
Abstract
Antibody drug conjugates (ADCs) represent a promising and an efficient strategy for targeted cancer therapy. Comprised of a monoclonal antibody, a cytotoxic drug, and a linker, ADCs offer tumor selectively, reduced toxicity, and improved stability in systemic circulation. Recent approvals of two ADCs have led to a resurgence in ADC research, with more than 60 ADCs under various stages of clinical development. The therapeutic success of future ADCs is dependent on adherence to key requirements of their design and careful selection of the target antigen on cancer cells. Here we review the main components in the design of antibody drug conjugates, improvements made, and lessons learned over two decades of research, as well as the future of third generation ADCs.
Keywords
Introduction
For several decades, chemotherapy has remained the preeminent modality for the treatment of cancer [1, 2]. The earliest category of chemotherapeutics tested in humans were chlorambucil and cyclophosphamide, which were reported to induce cellular cytotoxicity by DNA alkylation. The recognition that folic acid stimulates cancer cell growth led to the synthesis of antifolates, such as methotrexate, as antitumor agents. This was followed by nucleoside analogues, such as 5-fluoruracil, and cytosine arabinoside (ara-C), which essentially interfere with DNA synthesis. DNA inter-chelating agents such as cisplatin, actinomycin D, anthracyclines (e.g., Doxorubicin) and tubulin targeting molecules (Taxanes, Vinca alkaloids) subsequently came into play. The triumph of eliminating cancer cells was unfortunately retarded by a lack of tumor selectivity, in which chemotherapy ended up killing normal cells, eventually leading to unforeseen systemic toxicities. However, it was soon discerned that therapeutics with non-overlapping toxicity profiles and different mechanisms of action could be combined at different dose regimen to yield synergistic antitumor activity. Combinatorial chemotherapy thus emerged as a modality for the treatment of most cancers [3]. Clinicians worked with a plethora of individual drugs and their combinations to define the maximum tolerated dose (MTD), minimum effective dose (MED) and later, metronomic dose (MET) in order to reach an improved therapeutic index.
Antibody Drug Conjugate. (A) General depiction of the structure of an antibody drug conjugate (ADC). The cytotoxic payload is conjugated via variable types of linkers to a human, humanized, or hybrid monoclonal antibody (mAb). The antigen binding region of the mAb is indicated in light green. (B) Mechanism of action of ADCs. The circulating ADCs binds to cancer specific antigens and gets internalized through receptor mediated endocytosis (RME). Once internalized, the linker is either cleaved by a change from the bloodstream to the cellular environment, or by active degradation by lysosomes. The cytotoxic compound then diffuses through the cytoplasm to disrupt microtubule assembly or into the nucleus to induce DNA damage, finally resulting in cell apoptosis.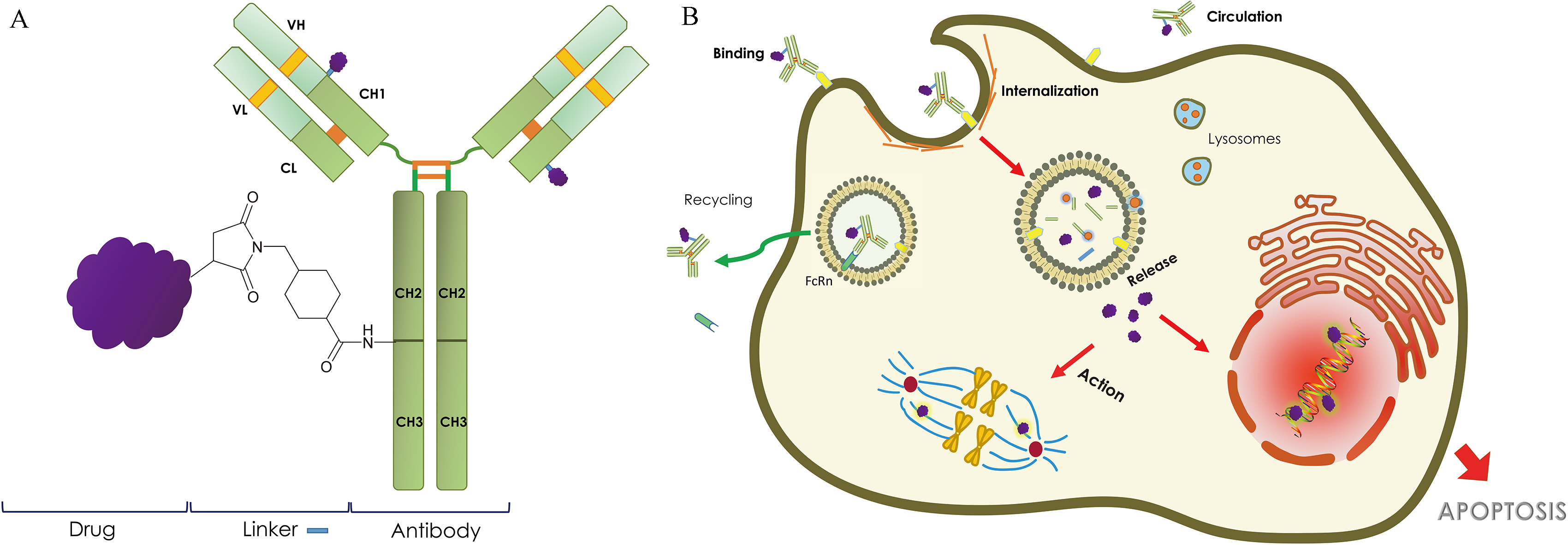
