
Abstract
Intrabodies are antibodies that are not secreted but bind to their antigens inside the cell producing them. Intrabodies targeting antigens in the endoplasmatic reticulum were successfully used in vitro and in vivo. However, many target antigens interesting for research or therapy are located in the reducing environment of the cytosol, where correct folding and formation of disulfide bonds cannot be ensured. The majority of different scFv fragments, when expressed in the cytosol of the cell, do not fold correctly, are not stable or cannot bind their antigen. Such scFv antibodies are therefore not suited as intrabodies.
In this study, we evaluated fast and simple screening methods to identify scFv fragments that are stable and functional in the cytosol. We analyzed various phage display derived human scFv antibodies recognizing extracellular signal-regulated kinase 2 (Erk2) for stability and antigen binding under reducing and non-reducing conditions. Further, we developed an assay allowing to measure the interaction of the scFv intrabodies with their antigen in the cytosol of in living cells, by using a Split-Luciferase (Split-Luc) assay. ScFv fragments showing antigen binding in the cytosol could successfully be identified.
Introduction
Intrabodies are antibodies which are expressed intracellularly and which can target intracellular antigens not accessible to serum antibodies [1]. Their activity depends on correct protein folding and stability in intracellular compartments to enable antigen binding [2]. Active intrabodies can interfere with protein-protein interaction and other protein functions or can be used to knockdown proteins on a post-translational level [3, 4]. Targeting proteins on a conformational or post-translational level opens up new opportunities and novel applications, beyond the capability to nucleic acid based methods such as RNA interference [5, 6] or knock out i.e. by CrispR/Cas9 [7]. An example is the knock down only in a part of the subcellular compartment. Zehner et al. knocked down Sec61 on the protein level within the post-endoplasmic reticulum (ER) compartments only, while its presence in the ER was not affected [8].
Complete antigen specificity of an antibody can be provided by single chain Fv fragments (scFv). These small scFv based intrabodies can be targeted into different compartments of the cells using different signal peptides [9]. Most intrabodies have been targeted into the ER to retain and functionally knock-down secreted and cell surface proteins. These retained ER-intrabodies have been also used to generate transgenic VCAM-1 knockdown mice, which have not been achieved before using nucleic acid level knock-out technologies [1, 10].
As ER retained intrabodies are produced- and find their antigen- in the normal secretory pathway of antibodies, the oxidizing ER environment and additional chaperons ensure the correct folding and formation of the domain disulfide bonds [11]. In all other cellular compartments, the redox potential of the environment is shifted to reducing conditions and many disulfide bonds cannot be formed, which can lead to inactive intrabodies [2]. It was estimated, that only 1–10% of the described scFv-fragments are also functional cytosolic intrabodies [12]. However, since many interesting target proteins are located in the cytosol, many different approaches have been proposed to generate stable cytoplasmic intrabodies.
The first approach focuses on the structural modification of the scFv-fragment, for example by changing to a cytoplasmic stable framework [13], by adding soluble fusion proteins like maltose binding protein (MBP) to stabilize the intrabody [14] or by using the smaller and in some cases more stable camelid V
However, these methods are time consuming, may not work for all antigens and antibody combinations or are imprecise. For example, any modification of structure or framework of an scFv-fragment may have a dramatic impact on its affinity [9]. Only 20–60% of the scFv-fragments assumed to be stable intrabodies when measured in Yeast-2-Hybrid assays were also stable in Mammalian-2-Hybrid assays [19].
In this study, we established a simple and fast screening setup to detect scFv-fragments functional as cytoplasmic intrabodies. Hereto, seven different anti-Erk2 scFv-fragments were tested for antigen binding in different ELISA setups under reducing versus non-reducing conditions i) with E. coli supernatant of periplasmic produced scFvs, ii) with E. coli lysate of cytosolic produced and purified scFv and iii) with cell lysates of scFv producing human embryonic kidney cell (HEK) 293 cells. Finally, we also used Split-Luc protein complementation assay [21] that allows the measurement of functional interactions of scFv-fragments and their antigen Erk2 in living cells.
Our study suggest that simple ELISA screening is sufficient to identify scFv intrabody candidates that are stable and active under reducing conditions.
