance their bioactions, a fact suggesting that they are useful tools for the development of novel therapeutic modalities.
Arachidonic acid (ARA) is the usual substrate for eicosanoid synthesis. The COX pathways form prostaglandins (PGs) and thromboxanes (TXs), the LOX pathways form leukotrienes (LTs) and lipoxins (LXs), and the cytP450 pathways form various epoxy, hydroxy and dihydroxy derivatives. Eicosanoids are highly bioactive acting on many cell types through cell membrane G-protein coupled receptors, although some eicosanoids are also ligands for nuclear receptors. Because they are rapidly catabolised, eicosanoids mainly act locally to the site of their production. Many eicosanoids have multiple, sometimes pleiotropic, effects on inflammation and immunity. The most widely studied is PGE2. Many eicosanoids have roles in the regulation of the vascular, renal, gastrointestinal and female reproductive systems [16].
Bioactions of lipoxins
Vasoactive actions
Lipoxins display vasodilatory and counterregulatory roles in in vivo and in vitro models. LXA4 and LXB4 promote vasorelaxation and relax the aorta and pulmonary arteries. LXA4 reverses the precontraction of the pulmonary artery induced by prostaglandin F2 and endothelin-1. The mechanisms of LXA4- and LXB4-induced vasodilatation involve endothelium-dependent vasorelaxation and involve prostaglandin-dependent and -independent pathways. In certain tissues, lipoxin can stimulate the formation of, for example, prostacyclin by endothelial cells, which can contribute to vasodilatation. These prostanoid-dependent actions of lipoxin are inhibited by COX inhibitors and indicate that lipoxins can stimulate the biosynthesis of a second set of mediators. These also include lipoxin-stimulated generation of nitric oxide, which may mediate a component of lipoxin-regulated vascular tone.
Immunomodulatory actions
The actions of lipoxins contrast with those of most other lipid mediators that are primarily proinflammatory, such as leukotrienes, platelet-activating factor (PAF) and prostanoids. It is now appreciated that lipoxins, LXA4 in particular, are potent counterregulatory signals in vitro and in vivo for endogenous proinflammatory mediators, including lipids (leukotrienes, PAF) and cytokines (TNF- α, IL-6), resulting in inhibition of leukocyte-dependent inflammation. Lipoxins display selective actions on leukocytes inhibition of neutrophil chemotaxis, b) transmigration through epithelial cells, and c) adhesion and transmigration with endothelial cells. LXA4 and ATL inhibit PMN-initiated second-organ injury in an ischemia-reperfusion model using LTB4 receptor transgenic mice, suggesting an endogenous compensatory or protective role to limit PMN trafficking and PMN-mediated damage.
LXB4 has not been studied as extensively as LXA4; it is chemically and biologically less stable because it isomerizes rapidly in vitro. Therefore, it has been more difficult to handle previously, but now stable LXB4 analogs have been prepared that will help to expand the evaluation of their biological roles. There are specific and potent actions attributed to LXB4, including stimulating proliferation and differentiation of granulocyte-monocyte colonies from human mononuclear cells and sleep induction. In addition to its specific actions, LXB4 also shares actions with LXA4, selectively stimulating human peripheral blood monocytes and inhibiting human neutrophil transmigration and adherence as well as PMN-mediated increases in vascular permeability in mice.
A considerable amount of data has well documented that lipoxin actions are closely linked with cytokine networks. In human enterocytes, LXA4 and LXA4 analogs inhibit IL-8 release at the gene transcriptional level. This report is consistent with recent findings showing that synthetic lipoxin analogs abolish the allergen-induced eotaxin formation and suppress TNF- α stimulated macrophage inflammatory peptide-2 and IL-1ß release but also concomitantly stimulate IL-4 in in vivo models. It is thus likely that lipoxin bioactions in vivo are up-regulated by cytokines and that lipoxins directly modulate the cytokine composition in a local inflammatory milieu, a concept supported by the findings showing that LXA4 may be involved in a negative feedback loop opposing inflammatory cytokine-induced activation of human synovial fibroblasts.
