
Abstract
ImmunoRNases combine tumor targeting by antibodies with the cytotoxic action of ribonucleases from the RNase A superfamily. This study investigated for the first time all catalytic active human RNase A family members (1 to 8) as effector components of antibody fusion proteins. ImmunoRNase fusion proteins were constructed using the CD30-specific bivalent recombinant scFv-Fc antibody SH313-B5. Production of the resulting entirely human immunoRNases 1 to 8 was done in mammalian cells by secretion of active forms. The immunoRNases mediated CD30-specific cell binding and showed ribonucleolytic activity. Interestingly, immunoRNases 1 and 2 were active in the presence of up to 5-/20-fold molar excess of the pancreatic RNase inhibitor (RI), which is supposed to efficiently inhibit all human RNase A activity. ImmunoRNases 3, 4, 6 and 7 were only inhibited by several fold molar excess of RI, whereas immunoRNases 5 and 8 were already completely inactive at equimolar RI concentrations. Compared to free RNases, activity and RI sensitivity were not significantly changed by antibody fusion or dimerisation. ImmunoRNase3 and 5 mediated tumor growth inhibition at low nanomolar concentrations. Anti-tumor activity was antigen-specific and did not show any correlation with ribonucleolytic activity or RI sensitivity.
Keywords
Introduction
In the last two decades, monoclonal antibodies (mAbs) became one of the fastest growing class of biological therapeutics on the market [61]. However, the success of unconjugated mAbs for cancer therapy has lacked behind initial hopes in respect of clinical efficacy. Therefore, antibodies have been intensively studied as vehicle to deliver highly cytotoxic payloads into tumors, but only recently, the first targeted toxins have been approved for therapy. The first respective drug, Mylotarg (Gentuzumab ozogamicin) was withdrawn from the market after no improvement in clinical benefit was observed in a post-approval study. The addition of a cytotocic payload (microtubule inhibitor DM1) improved the efficacy of trastuzumab, and Brentuximab vedotin is approved for the treatments of lymphomas. Key factors for such a reagent are efficient targeting and intracellular uptake, which are both depending on the antigen. Brentuximab, which recognises human CD30, has demonstrated that CD30 target, also referred to as Ki-1 [14, 16], is a suitable target to study human “immunotoxin”-like antibody fusion proteins. While unconjugated anti-CD30 mAbs achieved disappointing results in clinical trials of relapsed and refractory HL patients [17, 18, 59], The antibody drug conjugate (ADC) brentuximab vedotin was clinical effective, displayed manageable toxicities and was approved for treatment of CD30
In this study used oligonucleotides
In this study used oligonucleotides
Alternatively to ADCs, immunotoxins made of of mAbs conjugated or fused to bacterial or plant toxins like diphtheria toxin, Pseudomonas exotoxin A, saporin or ricin have been studied for more than two decades [19, 36, 43, 58, 61]. However, they frequently suffered from high unspecific toxicity and immunogenicity [5, 23, 58]. To overcome toxicity and immunogenicity issues, a number of mammalian and human enzymes have been investigated to replace heterologous toxin component [40, 53, 55, 56, 66, 72, 73]. Particularly, vertebrate RNase A family enzymes have been considered as alternative effector moiety. Human RNase A members are present in nearly all organs, tissues and body fluids which should reduce the risk of non-specific toxicity and immunogenicity [58]. Even heterologous bovine RNase A or ranpirnase (Onconase
Therefore, RNase A proteins were tested in conjugation with mAbs or fusion proteins, which are referred to as immunoRNases [54]. The amphibian Onconase, human RNases 1, 2 or 5 in combination with mAbs achieved strongly increased cytotoxicity and demonstrated the therapeutic potential of immunoRNases [10, 36, 58, 80]. The proposed cytotoxic mechanism of immunoRNases requires internalization and release of the active RNase moiety into the cytosol, where it degrades RNA substrates and inhibits protein synthesis [11, 58]. CD30 specific immunoRNase fusion proteins employing human pancreatic RNase (RNase1) or angiogenin (RNase5) inhibited CD30
In this study all catalytic active human RNase A members (RNases 1 to 8) were compared for their effects when fused to the described CD30-specific human bivalent antibody SH313-B5 [78]. The resulting immunoRNases 1 to 8 are entirely of human origin and were tested for antigen and cell binding, anti-tumor effect, ribonucleolytic activity and the sensitivity against the cytosolic human RNase-inhibitor (RI). RI is ubiquitously expressed in mammalian cells and binds RNase A with an extremely high affinity [46, 58]. It was previously proposed that the RI could abolish the anti-tumor effect of human immunoRNases by inhibiting catalytic activity of all human RNase A proteins [12, 35, 42, 46]. Thus, this study gives the first comprehensive overview about immunoRNases employing all catalytic active members of human RNase A superfamily including ribonucleolytic activity, RI sensitivity and potential anti-tumor properties.