Material and methods
Constructions of the expression vectors
Phage display with the naive libraries HAL7/8 [22] was used to select binders against recombinant human Erk2 (Uniprot number P28482) as described previously [23]. These selected scFv-fragments were re-cloned from the pHAL14 vectors into the pOPE101-Cyto-XP (intracellular E. coli expression), pCSE2.5-Cyto-XP (intracellular mammalian expression) and the Split-Luc vectors (intracellular mammalian expression and fusion to either C-or N-terminal part of luciferase) by NcoI and NotI digestion. pOPE101-Cyto-XP and pCSE2.5-Cyto-XP are variations of pOPE101-XP [24] and pCSE2.5-XP [25] respectively without the signal peptide. The Split-Luc vectors were constructed by adding either the N-(aa 2–416) or C-terminal part (aa 399–550) of the Red firefly Luciferase as an N-or C-terminal fusion with a SGGSGGSG serine-glycine linker into the open reading frame of the pCSE2.5-Cyto-XP.
Production in E. coli
Production of soluble scFv fragments
Heat shock competent E. coli XL1-Blue MRF’(Stra-tagene) were transformed with the pHAL14 expression plasmid and incubated overnight in 96 well MTP (Greiner bio-one) at 37
Production in cytoplasm of scFv intrabodies
In contrast to the soluble production approach, pOPE101-Cyto-XP was used and the intracellular production was harvested after 3 h cultivation at 30
Densitometric protein quantification
The concentration of scFvs in E. coli lysate was determined by densitometric protein quantification. Samples and a standard dilution series of a purified scFv were separated on an SDS gel, blotted and detected by an antibody directed to the His tag (mouse anti-penta-His, 34660 Qiagen, dilution 1:5000) and a secondary goat-anti-mouse Horse Radish Peroxidase coupled antibody (A0168 Sigma, dilution 1:80.000). Afterwards, the resulting chemiluminescence was measured in ChemiDoc™ MP and samples were calibrated to the scFv standard to determine the concentration.
Intrabody production in HEK293-6E
Cells were cultivated in F17 medium (Invitrogen Life Technologies) supplemented with 7.5 mM L-glutamine, 0.1% Pluronic-F68 (PAN-Biotech) and25
HEK293-6E cell lysis