Unlike PMN and eosinophils, lipoxins are potent stimuli for peripheral blood monocytes. While LXA4 and LXB4 stimulate monocyte chemotaxis and adherence, these cells do not degranulate or release reactive oxygen species in response to lipoxins, suggesting that the actions of these lipoxins are specific for locomotion and may be related to the recruitment of monocytes to sites of injury. These monocyte activities may be host-protective in view of the important role of these cells in wound healing and resolution of inflammatory sites. Along with these suggestions, LXA4 and LXA4 analogs were shown to accelerate the resolution of allergic pleural edema and enhance phagocytosis of apoptotic PMN by monocyte-derived macrophages. It is increasingly appreciated that the resolution of inflammation is a dynamically regulated process and these different observations raise the possibility that lipoxins play pivotal roles in the resolution phase of PMN-mediated inflammation.
Lipoxins and disease
Lipoxins are generated in human organs and are associated with a variety of inflammatory events. The first demonstration of lipoxin in clinical inflammation was in bronchial lavage [16]. Furthermore, LXA4 and LXB4 are formed in nasal polyps, and LXA4 is generated in nasal lavage from aspirin-sensitive asthmatics and in experimental nephritis. It has also been proposed that lipoxins are useful biomarkers of asthma and long-term clinical improvement in arthritic patients. Rupture of atherosclerotic plaque leads to rapid generation of LXA4 in the intracoronary artery [17]. Lipoxins are also generated by normal human bone marrow, and, during chronic myelocytic leukemia, platelets lose 12-LO. They lose their ability to generate lipoxins, and this finding may be related to the blast crisis observed in chronic myelocytic leukemia. To determine whether 15-epi-LXA4 could be detected in animal experimental models or in patient-derived materials, experiments were carried out with a mouse peritonitis model [18]. In this model, COX-2 protein levels were up-regulated by intraperitoneal injection of lipopolysaccharide (LPS) aspirin. The results revealed a low level of 15-epi-LXA4 generation in these LPS-treated animals, and the presence or absence of aspirin did not make any significant difference. This study suggested that LPS alone is not sufficient to elicit neutrophil infiltration. Furthermore, these experiments also demonstrated that aspirin administration in murine peritonitis yields inflammatory exudates that generate 15-epi-LXA, in appreciable levels from endogenous substrates.
When applied topically to mouse ears, these LX stable analogues inhibit both PMN infiltration and vascular permeability changes in a concentration-dependent fashion [19, 20]. At 130 nmol per ear, the degree of inhibition of PMN infiltration was more than 90% for both analogues, with apparent IC50S noted at 13–26 nmol per ear range for each analogue. In the same concentration range, these two LXA4 stable analogues also inhibited vascular permeability, namely, extravasation of Evans blue. At 130 nmol per ear, the inhibition of vascular permeability change was > 98% for 15(R/S)-methyl-LXA4 and 87% for 16-phenoxy-LXA4, respectively, and their impact was noted visually. The inhibition of vascular permeability changes paralleled inhibition of PMN infiltration with both the ATL and LX analogues.
In humans, ATL and LXA4 formation was studied in aspirin-tolerant and aspirin-intolerant asthmatics. The aspirin-tolerant subjects generated both LXA4 and ATL, but the aspirin-intolerant patients proved to have a diminished capacity to generate ATL and lipoxins upon aspirin challenge [21]. Furthermore, a reduction and alteration in lipoxin generation were found in patients with chronic liver disease and chronic myelogenous leukemia. In contrast to these disease states, LXA4 production is up-regulated, following atherosclerotic plaque rupture, and with nasal polyps.
Anti-inflammatory signaling and lipoxin-specific receptors
Resolution of inflammation is dynamically regulated and coupled to the generation of bioactive mediators including lipids and peptides [22]. The components of both the innate and adaptive immune systems are regulated by lipoxins which include neutrophils, macrophages, T-, and B-cells. Lipoxins are capable of modulating levels of various transcription factors such as nuclear factor κB, activator protein-1, nerve growth factor-regulated factor 1A binding protein 1, and peroxisome proliferator activated receptor γ and control the expression of many inflammatory genes [23].
Lipoxin actions are cell type, species and organ specific. These actions can be assigned to one or a combination of three mechanisms: a) lipoxins act at their own specific cell surface receptors (i.e., LXA4 specific and separate LXB4 receptor); b) LXA4 interacts with a subclass of the peptido-leukotriene (LTC4 and LTD4) receptor that also binds LXA4 [14], and c) lipoxins can act on intracellular targets after lipoxin transport and uptake or within their cells of origin.