Production and purification of immunoRNases
Sixteen different immunoRNases were constructed by fusing one of the human RNases 1 to 8 to the carboxy terminus either of the CD30-specific bivalent human scFv-Fc antibody SH313-B5 or the human negative control antibody MS112-IIB1. All immunoRNases were produced in mammalian cells by secretion in soluble form and purified by protein A affinity chromatography resulting in volumetric yields between 3 mg/L and 60 mg/L (Table 3). SDS-PAGE analysis revealed high purity of all immunoRNases (Fig. 1). The CD30-specific immunoRNases corresponded to a molecular weight of 70–75 kDa except for immunoRNases 2 and 3, which migrated at 80–85 kDa. Moreover, immunoRNases 1, 2, 3, 7, and 8 showed blurry protein bands suggesting heterogeneous glycosylation. ImmunoRNase7 displayed some proteolysis, as a faint additional band at
Production and purification of free human RNases
Free human RNases 1 to 8 without antibody moiety were produced for reference in respect of catalytic activity and RI inhibition. The expression of free RNases in mammalian cells resulted only in very low yields of recombinant protein (data not shown). However, all human RNases 1 to 8 could be successfully produced as Fc-TEV
Molecular masses and glycosylation of human RNases
Molecular masses and glycosylation of human RNases
SDS-PAGE analysis of immunoRNases 1 to 8. A total of 1 
Volumetric yields of immunoRNases 1 to 8 and free RNases 1, 4, 5 and 7
Production and purification of free recombinant human RNase 1 to 8. The human RNases 1 to 8 were produced as Fc-TEV
Ribonucleolytic activity was analyzed using two different substrates (rU, rC), which both contained only a single RNase cleavage site after C and U, respectively. Cleavage of the substrate resulted in a time-dependent increase of fluorescence as shown for RNase1 (Supplement 2). The negative control without RNase did not reveal any increase of fluorescence, which excludes potential contaminations of other RNases during production, purification and the activity assays. Enzyme efficiencies (
Enzyme efficiencies of immunoRNases 1 to 8 and of free RNases 1, 4, 5 and 7
Enzyme efficiencies of immunoRNases 1 to 8 and of free RNases 1, 4, 5 and 7
The highest in vitro ribonucleolytic activity was measured for free human RNase1, while the activities of the immunoRNases1 were
RI mediated inhibition of immunoRNases 1 to 8 and free recombinant RNases 1, 4, 5 and 7. Ribonucleolytic activity was measured with rU substrate in the presence of up to 20-fold molar excess of RI. The concentration of active RNases was normalized to samples incubated without RI (100%). Average values and standard deviations from triplicate experiments are given. n.d.: not determined.