In more recent decades, an increased understanding of cancer biology has spawned a new era of targeted cancer treatment by exploiting certain properties of tumor cells in contrast to normal cells [4]. These salient features, popularly coined the ‘hallmarks of cancer’ by Robert Weinberg and Douglas Hanahan, enable tumor cells to survive, multiply, and metastasize by altering several cellular mechanics, which include: attaining an autonomy in growth signals, insensitivity to anti-growth signals, evasion of apoptosis, infinite replicative potential, sustained angiogenesis, tissue invasion and metastasis, reprogramming cellular metabolism and evasion of immunological destruction. Targeted therapy of the ‘hallmarks of cancer’ promises selective cytotoxicity to the tumor cells, which ensures overall lower toxicity to the patient and yields a higher therapeutic index compared to traditional chemotherapy.
Cancer cells differ from normal cells due to genomic instability causing mutations in oncogenes and/or tumor suppressor genes [5]. Once the integrity of the genome is compromised, cells are more likely to develop additional genetic faults; some of which may give rise to tumor-specific antigens (found only on the surface of tumor cells) or tumor-associated antigens (overexpressed on tumor cells, but also present on normal cells) [6]. Ongoing research has demonstrated that several human cancers express unique tumor-specific or tumor-associated cell surface antigens, which are of great value as targets for monoclonal antibody (mAb)-based therapy [7]. Besides developing small and large molecule based receptor-mediated targeted cancer therapy, enormous efforts have also been directed towards developing an entirely new class of tripartite anti-cancer drugs, namely, antibody-drug conjugates (ADCs). These drugs are comprised of a tumor-specific mAb conjugated to a potent cytotoxin via a ‘stable’ linker (Fig. 1). In this review we summarize clinical advancements, challenges and future perspectives in the development of ADCs.
ADCs are one of the most intricate drug delivery platforms in the existing range of oncology medications. They are immuno-conjugates composed of recombinant monoclonal antibodies (mAbs), grafted to a small molecule cytotoxic agent via covalent linker [8]. However, some major concerns impeding their clinical success, namely: (i) the expression of the target antigen on the tumor versus normal tissues, (ii) the rate of internalization of the payload, (iii) the selection of payload for the tumor indication, and (iv) the linker stability and off-target toxicities [9]. It is therefore not surprising that we have only few commercially available agents despite over one hundred clinical trials evaluating this platform.
The journey began with promising preclinical data from a study demonstrating the antitumor effect of BR96-doxorubicin on breast cancer xenografts, where doxorubicin was conjugated to an IgG
Leading antibody drug conjugates in the market and in clinical trials
Leading antibody drug conjugates in the market and in clinical trials
(FDA) was gemtuzumab ozogamicin, an anti-CD33 mAb conjugated to calicheamicin, originally developed and marketed by Wyeth for the treatment of patients with acute myeloid leukemia (AML). Following a post-approval study in which gemtuzumab ozogamicin failed to exhibit improved survival and was associated with higher toxicity than chemotherapy, the ADC was removed from the market by Pfizer in 2010 [13].