Cells were centrifuged for 10 min at 500 x
Enzyme-linked immunosorbent assay (ELISA)
ELISA was performed in 96 well Micro Titer Plates (MTP, Costar, high binding) and 200 ng Antigen or BSA (negative control) in 1xPBS per well were immobilized overnight at 4
ELISA under reducing conditions
For an ELISA under reducing conditions, different amounts of dithioerythrit (DTE) were added to the scFv-fragments produced in the supernatant of E. coli to adjust to a final concentration of 0, 12.5, 25, 50, 100 or 200 mM.
ELISA under reducing conditions with scFvs from periplasmic expression. Stability of the Erk2 antigen and activity of the periplasmic produced scFvs in ELISA were tested in presence of 0 mM-200 mM DTE.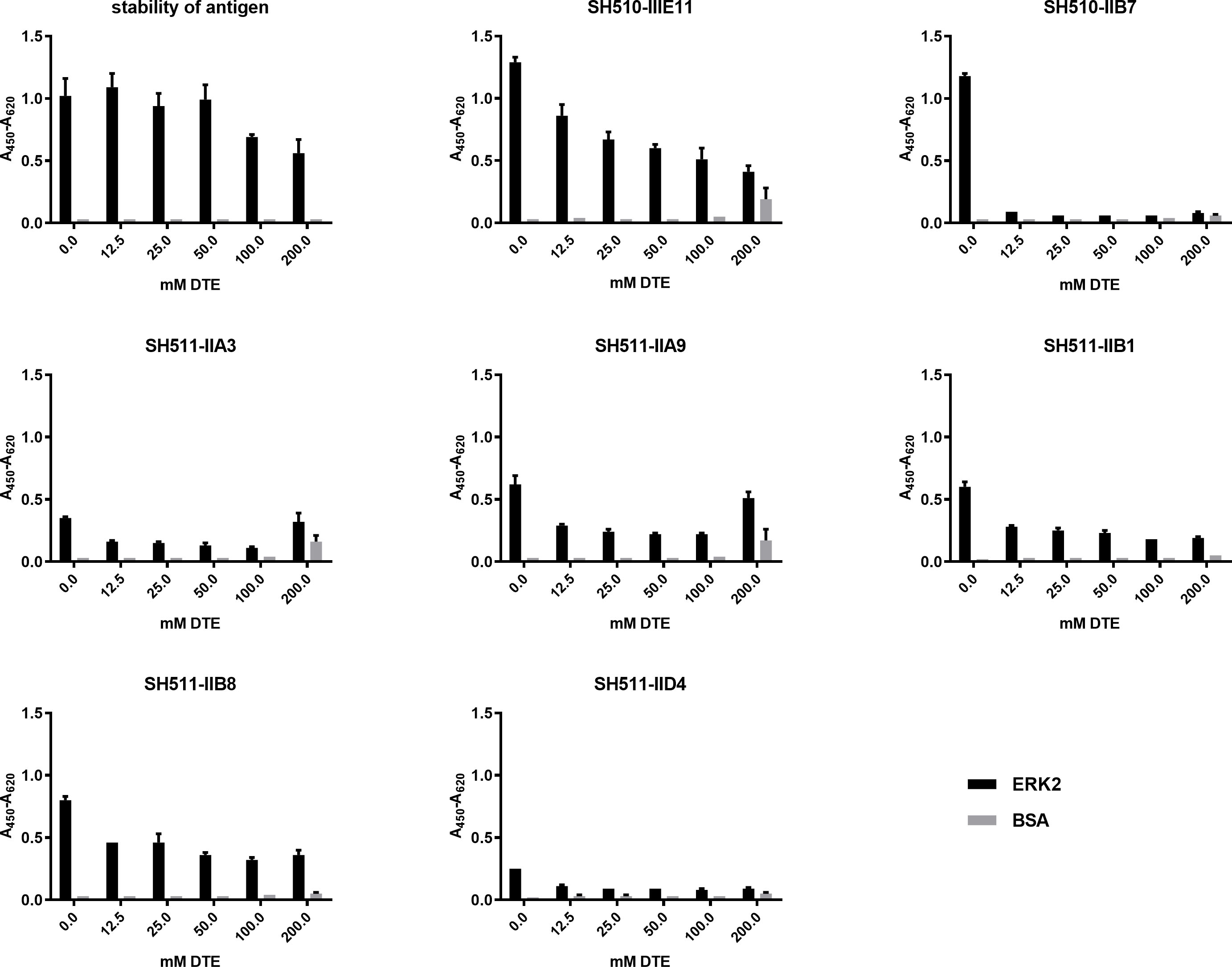
Expi293F cells (Thermo Fisher) were passaged one or two days before transfection to 1 or 0.5
Results
Binding activity decreased for soluble scFv fragments under reducing conditions
In a first assessment of the capability of different scFvs to be suitable as intrabody, their antigen binding activity was tested under reducing conditions. To exclude that the ELISA signal itself was effected by the reducing conditions Erk2 was incubated with the indicated DTE concentrations (0 mM–200 mM) and after washing detected by its His-tag (Fig. 1). Only at high concentrations of 100 mM–200 mM DTE the signal decreased to 50–60% compared to the control without DTE supplementation. The Erk2 protein was not proteolytically degraded under reducing conditions, while five out of seven tested anti-Erk2 scFvs already lost more than half of their original ELISA signal at the lowest tested DTE concentration of 12.5 mM. The scFv SH510-IIB7 did not show any binding even at this lowest DTE concentration. Three scFv-fragments (SH510-IIIE11, SH511-IIA9 and SH511-IIA3) showed an increase of unspecific binding to BSA at 200 mM DTE, indicating some denaturation of these antibodies. Compared to the other scFv candidates, SH511-IIB1 and SH511-IIB8 were stable and did not bind unspecific even at the highest DTE concentration. Thus, they were deemed the most promising intrabody candidates from this assay.