LXA4 and LXB4 act at two distinct sites, and in some cell types, they evoke similar actions, but their actions are distinct in others. 3H-LXA4 specifically binds to intact PMN, the binding being modulated by stable guanosine analogs, suggesting that LXA4-triggered responses in PMN are mediated by classical G-protein coupling of cell surface receptors. The bioactions of LXA4, 15-epi-LXA4 and stable analogs are transduced by this high affinity receptor (lipoxin A4 receptor, ALXR) that has been sequenced and cloned for both mouse and human leukocytes, as well as human enterocytes and, more recently, for mesangial cells. In addition, LXA4 actions on vascular endothelial cells are mediated via a distinct non-myeloid receptor that remains to be cloned.
Mouse ALXR isolated from a spleen cDNA library had a characteristic sequence of seven transmembrane spanning G protein-coupled receptors and its homology to the human ALXR was 73% at the deduced amino acid level. Mouse ALXR showed high affinity binding to 3H-LXA4 (Kd 1.5 nM), with values similar to those obtained with the human receptor (1.7 nM) expressed in CHO cells. CHO cells stably transfected with mouse ALXR and exposed to LXA4 selectively hydrolyzed guanine triphosphate, indicating that LXA4 stimulates functional coupling of ALXR and G protein. Tissue distribution of mouse ALXR mRNA paralleled the appearance of human ALXR mRNA, and this mRNA was most abundant in mouse neutrophils, followed by spleen and lung.
In several tissues and cell types other than leukocytes, results of pharmacological experiments have indicated that LXA4 can also interact with a subclass of peptido-leukotriene receptors (cysLT1) as a partial agonist mediating their actions. Along these lines, both LTC4 and LXA4, albeit at high concentrations (> 1μM), induce contractions of guinea pig lung parenchyma and release of thromboxane A2 which is sensitive to cysLT1-receptor antagonists, an event which is not likely to be a physiologic action of LXA4 [24].
In addition to specific binding to membrane surface receptors, specific binding of labeled LXA4 is associated with subcellular fractions including granules and nucleus. In this regard, it was recently reported that LXA4 binds to and activates the aryl hydrocarbon receptor, a ligand-activated transcription factor, in a murine hepatoma cell line.
Our current understanding of the LXA4 receptor’s intracellular down-regulatory signals remains incomplete. In neutrophils, for example, lipoxins do not promote a sustained mobilization of intracellular Ca2 +, acidification of the intracellular milieu or generation of cAMP. But, LXA4 binding to its receptor triggers the activation of GTPase, phospholipase A2 and phospholipase D, responses that are inhibited by pretreatment of the cells with pertussis toxin. In addition, lipoxins are not receptor level antagonists for inflammatory stimuli such as fMLP or LTB4. For example, lipoxins inhibit LTB4 responses in neutrophils perhaps by uncoupling LTB4 receptor-initiated proinflammatory signaling, as evidenced by downregulation of CD11b/CD18, decreased IP3 formation and changes in intracellularprotein kinase C distribution. Recently, a novel polyisoprenyl phosphate signaling pathway was identified with one of its components, pre-squalene diphosphate (PSDP), being a potent negative intracellular signal in PMN. Activation of ALXR inhibits PSDP remodeling, leading to an accumulation of PSDP and potent inhibition of both phospholipase D and superoxide anion generation in PMN.
The receptor coupling in monocytes and PMN is similar to G-protein activation, being critical in both cell types. However, there could be different G-protein subtypes that diverge downstream in the signal transduction pathway to stimulate monocytes and inhibit PMN.
It was shown that small peptides selectively compete for specific 3H-LXA4 binding with PMN and recombinant human ALXR, increasing extracellular acidification rates and inducing cell chemotaxis and migration in vivo [25].
Chimeric receptors constructed from receptors with opposing functions, namely ALXR and LTB4, revealed that the seventh transmembrane segment and adjacent regions of ALXR are essential for LXA4 recognition, and additional regions of this receptor are required for high affinity binding of the peptide ligands. It appears, however, that the G-protein interactions evoked by ligand-receptor binding and their intracellular amplification mechanisms are different for peptide versus lipid ligands of ALXR, and hence they can dictate different functional responses.