Subsequently, two of the next-generation ADCs (brentuximab vedotin and trastuzumab emtansine) received their FDA approval in 2011 and 2013, respectively (Table 1). First, brentuximab vedotin, a conjugate of a chimeric IgG1 mAb specific for CD30 and four monomethyl auristatin E (MMAE) moieties per IgG (Seattle Genetics), for the treatment of relapsed or refractory Hodgkin lymphoma (HL) and systemic anaplastic large cell lymphoma (ALCL). Secondly, trastuzumab emtansine, a conjugate of a humanized IgG1 mAb specific for human epidermal growth factor receptor 2 (HER2) and 3–4 DM1 (N2’-de acetyl-N2’-(3mercapto1oxopropyl)-maytansine) moieties per IgG, for use in HER2 positive metastatic breast cancer (Roche). These two were also approved by the European Medicines Agency (EMA) [14, 15]. Since 2013, the field has made vigorous progresses, highlighted by over 60 ADCs currently in clinical trials [16]. The latest breakthrough in the field was the advent of inotuzumab ozogamicin (Pfizer) in 2017, approved together by the FDA and EMA for the treatment of adults with relapsed or refractory CD22-positive B-cell precursor acute lymphoblastic leukemia (ALL) [17, 18]. Herein, we will reflect upon some of the lessons learned over the last two decades in the development of ADCs.
Selection of target antigen
Target antigens expressed on the surface of tumor cells are preferentially recognized by the monoclonal antibody of ADCs and thus play a pivotal role in accumulating the cytotoxic agent at the tumor site. A higher binding affinity ensures higher tumor target engagement, but some in vivo studies have also suggested that antibodies with lower binding affinity penetrate solid tumors to a greater extent [19]. Besides efficiently binding to the surface of tumor cells, the ability of the antibody-antigen complex to become internalized into the cell and enable intracellular delivery of the attached cytotoxic payload is an equally important factor. This receptor-mediated endocytosis, in principle, is governed by the nature of the antigen; for example, EGFR is known to be well internalized [20]. Non-internalized ADCs can induce significant systemic toxicity, in many cases a ‘bystander effect’ [21].
Identification and validation of adequate antigenic targets for the mAb component still remains an arduous obstacle in the clinical success of ADCs. Oftentimes, tumor antigens are not restricted to tumor cells and are expressed on normal tissues as well; rendering them tumor-associated and not tumor-specific [22]. The bio-distribution and therapeutic index of an ADC, therefore, crucially depends on the relative expression and distribution of the target antigen on tumor as well as normal tissue. For example, the clinical emergence of BR96-doxorubicin was impeded by significant on-target toxicities, including eventually hemorrhagic gastritis, at levels even higher than systemically delivered free doxorubicin [23]. The latent source of the hemorrhagic gastritis ultimately was revealed to be a formerly unrecognized expression of Lewis
It is worth mentioning that the target antigen of an ADC does not necessarily need to be a target for which the naked mAb shows activity. For example, mAb trastuzumab was first developed for targeting HER2 expressed in breast cancer cells (Roche, 1998). Fifteen years later, the ADC trastuzumab emtansine was developed with same mAb (trastuzumab) coupled with cytotoxic drug component maytansine derivative DM1 (emtansine) for the treatment of breast cancer. In contrast, approved ADC brentuximab vedotin is active in various lymphoproliferative diseases, even though the antiCD30 antibody brentuximab showed modest activity in ALCL in clinical settings [27]. Similar observations were made with antiCD138 mAb [28]. Therefore, the validation of activity of the naked mAb is not a requirement for the development of an active ADC.
Interestingly, an attempt has been made by Pfizer to develop an ADC (PF06647263, in phase I clinical trial) directed towards triple negative breast cancer (TNBC), for which no naked mAbs are approved. Comprised of a humanized mAb directed against the breast cancer antigen ephrin A4 and conjugated to calicheamicin, this ADC (PF06647263) conferred sustained tumor growth inhibition in preclinical experiments with both TNBC and ovarian cancer xenografts [29]. In another case, Glembatumumab vedotin (CDX-001) an ADC which targets cancer cells expressing transmembrane glycoprotein NMB (GPNMB), was found to prolong progression-free survival in patients with advanced TNBC [30].