Production levels and binding activity of scFvs produced in the cytosol of E. coli. A) production levels of the different scFv-fragments determined by densitometric quantification. B) Titration ELISA of these scFv-fragments to determine antigen binding to the antigen Erk2 and the control protein BSA.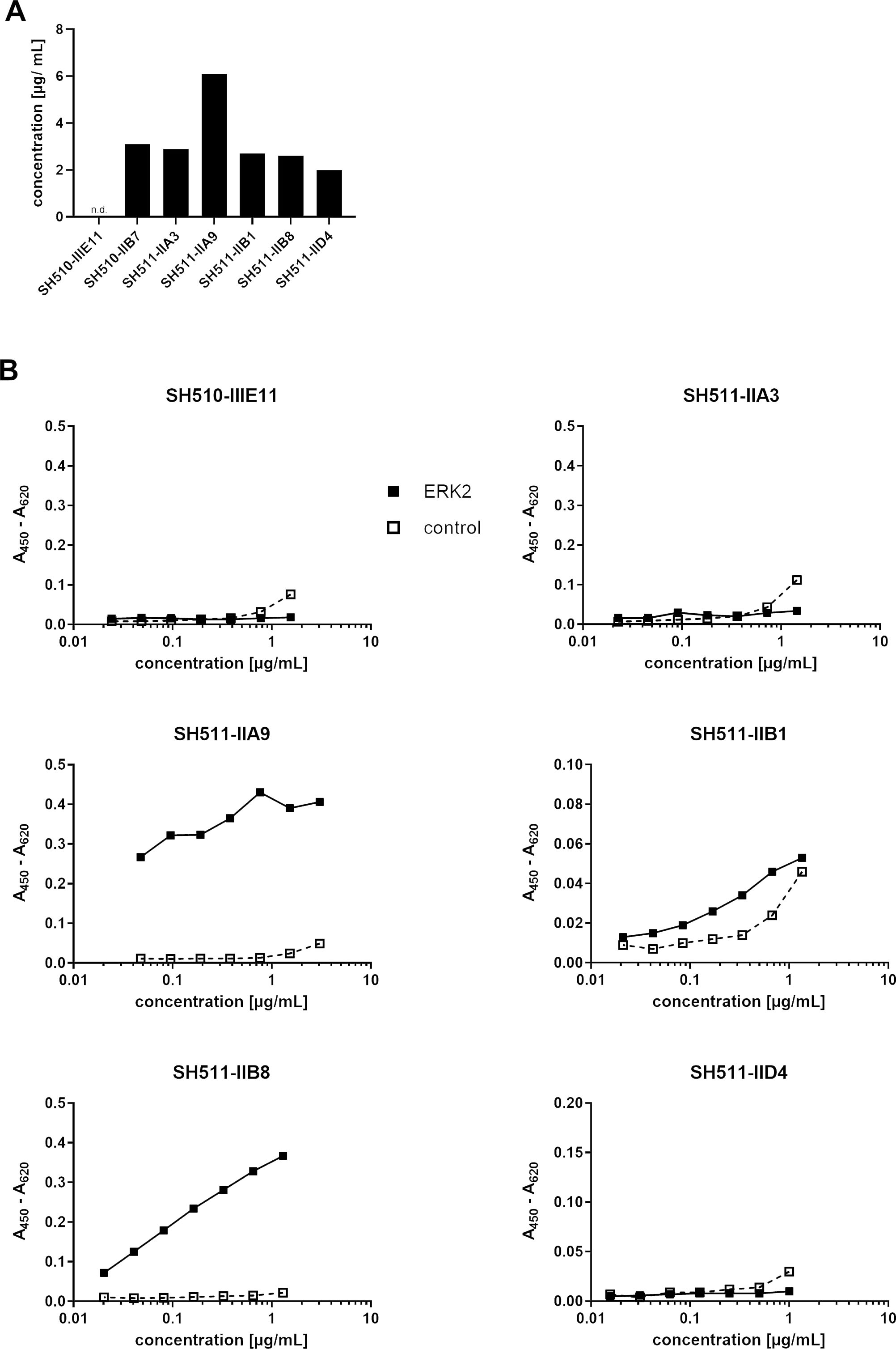
Binding activity of anti Erk2 scFvs produced in the cytosol of HEK293 cells. Lysates of transfected HEK293 cells were tested in ELSIA for binding to Erk2 and BSA (control).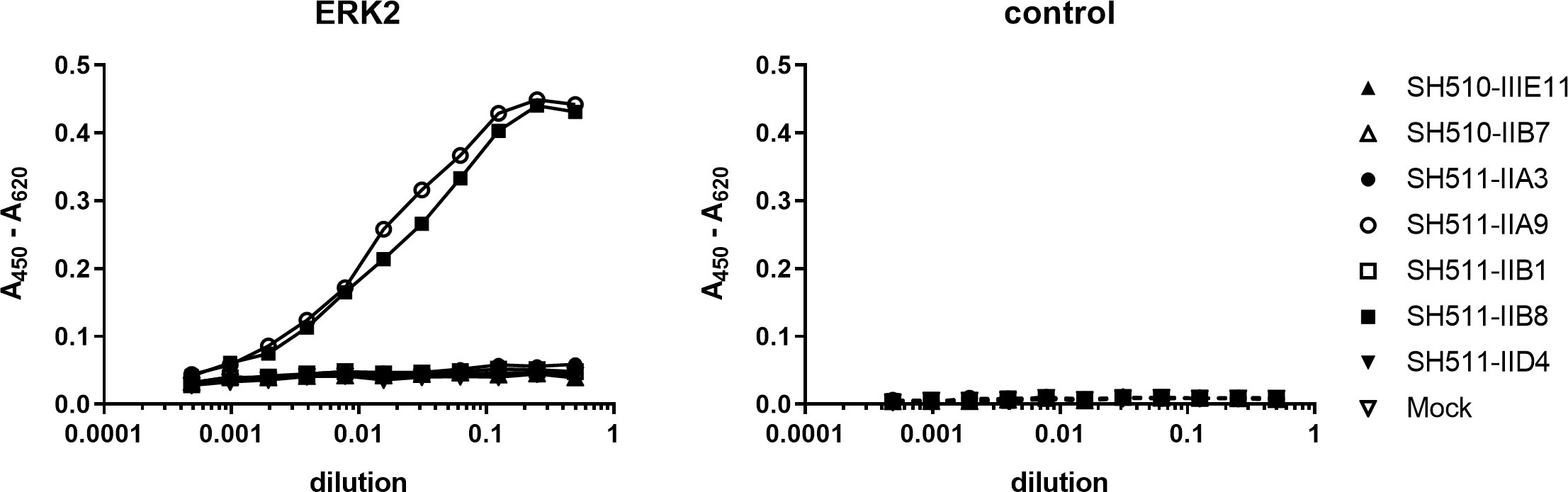
Periplasmic production is the classical way to generate functional scFv antibodies in E. coli, but may not reflect the situation of cytoplasmic intrabodies in vivo, where expression and folding takes place under reducing conditions. Therefore, the candidate scFv fragments were produced in the cytoplasm of E. coli by removal of the signal peptide from the expression vector. Production of the intracellular produced scFvs was determined by chemiluminescence quantification of the respective band on an Immunoblot blot. One out of the seven scFv-fragments was not produced at all in the cytoplasm of E. coli (Fig. 2A). All other scFv candidates showed nearly identical expression levels except clone SH511-IIA9, which was produced nearly twice as much. The titration ELISA (Fig. 2B) of three scFv candidates (SH510-B7, SH511-A3 and SH511-D4) revealed no specific binding to the Erk2 antigen. SH511-IIB1 bound to Erk2 but showed unspecific binding to BSA. Only the scFv fragments SH511-IIA9 and SH511-IIB8 specifically bound to Erk2 after intracellular production in E. coli. In conclusion, two out of 6 tested scFv-fragments were successful expressed and folded correctly in the cytosol of E. coli.
Two scFv-fragments are active after production in the cytoplasm of HEK cells
Most intrabody applications are in mammalian, particularly human cells, which have a different protein expression apparatus. Since cytoplasmic expression differs in many ways from those in E. coli, the seven intrabody candidates were produced in the cytosol of HEK293 cells. Their antigen binding activity was then assessed in ELISA using lysates of the transfected HEK293 cells (Fig. 3). Again, only SH511-IIA9 and SH511-IIB8 showed specific binding to Erk2. Interestingly, in contrast to the cytoplasmic production in E coli, SH511-IIB1 showed no unspecific binding to BSA when produced in HEK293 cells.