Analytic SEC of immunoRNases 1 to 6 and 8. PBS was used as running buffer. ImmunoRNase 7 was not analyzed due to instability of the fusion protein.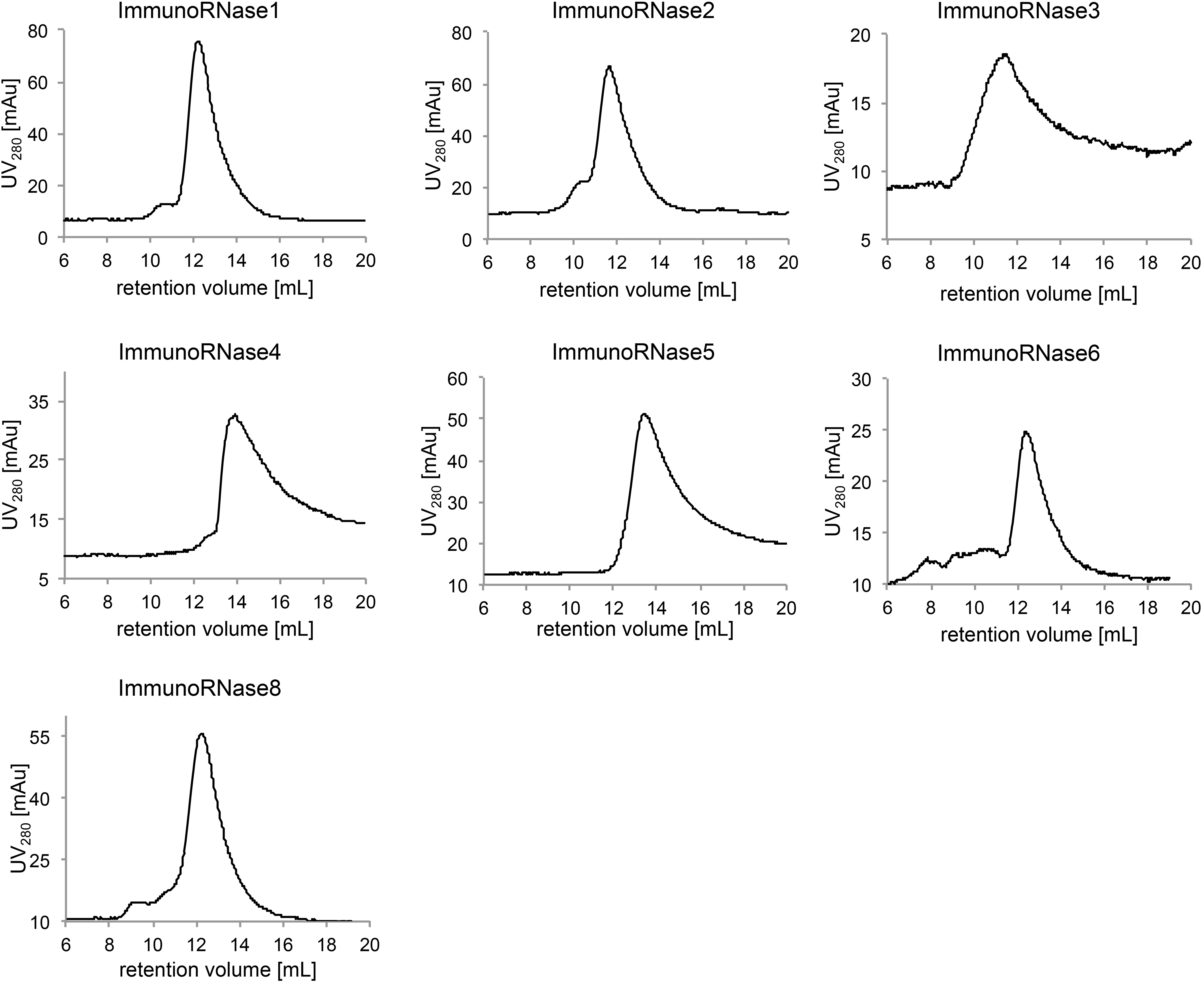
The inhibition of the ribonucleolytic activity of the CD30-specific immunoRNases 1 to 8 and the free recombinant human RNases 1, 4, 5 and 7 was analyzed in the presence of the human cytosolic RI, which was added in up to 20-fold molar excess 15 minutes before the substrate (Fig. 3). The RI had very different inhibitory effect on the catalytic activity of the tested free RNases 1, 4, 5 and 7 and the immunoRNases 1 to 8, which can be arranged into three groups. The first group is not or only slightly inhibited at a 5- to 20-fold molar excess of RI like immunoRNase1 and the corresponding free RNase1. ImmunoRNase 2 also belongs into this group, but may show higher RI sensitivity than RNase1 at higher excess of the inhibitor. ImmunoRNases 3, 4, 6 and 7 as well as the recombinant free RNases 4 and 7 belong to the second group, which is not completely inhibited at equimolar concentration of the RI, but the ribonucleolytic activity is strongly reduced in the presence of 5- to 20-fold molar excess of the RI. Both, immunoRNase4 and free RNase4 were completely inhibited at 20-fold molar excess of RI, whereas immunoRNase 3, 6 and 7 still had significant (23–29%) residual ribonucleolytic activity. The third group comprising immunoRNases 5 and 8 and free RNase5 was completely inhibited in the presence of equimolar concentrations of RI. Titration of RNase5 revealed a highly efficient strong 1:1 interaction with RI [32].