Braune S et al. studied the effect of prostanoids on the bioactive lipid mediators and take part in many physiological and pathophysiological processes in practically every organ, tissue and cell, including the vascular, renal, gastrointestinal and reproductive systems. The function of platelets is influenced by mediators in the blood and the vascular wall. Activated platelets aggregate and release bioactive substances, thereby activating further neighbored platelets, which finally can lead to the formation of thrombi. Prostanoids regulate the function of blood platelets by both activating or inhibiting and so are involved in hemostasis. Each prostanoid has a unique activity profile and, thus, a specific profile of action [26].
Lipoxins in periodontal diseases
Neutrophils are within the first line of host defense, and, by their ability to phagocytose microbes; they can protect the host from infection. They can also give rise to neutrophil-dependent vascular injury and contribute to increased vascular permeability, edema, and further release of chemo-attractants, with a net pro-inflammatory effect. The involvement of the inducible cyclooxygenase isoform (COX-2) and the role of novel lipid mediators in the pathogenesis of periodontal disease are under study, and data derived from these observations have showed that periodontitis represents an important inflammatory model for the investigation of lipid mediators. The working hypothesis is that COX-2 could have multiple role(s) in the development and progression of the periodontal disease. First, crevicular fluid samples from localized aggressive periodontitis (LAP) patients were examined and found to contain prostaglandin PGE2 and 5-LO-derived products, LTB4 and the biosynthesis interaction product, lipoxin UA, As opposed to early suggestions that monocytes and macrophages were the major source of PGE2 production in periodontal disease [27], neutrophils were found to generate considerable arachidonic acid metabolites in this study. These finding suggests that neutrophils contribute to the pathogenesis of periodontal disease in ways that were not previously anticipated. Furthermore, neutrophils from peripheral blood of LAP patients, but not from healthy volunteers, also generated LXA4, suggesting that this immunomodulatory molecule may also have a role in periodontal disease (Fig. 5).

Effect of lipoxin on PMNs and monocytes.
The role of lipid mediators in the neutrophil response to Porphyromonas gingivalis was also characterized in an animal model. When P. gingivalis was introduced into murine dorsal air pouches, leukocyte infiltration was initiated. Elevated PGE2 levels in the cellular exudate and up-regulated COX-2 expression in the leukocyte infiltrate accompanied neutrophil accumulation. In addition, human neutrophils exposed to P. gingivalis also demonstrated up-regulation of COX-2 mRNA expression. Interestingly, P. gingivalis introduced into the epithelium-lined air pouch caused significant increases in the murine tissue levels of COX-2 mRNA associated with both heart and lung, supporting a potential role for this oral pathogen in the evolution of systemic events. The administration of metabolically stable analogues of lipoxin and of aspirin-triggered lipoxin potently blocked neutrophil traffic into the dorsal pouch cavity and lowered PGE2 levels within exudates without allowing infection to spread. These results show that neutrophils can provide an important source of PGE2 in periodontal tissues.Moreover, they provide strong support for the notion that lipoxin can have a protective role in periodontitis, limiting further neutrophil recruitment and neutrophil-mediated tissue injury that can lead to loss of inflammatory barriers that prevent tissue invasion by oral microbial pathogens. It has been further proposed that lipoxin generation and its relationship to PGE2 and LTB4 can be important markers for the pathogenesis of periodontal disease. Indeed, activated neutrophils from LAP patients produced lipoxin, whereas healthy neutrophils did not. Lipoxins have also been shown to stimulate proliferation and migration of stem cells of the apical papilla -the main source of dentin-like structures- as well as inhibiting their pro-inflammatory activation, which may indicate LXs have also regenerative potential other than their anti-inflammatory properties [27].
Bioactive lipids play a major role in the fine-tuning of this dynamic process in a timely manner. During inflammation and its resolution, polymorphonuclear cells (PMNs) and macrophages switch from producing pro-inflammatory prostaglandins and leukotrienes to specialized pro-resolving lipid mediators (SPMs), namely, lipoxins, resolvins, protectins, and maresins, which are operative at the local level to limit further inflammation and tissue injury and restore homeostasis [28].