Split-Luc protein complementation assay to screen intrabody: antigen interaction. A) Nomenclature of the different co-transfections. Erk2 and every intrabody was fused either N-or C-terminal to either the N-or the C-terminal part of the Red Firefly luciferase. All eight possible combinations were tested. B) Luciferase signal of the Intrabodies or Erk2 fused C-terminally to full-length Red Firefly luciferase, indicating the expression level of the different intrabodies and Erk2. C) Resulting Split-Luc signal of the eight possible combinations for each Erk2-intrabody and the anti-myosin scFv intrabody SF9 for negative control.
Antigen specific interactions between intrabody and antigen were observed in living HEK293 cells by Split-Luc protein complementation assay (Fig. 4). Either the C-or the N-terminal fragment of the Red Firefly luciferase was fused to either the intrabody or the antigen in C-or N-terminal configuration. In total, eight combinations are possible to co-express intrabody and antigen fused to parts of the luciferase in a way to reconstitute luciferase catalytic activity. All of these combinations had to be tested to identify potential false negative results due to a non-reconstituted luciferase, for example, by sterical hindrance of the fusion partners or by suboptimal orientation and distance of the Split-Luciferase moieties. Figure 4 shows the results of the Split-Luc assay for the Erk2 intrabodies and the negative control anti-Myosin intrabody SF9, which is described to be a functional intrabody but does not bind ERK2 [26]. The observed Split-Luc signal for SF9 was low in all possible eight combinations. No interaction signals were measured for the anti-Erk2 intrabodies SH510-IIIE10 and SH510-IIB7. In contrast, SH511-IIB8 resulted in the highest Split-Luc signal of all intrabody-antigen Split-Luc combinations, in particular, when it was fused to the N-terminal part of the Luciferase (Fig. 4B, combinations A, B, C, D). Fusions to the C-terminal part of the Luciferase were not favored for this intrabody and resulted in low signals. The intrabodies SH511-IIA9, SH511-IIB1 and SH511-IID4 reached a maximal Split-Luc activity in a comparable range. The maximal Split-Luc signal of SH511-IIA3 was approximately less than half of that of the other functionally active intrabodies.
In addition, expression levels of the intrabodies and Erk2 were determined by C-terminal fusion of the full-length luciferase (Fig. 4B), as expression could influence the signal of the Split-Luc assay. No or low expression of the intrabody enzyme fusion may cause false-negative results regarding a non-detected intrabody antigen interaction. Still, such low-expressing intrabodies are probably not desired and might be as well excluded from further studies by this expression test. However, expression does not have to correlate with the Split-Luc signal as the antigen might not interact. Indeed, the SF9 intrabody expression was high, as expected from previous publications [26], but no significant Split-Luciferase signals were detected.
Similar results observed for SH510-IIIE11 indicate that this scFv candidate was expressed, but possible not folded correctly, as it could not bind Erk2. SH510-IIB7 did not show any functional expression of the intrabody full-length Luciferase fusion protein nor any intrabody to antigen Split-Luc interaction.
The SH511-IIB1 scFv was moderately expressed while the Split-Luc signal for most intrabody antigen combinations accept combination A was low. The highest intrabody full-length Luciferase expression was observed for SH511-IIA3. However, normalization of the Split-Luc signal to this production level revealed that only less than 5% luciferase signal could be reached in Split-Luc assay. In comparison, the best intrabody SH511-IIB8 could reconstitute
The luciferase assays allowed for the detection and ranking of intrabodies according to their production levels as well as their capability to specifically interact with their antigen in the cytosol of living cells. Yet, the assay requires all of the eight possible fusion protein combinations to avoid false-negative results.
Comparison of the results of the four different screening assays. “Yes” indicates that the assay deemed the scFv-fragment suitable as an intrabody, “No” as unsuitable
Comparison of the results of the four different screening assays. “Yes” indicates that the assay deemed the scFv-fragment suitable as an intrabody, “No” as unsuitable
* unspecific binding to BSA detected; ** no expression of (scFv or) intrabody detected; *** low signals.