RI-mediated inhibition of immunoRNases 1, 4, 5 and 7 was similar compared to the corresponding free RNases 1, 4, 5 and 7, respectively. Therefore, the fusion of RNases and antibody did not affect the RI-mediated inhibition of these immunoRNases. Moreover, the Fc-mediated dimerization did not have any major influence on the RI-mediated inhibition.
SEC analysis of immunoRNases
The CD30 specific immunoRNases were analyzed by SEC (Fig. 4) and revealed one major peak except for immunoRNase7, which showed a pattern of degradation (data not shown). Homodimeric proteins were expected due to the dimerization of the Fc-part of the antibody moiety. The analyzed immunoRNases showed two generally different distributions. ImmunoRNases 4 and 5 had their major peak at about 130–150 kDa, which corresponded to a homodimeric, unglycosylated scFv-Fc-RNase form. In contrast, immunoRNases 1, 2, 3, 6 and 8 migrated at higher molar masses, indicating substantial glycosylation.
Cell binding of immunoRNases 1 to 8. Purified immunoRNases 1 to 8 were tested for binding to CD30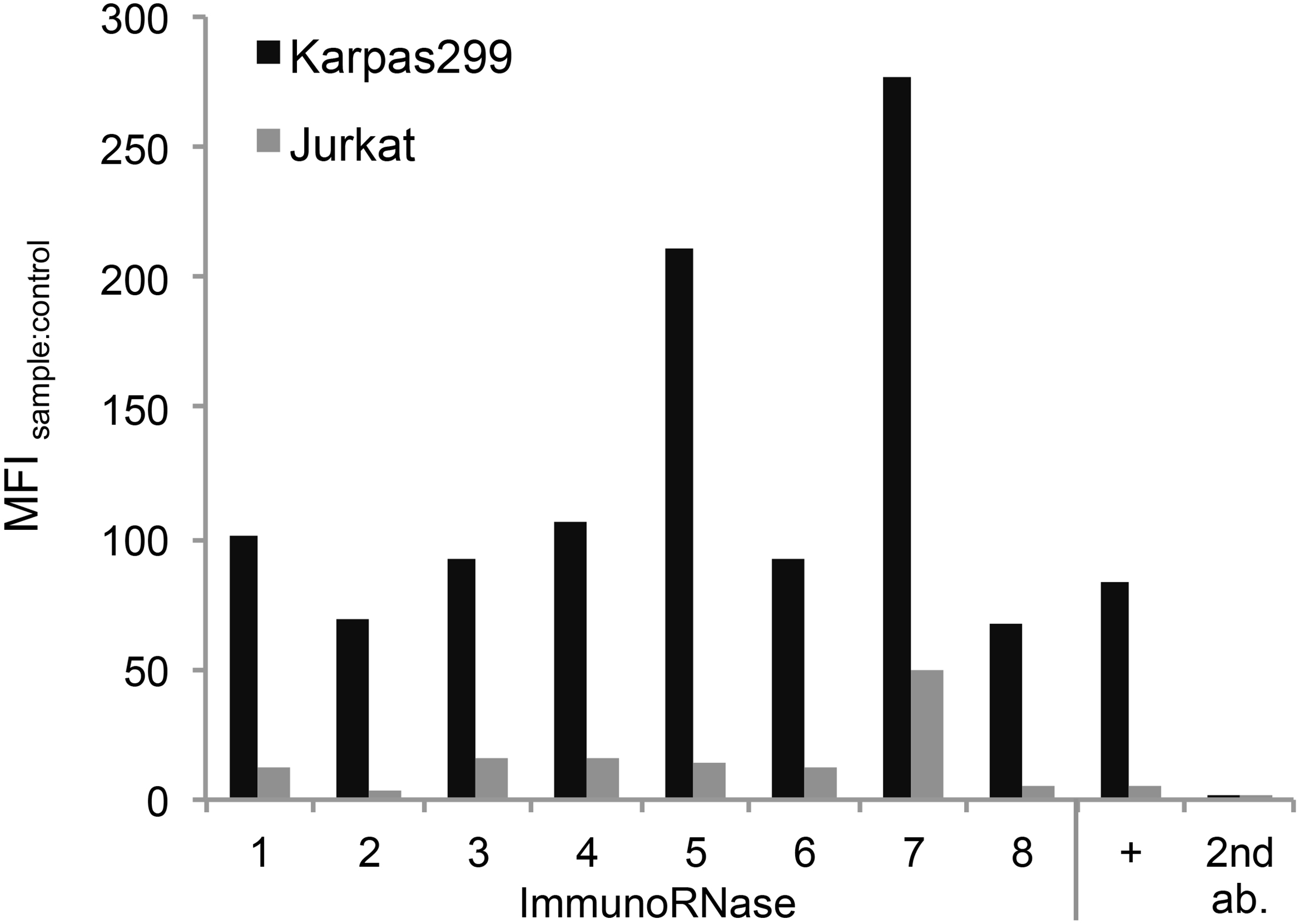
The binding of the immunoRNases to CD30
Tumor growth inhibition by immunoRNases 1 to 8. A) CD30 specific immunoRNases 1 to 8 were incubated with CD30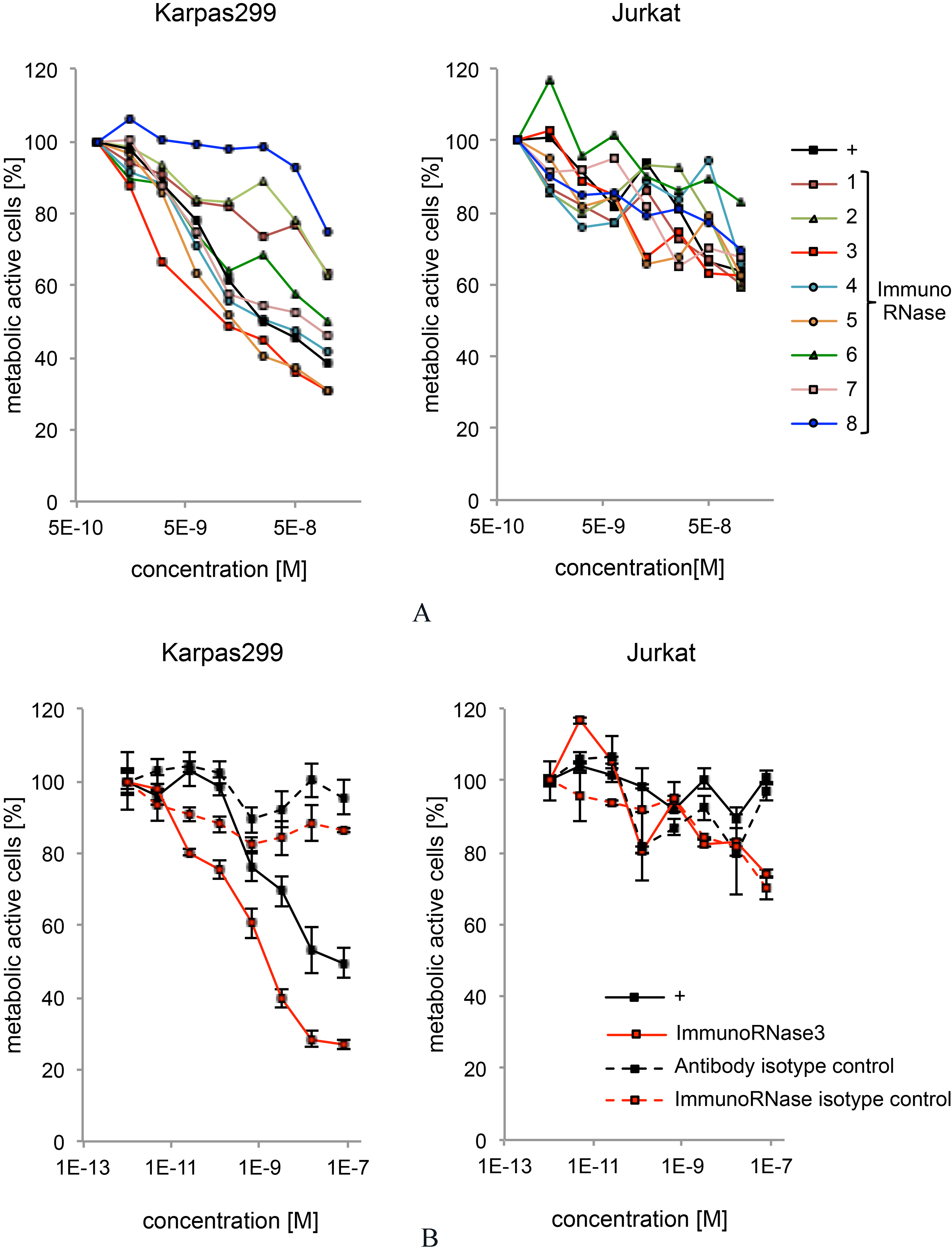
The anti-tumor effect of immunoRNases 1 to 8 was measured by an MTT assay using CD30
Discussion
ImmunoRNases consisting of human antibodies and human RNase A superfamily members represent an alternative to ADCs or immunotoxins for treatment of tumors, because they promise to overcome low efficacy, liver or kidney toxicity as well as immunogenicity and off-target binding [5, 23, 58]. Since ribonucleolytic activity has been proposed to be of central importance for the cytotoxic function of immunoRNases [36, 54, 58], the catalytic active human RNases 1 to 8 are of particular interest for tumor therapy. Consequently, in this study the human RNases 1 to 8 were