Selection of cytotoxic drug
A prevalent, yet false assumption is “one size fits all” when it comes to drug selection: “all proliferating cells will become responsive to a specific drug if transported by an ADC”. This sets back years of clinical research experience and clinical trials, which point to specific chemotherapeutics, delivered at specific concentrations, leading to the treatment of specific indications. Antimicrotubule agents, such as vinca alkaloids, maytansines, auristatins, and dolastatins, were active in ALL, Hodgkin’s disease, and non-Hodgkin’s lymphoma, but showed intrinsic insensitivity toward colon, gastric, uterine, pancreatic, cervical, prostate, and hepatocellular carcinoma [33, 34, 35, 36]. In 2008, in a preclinical evaluation of SAR566658, an anti CA6 DM4 ADC, no tumor regression was observed in pancreatic cancer despite over expression of the target antigen [37].
Drug delivery by mAb is critically dependent on the following important factors: i) Drug to antibody ratio (DAR), i.e., number of drug molecules that can bind to a single mAb, ii) the number of antigen molecules on the cell surface to which the antibody can bind (typically 5,000 to 1
Many early ADCs reported a decrease in anti-proliferative potency compared to that of the parent drug molecule. This could possibly be attributed to the covalent conjugation of the compound to the mAb, as well as the different modes of cellular uptake of conjugated versus free drug [31, 32]. In general, the number of cytotoxic drug molecules required to commence cancer cell death could be very high (
In order to achieve clinical success, a hydrophobic drug must get properly solubilized in the aqueous sphere of the mAb, avoiding antibody aggregation to guarantee a prolonged half-life and to prevent rapid clearance from the body [38]. The site for chemical conjugation should be carefully selected so that the antitumor potency of the drug remains intact [39]. FDA approval of brentuximab vedotin and trastuzumab emtansine declares that cytotoxins used therein, a maytansinoid and an auristatin, satisfy these criteria. Besides these two classes of chemical scaffolds, a few others have also paved their way to clinical trials, namely, calicheamicins, irinotecan derivatives, tubulysins, indolinobenzodiazepines, pyrrolobenzodiazepines (PBDs), duocarmycins, and doxorubicin [39, 40].
Design of the linker
The ‘Linked-In’ chemistry plays the most crucial role in deciding the therapeutic index and safety of ADCs. An untimely release of payloads while in circulation can lead to off-target systemic toxicity. Effective linkers are, therefore, designed to sustain survival of ADCs in circulation and to release the drug by efficient cleavage at the cancer site [41]. Two major parameters governing the design of ideal linker are: i) cleavability of the linker and ii) conjugation chemistry [42]. Linkers are broadly classified into two groups: cleavable and non-cleavable linkers [43].
Cleavable linkers
Design of cleavable linkers harnesses the differences between the physiological conditions of circulation and the intracellular conditions within cancer cells, or in other words, tumor microenvironment, which elicits bond-breaking and release of active ingredients in the cytosol once the adduct trespasses [44]. There exist three types of cleavable linkers: hydrazone, disulfide, and peptide linkers, each of which gets sensitized differentially by intracellular conditions. Acid-labile hydrazone linkers remain stable in neutral pH and utilize the low pH within the endosome/lysosomes to cleave the conjugation and release the drug from the ADC [45]. Linkers present in gemtuzumab ozogamicin and inotuzumab ozogamicin are examples of this kind. Unfortunately, in clinical studies, such linkers were shown to be associated with non-specific breakdown and therefore significantly higher toxicity in comparison to standard therapy [46, 47]. Next, disulfide linkers emerged as a possible alternative. They exploit raised abundance of thiol molecules within tumors, especially generated during stress like hypoxia, for thiols are involved in survival and growth of tumor cells [48]. The steric hindrance of disulfide bridges can be optimized to control premature release inside the cell. Lorvotuzumab mertansine, coltuximab ravtansine and anetumab ravtansine are stock examples of this kind. The disulfide linker is initially cleaved to release the thiol compound, which is subsequently Smethylated by cellular methyltransferase activity [49]. The last type of linker is the enzyme labile peptide motif or amide bond, which is subject to fragmentation at low pH by lysosomal proteases such as cathepsin-B and plasmin [50]. A cogent example is brentuximab vedotin, where a split of valine-citruline bonds is followed by drug release [51]. When compared with the other two linker types, peptide linkers offered better stability, specificity and drug release pattern [52, 53]. Clearly, all of them rely on distinguishing cellular mechanics.