Lipoxin-stable analogs were studied for their clinical inhibition of inflammation. Several LXA4 and ATL stable analogs were synthesized by means of a recombinant dehydrogenase screen assay as a relatively inexpensive and rapid screen to design suitable analogs that might function in vivo. These analogs were generated by total organic synthesis and were tested for their ability to inhibit neutrophil infiltration and changes in vascular permeability In vivo in several murine models. Methylated 15(R/S)-LXA4 is an analog of both the aspirin-triggered 15-epi-LXA4 and native LXA. Likewise, 16-phenoxy-UA., which has a phenoxy group at the C-16 position, is an analog of native LXA4 that prevents enzymatic inactivation with recombinant 15-prostaglandin dehydrogenase in vitro. These analogs have been shown to act by competition for the LXA4 receptor [29].
When applied topically to mouse ears, the lipoxin-stable analogs inhibit both neutrophil infiltration and vascular permeability changes in a concentration-dependent fashion. The inhibition of vascular permeability changes paralleled inhibition of neutrophil infiltration with both the ATL and lipoxin analogs. In addition, the impact of LXB4 analogs that resist enzymatic inactivation was also tested in vitro [30]. Of the lipoxin-stable analogs tested in this inflammatory model, inhibitory actions of 15(R/S)-methyl-LXA4 0n neutrophil infiltration and vascular permeability changes were significantly greater than those of topically applied native LXA4. In addition, a 16-para-fluoro derivative of 16-phenoxy-LXA4 was also prepared to assess whether fluorination of the phenoxy ring could enhance potency. Results indicate that 16-phenoxy-LXA4 was also potent and retained activity at levels comparable with those of 16-parafluorophenoxy-LXA4. Both LXB4 analogs inhibited neutrophil infiltration and vascular permeability. These also proved to be potent inhibitors of neutrophil infiltration in the dorsal air pouch.
Lipoxins in the management of periodontal diseases
The new paradigm posed suggests that periodontitis results from the lack of resolution of inflammation at the site of chronic infection. Faulty resolution is likely the result of failure of endogenous counter-regulatory mechanisms leading to overt chronic inflammation, as seen in certain periodontal diseases. Neutrophil-mediated tissue injury followed by a chronic inflammatory lesion is the result. It is hypothesized that limiting the destructive aspects of the inflammatory response without ablation of host defense could be accomplished by treatment with lipoxins and their analogs, resulting in the prevention of the onset of periodontal lesions.
To test this hypothesis, recently a rabbit ligature model of periodontal infection with P. gingivalis that produced significant, reproducible, periodontal lesions was developed [9]. In this model, rabbit teeth were ligatured and P.g, placed on the ligatures three times a week for 6 wks. During the course of the experiment, the animals experience 50% bone loss on infected ligatured teeth. Ligature alone or concurrent treatment with metronidizole did not result in bone loss, demonstrating the infectious nature of the lesion. In a parallel design experiment, two groups of animals received ligatures and P.g. One group received lipoxin topically applied in vehicle (6μg in 6μL) three times a week, and the other group received ethanol alone following the same regimen. At 6 wks, animals were killed and jaws harvested for x-ray analysis of bone loss, histology, and direct bone measurements. Results revealed a marked and statistically significant reduction in all disease parameters in the lipoxin-treated group. Topical application of lipoxin inhibited bone loss > 90%, inhibited connective tissue destruction, and limited connective tissue infiltration by inflammatory and immune cells. However, there was no evidence of direct tissue invasion or damage from the topically applied P.g., suggesting that the microorganism was cleared normally and lipoxins have no innate antibacterial activity [31].
Lipoxins are readily generated by cells to limit leukocyte recruitment, chemotaxis to inflamed sites, and the progression of acute inflammation to chronic inflammation. Additionally, SPMs promote the clearance of leukocytes, fibrin, and exudates, which restore damaged tissue function. In general, SPMs work by activating repair mechanisms and returning inflamed (or diseased) tissue back to normal physiological function (or homeostasis). These induce apoptosis in PMNs and promote their removal by macrophages. More importantly, exogenous SPMs can be used to reverse inflammation in periodontal and peri-implant disease and return inflamed tissue to homeostasis [32, 33].