fused to the CD30-specific human IgG-equivalent scFv-Fc antibody SH313-B5 [78] for direct comparison. Yields varied depending on the employed RNase moiety, but were comparable to immunotoxin fusion proteins [29, 37, 58]. Significantly, all of the immunoRNases 1 to 8 were produced in active form and secreted to the supernatant by human embryonal kidney cells in concentrations in the mg/L range. The producing cells were not harmed, which confirms previous results suggesting very low unspecific cell toxicity obtained with another human immunoRNase1 variant [37].
In contrast to these immunoRNase fusion proteins, the recombinant production of free human RNases 1 to 8 in their mature form was found to be very inefficient in E. coli (data not shown), which may to be due to high content of disulfide bonds reducing the yield of functional protein. Mammalian production allows for functional expression including correct formation of the 3–4 disulfide bridges and glycosylation, which is described for RNases 1, 2, 3, 6 and 7 [3, 24, 48, 50, 51, 52, 64, 68, 70, 71, 75, 81, 82]. However, human RNases 1 to 8 were only inefficiently secreted from HEK293 cells, which may indicate cell-dependent secretory pathways. Such alternative secretion pathways are described, for example for the RNases 2 and 3, which are stored in specialized vesicle of eosinophiles and are released upon stimulation [49, 51, 75]. To enhance recombinant secretory production, the human RNases 1 to 8 were fused to the Fc-part, which can promote expression and secretion of other proteins in mammalian cell lines [9, 37]. RNases 1, 4, 5 and 7 were successfully purified after site-directed cleavage from the Fc-moiety using TEV protease [8, 9, 15, 22, 62], whereas RNases 2, 3, 6 and 8 were either sensitive to non-specific TEV cleavage or degradation.