Non-cleavable linkers
While all of the cleavable linkers rely on intracellular mechanics for drug release, non-cleavable linkers engage lysosomal degradation only. Lysosomal proteases disintegrate the mAb, producing a neonate, an amino acid-linker-drug adduct which in turn becomes the active ingredient. Non-cleavable linkers have shown enhanced stability in circulation with prolonged half-lives [54]. This understanding was translated in the clinical development of trastuzumab emtansine, where the thioether succinimidyl4- (Nmaleimido-methyl) cyclohexane1carboxylate linked to DM1 (SMCC-DM1) was catabolized to lysine-SMCC-DM1 as active drug [55]. Another ADC, depatuxizumab mafodotin, consisting of maleimi-docaproic acid linked to MMAF (mc-MMAF), yields cysteine-mc-MMAF upon catabolism [56].
Importantly, in the context of linker stability, special care should be taken while choosing the number of drugs to be conjugated to an antibody. An unstable linker and inferior ADC uptake demand a high DAR to reach expected cytotoxicity level. This can, however, cause immunogenicity and lead to faster clearance [57]. A study from Seattle Genetics, where ADCs with 2, 4, or 8 drugs per mAB (E2, E4, E8 respectively) were synthesized, revealed that in vivo efficacy of E4 was comparable to that of E8, while E2 required higher dosing. Single dose MTDs of E8, E4, E2 were 50 mg/kg, 100 mg/kg and 250 mg/kg respectively and therapeutic indices were 100 for E8, 200 for E4, and at least 250 for E2. Moreover, clearance value increased with increase of drug loading [58].
The bystander effect
Another important aspect of linker design is the presence or absence of bystander effect, which induces target-cell mediated killing of other cells in the vicinity. Generally, cellular efflux enables hydrophobic drugs to diffuse out of an antigen positive cell into adjacent antigen negative cells. Since target antigen expression in solid tumors mostly happens in a heterogeneous manner, bystander effect could well be useful to kill antigen negative cancer cells [59]. However, the bystander effect depends on the charge of the effector. The amino acid-linker-drug metabolite of non-cleavable linkers is charged and thus not capable of crossing the lipid bilayer of the plasma membrane. It is therefore only observed in the case of cleavable linkers [60]. For example, on cleavage, brentuximab vedotin releases MMAE, which is neutral and able to cross cell membranes and kill surrounding epithelial cells [61, 62]. On the other hand, cleavage of denintuzumab mafodotin results in a MMAF derivative with a charged carboxyphenylalanine residue, which does not spontaneously cross plasma membranes and is thus less toxic to bystander cells than MMAE. Moreover, trastuzumab duocarmazine (an ADC linked via reducible disulfide bond) showed bystander cytotoxicity in HER2 negative mixed with HER2 positive cells, while trastuzumab emtansine (linked via non-reducible thioether) did not [63].
Linkers with enhanced polarity to minimize MDR
An arduous obstacle for the treatment cancer is multi drug resistance (MDR): cancer cells routinely become impervious to drugs by up-regulating MDR1 protein content. MDR1 has been shown to preferably efflux hydrophobic rather than hydrophilic compounds. As necessitated, polar or charged linkers were designed and synthesized in order to get polar or charged derivatives from the ADC upon cleavage. This idea was supported by improved effectiveness against MDR1+ cells. For example, in a phase I trial with (FR
Next generation conjugation chemistry
Conventionally, most of the second-generation ADCs share a commonality in structural features, such as a linker which reacts with certain amino acids present in the mAbs [65]. For example, a linker was conjugated to the
The last few years, there has been a push to develop alternate technologies that involve the introduction of a specific reactive moiety to a selective position of the antibody, eventually expanding the therapeutic window of the ADC. Summarized below are a few such strategies.
Cysteine insertion
As of now, many pharmaceutical companies have investigated a new class of ADCs where additional cysteines were conjugated to different locations of the mAB to get rid of the heterogeneity and to gain a uniformity in DAR [70, 71]. As expected, these ADCs turned out to be better tolerated, having a wider therapeutic window than traditional ADCs in preclinical studies performed with rodents and non-human primates [72].