The next generation of cell and gene therapies and mRNA technologies provide different ways to efficiently deliver genes or transcripts into cells and tissues even in vivo. These therapies also open-up new opportunities to use intrabodies for the targeting of intracellular antigens, which were previously not accessible for antibody therapy. However, most antibody technologies have been focused on the development of antibody as protein and their production in the secretory pathway of cells. Only few data are available about intracellular expressed antibodies and some studies suggested, that only 1–10% of the antibodies are expressible in the cytosol of cells due to limited folding and formation of disulfide bonds [12].
Antigen ELISA of anti-ERK2 specific human scFv antibodies under non reducing (0 mM DTE) and reducing (12.5–200 mM) conditions revealed that five out of seven scFv antibodies retained their antigen specific binding at least after treatment with lower DTE concentrations. Three out of seven demonstrated function and stability even after having been exposed to higher non-physiological reducing conditions. However, this assay did not completely reflect the situation of in vivo intrabodies which need to be expressed and folded correctly within the reducing environment of the cytosol. After being produced as intrabody in the cytosol of E. coli, only two scFv fragments were still able to specifically bind their antigen. The same two scFv candidates SH511-IIA9 and SH511-IIB8 were also the only scFvs showing specific binding when produced as intrabodies in the cytosol of mammalian cells.
Finally, a Split-Luciferase protein complementation assay allowed to detect the intracellular interaction of the intrabodies with their antigens. Here, again SH511-IIA9 and SH511-IIB8 were confirmed as active intrabodies by binding to their antigen ERK2 in living cells. Surprisingly, three additional scFvs showed some activity as intrabodies but with low Split-Luc signals.
The comparison of all tested assays revealed that the ELISA under reducing conditions with periplasmic produced scFv is not a suitable assay to detect intrabody candidates (Table 1). Obviously, the protein stability is either not as strongly affected by reducing conditions once the scFv antibodies are correctly folded, or activity is reconstituted after removal from the reducing milieu. In contrast, cytosolic expression in both E. coli or human cells was highly predictive of the functional binding as measured by the Split-Luc complementation assay. The Split-Luc assay, despite demonstrating in vivo binding directly, requires considerable effort since four constructs of each intrabody plus four for each antigen have to be constructed. Antigen expression needs to be verified to efficient as well.
Nevertheless, this assays still only advances a candidate scFv half of the way towards validation as effective cytosolic intrabody, since some modulating physiological effect by antigen binding has to be demonstrated as well. The proof of these modulating effects, e.g. by steric hindrance of complex formation of the antigen with other intrinsic cellular proteins, blocking of active sites or other influence on the antigen function always requires functional cell assays individually developed for every particular antigen, pathway and function. This is the most labor-intensive step in the development of cytosolic intrabodies [27]. It explains why ER targeted antibodies, which typically provide a knock down phenotype by just binding to the antigen, have been used successfully in much larger numbers.
An antigen ELISA using crude lysates of HEK293 cells or E. coli expressing scFv cytoplasmically as described in this study may help to significantly shorten the list of scFv candidates to be evaluated in subsequent cumbersome modulation assays. While of course a general conclusion would require a higher sample number and the use of different antigens, our results suggest that ELISA after cytosolic expression may substitute binding assays in the cytosol of living cells as a first screening step to identify robust scFv which retain their binding function in the cytosol. This is an essential prerequisite for any intrabody before being tested for interference with antigen function in the cytosol. In addition to the expected reduction of the sample number to be tested for modulation, this simple ELISA assay further facilitates assessment of cross-reactivity or poly-reactivity, since many different antigens can be tested without much additional effort in parallel. Further, it mitigates the potential risk of false positive or false negative phenotypes in modulation assays by unspecific effects induced by denatured, aggregated and/or sticky intrabody candidates.
Footnotes
Acknowledgments
We are grateful to Stefan Dübel for his valuable comments during the entire project and for reading the manuscript. Parts of this work were funded by the EU programme “Affinomics”.