All immunoRNases 1 to 8 and the free recombinant human RNases 1, 4, 5 and 7 exhibited ribonucleolytic activity. Free RNases 1, 4, 5 and 7 revealed similar or slightly higher enzyme efficiencies compared to the much larger dimeric immunoRNases, which can be due to their higher diffusion rate and better accessibility of their active center [2]. Other groups also described lower activity of immunoRNase fusion proteins, e.g. only 6–13% activity of an immunoRNase2 fusion protein compared to free RNase2 [41].
In this study, the immunoRNases and recombinant free RNases revealed activity corresponding to previously published data. However, there were differences regarding the preference to cleave after U or C compared to previous publications, which may be explained by differences of the employed expression systems, activity assays and test substrates. Moreover, complex substrates with potential secondary structures were not investigated in this study. Among all tested immunoRNases, only immunoRNase/RNase 5 showed a significantly higher activity than described by any other group [31], which we assume is caused by the mammalian expression.
Mammalian cells express high levels of cytosolic RI, which is supposed to bind to all members of the RNase A superfamily with extremely high affinities [35, 42, 46] and to inactivate their enzymatic activity efficiently to protect cells from RNA damage by entrance of RNase A proteins [64]. Lowering affinities between RNases and RI enhanced the RNase-mediated cytotoxicity [4, 6, 35, 69, 74, 79]. Fusion with an antibody moiety or insertion of additional domains into the RNase structure can also lower RI interaction and inhibition of RNase activity [4, 69]. In this study, the RI had very different effect on the enzymatic activity of the tested immunoRNases and RNases. Since no major differences were observed between immunoRNases 1, 4, 5 and 7 and the corresponding free RNases 1, 4, 5 and 7, the fusion with the antibody moiety as well as the dimerization mediated by the Fc-moiety does not seem to influence the inhibition by the RI.