Conjugation via unnatural amino acids
In this method, using stop codons, non-native amino acids (UAAs) are inserted into the antibody during transcription. Common examples are selenocysteine (sulfur is replaced by selenium) and acetylphenylalanine (contains a ketone group) among others [73]; which despite these insertions are otherwise structurally similar to their wild-type counterpart. In 2013, Tian et al. prepared a novel site-specific ADC via oxime bond formation between ketones on the side chain of the incorporated non-native amino acid and hydroxylamine functionalized monomethyl auristatin D [74]. Compared with conventional cysteine conjugated ADCs, these newly derived ADCs demonstrated superior in vitro efficacy and selectivity, in vivo pharmacokinetics and efficacy in rodent models, and more interestingly, superior efficacy in lower antigen (2+)-expressing target cells.
Enzyme-aided conjugation
Another method employs bacterial transglutaminase, which catalyzes a covalent bond formation between a primary amine of a drug and specific number of glutamine side chains of a modified mAb [75]. Pfizer utilized this technology to develop a TROP2specific ADC RN927C for a microtubule inhibitor (MTI) linker-payload PF-06380101, which entered phase I clinical trials in patients with advanced and/or metastatic solid tumors [76].
Non-oncology applications
The therapeutic potential of ADCs has also been investigated beyond cancer. Of these, an immunosuppressive ADC is reported by Rongsheng E. Wang to selectively deliver dasatinib, an inhibitor of Src-family kinases and clinically used to treat BCR-ABL-dependent CML, to human T-lymphocytes highly expressing chemokine receptor 4 CXCR4 on its surface [77]. ADC potency has also been evaluated in infectious disease models, such as bacterial infections caused by Staphylococcus aureus. In 2015, a group of scientists from Genentech developed an antibody-antibiotic conjugate (AAC) where an anti-S. aureus antibody (THIOMAB) was site-specifically conjugated to a potent antibiotic (dmDNA31) which is activated only after it is released in the proteolytic environment of the phagolysosome (phase I trial, NCT02596399) [78].
Future Possibilities
It has become a matter of fact that highly effective targeted therapies, often observed in murine/rodent tumor models, seldom translate to clinic; the major reason being absence of target antigen in the murine tissue except the tumor xenograft. As a solution, bio-distribution profiles of the therapeutics using companion diagnostics such as radiolabeled imaging products are evaluated in both mice and humans [79]. Despite gathering profuse knowledge on highly selective antibody generation and their pharmacokinetic and pharmacodynamic properties, we still lack an understanding of tumor targeting properties of mAbs both at a microscopic as well as macroscopic level. Simultaneous efforts have thus been directed towards developing small molecule ligands which can target tumors rapidly and more efficiently [80].
Moreover, low single agent responses of some ADCs have prompted combinatorial trials of the active single agents. For example, in a phase 1/2 study (NCT02572167), the combination of brentuximab vedotin plus nivolumab has been proven to be efficacious and well-tolerated, which can actually provide patients with refractory/relapsed Hodgkin’s lymphoma a substitute to conventional chemotherapy [81]. Bio-orthogonal toxicity profiles of cytotoxins and cytokines has made their combinations very attractive for mAb-based delivery. Intending to gain insight about ADCs’ ability to promote anticancer immunity, Gutbrodt et al. performed a F8 antibody-based delivery of cemadotin, a potent cytotoxic drug and IL2, a strong proinflammatory cytokine in an immunocompetent syngeneic mouse model of AML. Combinatorial treatment resulted in tumor eradication mediated by CD8 (+) T cells and NK cells [82]. In another study, List et al. developed a novel immunocytokine-drug conjugate, where DM1 and IL2 were conjugated to F8 antibody in a site-specific manner and showed very potent anti-cancer activity [83].
Recent progresses in the field have revived an interest in identifying potent natural products with applicable toxicities. In the near future, clinical advancements of novel ADCs are likely to reveal new mechanisms of action for the anti-tumor payloads [84].
Conclusions
Regardless of impediments in their conceptualization and synthesis, ADCs have created one of the most sophisticated paradigms in cancer research. They hold promise as a large part of the future of precision medicine: as single agents in patients with refractory or relapsing disease, and in combinatorial modalities with other therapeutics as first line therapy. However, commercial and regulatory success will critically depend on contextual efficacy, toxicity and production cost.