Liu X, Wang G, Zhang T (2020) studied that the patients with AAD were often accompanied by systemic inflammatory responses, with inflammation-related cytokines (IL-6, TNF-α, CRP and MMP-9) and endotoxins levels significantly elevated. Combined monitoring of dynamic changes in inflammatory cytokines and endotoxins, as well as early interventions, have important clinical implications for evaluating the prognosis of AAD and reducing mortality [34].
Jordan PM, Werz O (2021) commented that the lipid mediators that are de-novo biosynthesized from polyunsaturated fatty acids (PUFAs) mainly by lipoxygenases and cyclooxygenases, critically regulate the initiation, the maintenance but also the resolution of infectious inflammation and tissue regeneration. The discovery of specialized pro-resolving mediators (SPMs) generated from omega-3 PUFAs stimulated intensive research in inflammation resolution, especially in infectious inflammation elicited by bacteria. SPMs are immune-resolvents that actively terminate inflammation by limiting neutrophil influx, stimulating phagocytosis, bacterial killing and clearance as well as efferocytosis of apoptotic neutrophils and cellular debris by macrophages. Moreover, SPMs prevent collateral tissue damage, promote tissue repair and regeneration and lower antibiotic requirement [35].
Nowadays, it is known that an excessive inflammatory response underlies the most prevalent human pathologies worldwide. Therefore, great biomedical research efforts have been driven toward discovering new strategies to promote the resolution of inflammation with fewer side-effects and more specificity than the available anti-inflammatory treatments. In this line, the use of endogenous specialized pro-resolving mediators (SPMs) has gained a prominent interest. Among the different SPMs described, lipoxins stand out as one of the most studied and their deficiency has been widely associated with a wide range of pathologies. In this review, we examined the current knowledge on the therapeutic potential of lipoxins to treat diseases characterized by a severe inflammatory background affecting main physiological systems, paying special attention to the signaling pathways involved [36].
Conclusion
Neutrophils play an important role in the mediation of inflammation. These cells, previously regarded as solely generators of enzymes and thus contributors of enzymatic killing mechanisms against pathogens, are gaining more significance in terms of their role at various stages of inflammation. In addition to the recent findings on the generation of lipid mediators of inflammation, persistent activity of neutrophils also contributes to the tissue destruction. Through this mechanism, the protective role of these cells turns into a deleterious action targeting the host itself. Periodontal disease is an important model in which various aspects of lipid mediators can be studied. It also represents a local phenomenon in which neutrophil-mediated tissue injury is shown to be a major contributing factor for progression of the disease. Therefore, studies aimed to elucidate the pathogenic mechanisms in periodontal disease will likely add to our understanding of various disease sequelae as well. Within this context, lipoxins and lipid mediators not only present an exciting area of research, but also have potential for the development of novel treatment strategies.
Our understanding of the pathogenesis of periodontitis has continued to evolve as our understanding of the underlying mechanisms of the inflammatory response has become more sophisticated. There have been major advances recently that have led to a change in the way we think about inflammation and the pathology that results from inflammation. The new paradigm is the resolution of inflammation as a natural step in the inflammatory process, which leads to the concept that tissue injury mediated by inflammation is a consequence of the inability of the host to resolve the inflammation, not the initial inflammation itself. This is an important distinction, because inflammation is necessary to protect the host from infection, but persistent inflammation can also cause disease-hence the notion that, in a disease such as periodontitis, resolution of inflammation in a timely fashion will protect the host from tissue injury, while the infectious agent is still cleared. The working hypothesis is that periodontal disease reflects an inability to resolve inflammation, which also may have a genetic phenotype, and predisposition.
Lipoxins are a series of oxygenated arachidonic acid derivatives formed by interactions between individual BLO and appear to function as endogenous anti-inflammatory mediators. Recently, it was demonstrated that lipoxins are produced by peripheral blood neutrophils from patients diagnosed with aggressive periodontitis. In addition, lipoxins have also been found in the GCF of these individuals. In a mouse model, it was shown that administration of metabolically stable analogues of lipoxins blocked P. gingivalis elicited neutrophil infiltration and also reduced PGE2 levels. These results support the concept that lipoxins may be involved in the regulation of local acute inflammatory responses in periodontal disease. However, additional studies are needed to elucidate the role of lipoxins in the pathogenesis of periodontitis.