Three major groups of immunoRNases (and RNases) were identified according to different inhibition characteristics in the presence of the RI. The highly efficient inhibition of immunoRNases 5 and 8 and the corresponding free RNase5 indicates a 1:1 inhibitory interaction with the cytosolic RI, which was previously described with inhibitory concentrations in the femtomolar range [24, 32, 64, 68]. In contrast, immunoRNases 3, 4, 6 and 7 as well as the free RNases 4 and 7 were only partially inhibited at equimolar concentrations of RI. ImmunoRNases 1 and 2 as well as free recombinant RNase1 were not or only slightly inhibited by the RI even at 5- to 20-fold molar excess of RI, which was quite unexpected [4, 6]. However, very recently, several IgG-based immunoRNases1 fusion proteins were described to be not inhibited by a 50-fold molar excess of RI in an anti-tumoral study [57], which may require a new view on RI:RNase1 interaction and its physiological importance. It should be noticed that all immunoRNases and free RNases were incubated with the RI 15 minutes before addition of the substrate to allow to reach the equilibrium of RI:RNase complex formation before starting the activity assay. It cannot be entirely excluded that previous studies differ from these results due to different substrates and RNase activity assays, because small hypersensitive fluorogenic substrates [31] may still access the active site after RI-binding. However, in this case different models would be required to explain the interaction of RI with individual RNase A family members.
The immunoRNases 1 to 8 formed stable disulfide-linked scFv-Fc-RNase homodimers. Only immunoRNase7 showed a tendency of degradation. Moreover, immunoRNases 1, 2, 3 and 6 showed a pattern of high glycosylation, which was dependent on the RNase moiety, because all fusion proteins shared the same scFv-Fc antibody moiety. Glycosylation of RNases 1, 2, 3, 6 and 7 is in accordance with previous investigations [20, 63, 64]. ImmunoRNases 4, 5 and 8 did not show any signs of high or diverse glycosylation, which was confirmed for the corresponding free RNases 4, 5 and 8 by other groups [20, 64].
Each of the immunoRNases 1 to 8 containing the anti-CD30 antibody SH313-B5 specifically bound to CD30
The CD30-specific scFv-Fc antibody SH313-B5 inhibited the growth of CD30
Since RNase3 did not mediate the highest RNase activity and was at least partially sensitive to the RI, other mechanisms of inhibitory action have to be considered. For example, immunoRNase3 was strongly glycosylated [64], which may enhance cell interaction and internalization. RNase3 is also described to destabilize cell membranes by formation of pores, which can lead to high cytotoxicity [51, 75].
This study further provides the first comprehensive comparison for all catalytically active human RNase A members produced in the same way and using the same substrates and assays. Moreover, the catalytic activities of immunoRNases 1 to 8 and free RNase 1, 4, 5 and 7 were tested in the presence of RI. RI mediated inhibition was quite different among the different members of the gene family. Interestingly, immunoRNases 3, 4, 6 and 7 were only partially inhibited at equimolar RI concentrations, whereas immunoRNases 1 and 2 were not or only slightly inhibited even in 5- to 20-fold molar excess of RI, which draws new light onto the RI:RNase interaction and its biological function. ImmunoRNases 1 to 8 mediated inhibitory effects on CD30
Material and methods
Cell lines and cultivation
HEK293-6E cells (NRC, BRI, Montreal, Canada) were cultivated at 37
The CD30
Isolation of RNase genes
The cDNA encoding human RNases 1, 4, 5 and 6 were kindly provided by Zoltán Konthur (Max Planck Institute of Molecular Genetics, Berlin) from the ORFeome library (
Cloning, production and purification of immunoRNases
The gene fragments of the CD30-specific single chain (sc)Fv antibody SH313-B5 [78] and the DNA encoding the human IgG1-Fc moiety were fused resulting in a sequence encoding a bivalent scFv-Fc antibody. Gene fragments encoding a seven amino acid spacer (GSGGGTS) and the cDNA encoding one the mature human RNases 1 to 8 were added to result in a fusion to the carboxy terminus of the human scFv-Fc antibody in mammalian expression vector pCMV5.2-hIgG1-Fc-XP [45]. Oligonucleotides used for amplification of linker and RNases are summarized in Table 1: RNase1 (#968 and #827), RNase2 (#1118 and #849), RNase3 (#1119 and #851), RNase4 (#1120 and #853), RNase5 (#1121 and #855), RNase6 (#1122 and #857), RNase7 (#1123 and #859) and RNase8 (#1124 and #861). The resulting gene constructs will be referred to as immunoRNases 1 to 8 in this article. In addition to CD30-specific immunoRNases 1 to 8, control constructs were generated using the human scFv-Fc antibody MS112-IIB1 [60] that does not bind to human cells.
The expression was performed in HEK293-6E cells in 500 mL shaking flasks and 100 mL F17 medium. Cells were transfected at cell densities between 1.3 to 2.0
The elution fraction was analyzed for purity, integrity and degradation using sodium dodecylsulfate (SDS) polyacrylamide gelelectrophoresis (-PAGE) and Coomassie brilliant blue staining. Concentrations were determined using NanoDropND-1000. All fractions were stored at 4
Cloning, production and purification of free human RNases
The gene fragments encoding the mature form of human RNases 1 to 8, i.e. without leader sequence, were amplified with oligonucleotides according to Table 1: RNase1 (#826 and #827), RNase2 (#848 and #849), RNase3 (#850 and #851), RNase4 (#852 and #853), RNase5 (#854 and #855), RNase6 (#856 and #857), RNase7 (#858 and #859) and RNase8 (#860 and #861) and sub-cloned into the expression vector pYD5 [76] (National Research Council, Biotechnological Research Institute, Montreal, Canada). The resulting gene constructs encoded fusion proteins consisting of human IgG1-Fc part, TEV protease cleavage site (TEVcs) and one of the RNases 1 to 8.
The expression, purification and analysis of elution fractions of the Fc-TEV
TEV protease was kindly provided by Konrad Büssow (Helmholtz Centre for Infection Research, Braunschweig, Germany). The cleavage of Fc-TEV
SDS-PAGE
Protein samples were prepared for 10 min at 95
Western blot
Protein samples were prepared for 10 min at 95
After washing the membrane twice with PBS containing 0.1% (v/v) Tween 20 and once with substrate buffer (100 mM Tris-HCl, 0.5 mM MgCl
RNase activity assay
The RNase activity assay was modified from Kelemen et al. [31]. The two different substrates contained 6-carboxyfluorescein (6’-FAM) as fluorophor and Black-Hole-Quencher1 (6’-BHQ1) as quencher. They were linked to the 5’ and 3’ termini of a single stranded chimeric DNA/RNA oligonucleotide sequence, which consisted of one desoxy-adenine (dA), a single uridine (U) or cytidine (C) representing the single scissile bond and two downstream dA. The 6’-FAM-dA-rU-(dA)
The free RNases or immunoRNases were diluted in 150
Inhibition of RNase activity by human RNase inhibitor (RI)
Serial dilutions of human RI (RNasin, Promega) were added to the free recombinant RNases or immunoRNases in 150
Size exclusion chromatography (SEC)
The molecular weights of the immunoRNases were determined via analytical SEC with Äkta purifier system (GE Healthcare). The column Superdex200 10/300 GL (GE Healthcare) was used with a flow rate of 0.3 mL/min. PBS was used as running buffer and 100
Binding studies using flow cytometry
A total of 1
MTT viability assay
A total of 5
Footnotes
Acknowledgments
We acknowledge Michael Hust and Saskia Helmsing for the provision of the CD30 specific antibody genes. We also thank Konrad Büssow (Helmholtz-Center for Infection Biolology, Braunschweig) for providing the TEV protease and Zoltán Konthur (Drug Response Dx GmbH, Hennigsdorf, and Max Planck Institute of Colloids and Interfaces, Berlin, Germany) for providing RNase 2, 3, 4, 5, and 6 ORF vectors.
Abbreviations:
Appendix
SEC purification of RNase 1 from Fc-TEV
RNase activity assay with free RNase1 and 5 nM rU substrate in 200 