
Abstract
The neuroanatomic specificity with which Alzheimer’s disease (AD) progresses could provide clues to AD etiopathology. Magnetic resonance imaging studies of AD clinical progression have confirmed general conclusions from earlier studies of AD neuropathological progression wherein neurofibrillary tangle pathology was observed to spread along a well-defined sequence of corticocortical and corticosubcortical connections, preferentially affecting certain cell types, while sparing others. Identical and non-identical twin studies have consistently shown AD has mixed (environmental and genetic) etiopathogenesis. The decades-long prodromal phase over which AD develops suggests slow but progressive accumulation of a toxic or infective agent over time. Major environmental candidates are reviewed to assess which best fits the profile of an agent that slowly accrues in susceptible cell types of AD-vulnerable brain regions to toxic levels by old age, giving rise to AD neuropathology without rapid neuronal lysis. Chronic aluminum neurotoxicity best matches this profile. Many humans routinely ingest aluminum salts as additives contained in processed foods and alum-treated drinking water. The physical properties of aluminum and ferric iron ions are similar, allowing aluminum to use mechanisms evolved for iron to enter vulnerable neurons involved in AD progression, accumulate in those neurons, and cause neurofibrillary damage. The genetic component of AD etiopathogenesis apparently involves a susceptibility gene, yet to be identified, that increases aluminum absorption because AD and Down syndrome patients have higher than normal plasma, and brain, aluminum levels. This review describes evidence for aluminum involvement in AD neuropathology and the clinical progression of sporadic AD.
Keywords
INTRODUCTION
One of the most important and challenging problems in contemporary clinical neuroscience is to explain the neuroanatomic specificity of neurodegeneration that occurs in Alzheimer’s disease (AD) [1]. That is, to understand why certain large neurons, particularly in the superficial entorhinal cortex, are among the earliest and most severely-damaged cell populations to be affected in AD. An understanding of this selective damage may provide important clues for unraveling AD causality [2].
Epidemiological studies for AD, based on identical and non-identical twin pairs, have consistently shown that AD causality has significant environmental and genetic components. If one member of an identical twin pair develops AD while the other remains AD-free, the discordance is attributable to an environmental component of AD causality. If identical twins show a higher rate of concordance for AD than non-identical twins, this provides evidence for a genetic component of AD causality.
A nationwide Finnish twin cohort study [3] showed more than two-thirds (68.7%) of identical twin pairs were discordant for AD, indicating a significant environmental component for AD causality. The same study showed a probandwise concordance rate for AD of 31.3% in identical twin pairs compared to 9.3% in non-identical twin pairs, also indicating genetic involvement. Twin-based studies have consistently shown that AD has mixed causality with significant environmental and genetic components [3–5].
Many AD researchers have focused on the genetic component of AD which is currently believed to involve aberrant metabolism of the amyloid-β protein precursor (ΑβΡΡ) and its amyloid-β (Αβ) peptide cleavage product. Attention has more recently been directed toward the environmental component of AD. Conventional viruses, prions, and metal neurotoxicants, in particular soluble forms of lead, mercury, and aluminum, have all been proposed for this role. We will consider how well these environmental candidates fit the profile as the environmental component of AD etiopathogenesis and attempt to understand how that component relates to AD progression.
The present review article has five parts, describing: (1) the anatomical sequencing of interconnected brain regions involved in clinical and neuropathological AD progression; (2) cell and tissue features proposed to enhance the propensity of certain cell types in AD-vulnerable brain regions for neurofibrillary tangle (NFT) formation; (3) a comparison of the major candidates for the environmental component of AD; (4) evidence that the environmental component participates in AD neuropathology; and (5) how the environmental component may interact with cell and tissue features to increase neuronal propensity for NFT formation and its role in AD progression.
LONGITUDINAL STUDIES OF AD CLINICAL PROGRESSION
Magnetic resonance imaging (MRI) allows specific brain regions to be mapped in living patients. Brain mapping software, in particular voxel (3D pixel) based morphometry (VBM) [6], can effectively combine multiple MRI scans from a single individual, taken on several occasions over a period of time, to detect rates of atrophy in particular regions of the individual’s brain. Alternatively, MRI/VBM can combine brain scans from a population of patients at particular stages of a disease to determine average gray matter volumes at those stages. For AD, cognitive testing of the same subjects is generally carried out close in time to their MRI scans.
Three examples are described here of longitudinal MRI/VBM studies carried out at different stages of AD. The first of these studies takes place in the prodromal phase of AD, comparing brain scans of apparently normal individuals with a family history of AD, which confers an increased risk for this condition, and apparently normal individuals without a family history for AD [7]. The second study takes place near the end of the prodromal period and compares MRI scans of subjects with (amnestic) mild cognitive impairment (aMCI) before and after they convert to overt AD, with scans performed over the same time period from healthy controls [8]. The third study compares two brain scans, separated by at least one year, from groups of patients diagnosed with probable AD, and age-matched non-demented controls, to demonstrate the gray matter atrophy that occurs over time in specific regions of AD-affected brains [9]
Family history and increased risk of AD
An MRI/VBM study aimed to measure atrophic change in gray matter of cognitively-normal individuals with increased risk for AD due to family history (FH+) [7]. The study included 11 subjects with a maternal history of AD (FHm), 10 with a paternal history of AD (FHp), and 32 without any parental history of AD (FH-). Offspring with a family history of AD, particularly from the maternal side, have been shown to have a 4- to 10-fold increased risk for late-onset AD compared to those without a family history of AD [10].
The prospective subjects, recruited from a referral-based memory clinic, were given a standard diagnostic evaluation. A neurologist used the Clinical Dementia Rating (CDR) scale and the Mini-Mental State Examination (MMSE) to assess the subjects for dementia. Subjects were aged 63–83 years at baseline and had at least 12 years of education. Controls without a family history for AD had a CDR score of zero and a MMSE score of 28 or higher. The subjects were given medical, neuropsychological, and MRI assessments at baseline and at the 2-year follow-up. Gray matter, white matter, and cerebrospinal fluid (CSF) volumes were measured on both occasions and VBM was used to superimpose the MRI scans in order to assess them for change.
Baseline MRI data already revealed significantly greater loss of gray matter and CSF expansion in FH + subjects than in FH− subjects [7]. The FH + subjects had significantly more gray matter atrophy than FH− subjects in the parahippocampus (including the entorhinal cortex), hippocampus, anterior cingulate, medial frontal cortex, and precuneus (p < 0.001). When the family history group was split into FHm and FHp sub-groups, only the maternal group showed an association with increased whole brain atrophy. Those in the FHm group with an APOE4 allele exhibited significantly more atrophy in the frontal cortex [7].
At the 2-year follow-up visit, further atrophy was observed in the left precuneus and the left parahip-pocampal gyrus. Atrophy had also increased in the right hippocampus, right precuneus, bilateral middle temporal gyrus, anterior cingulate, and posterior cingulate of those with a positive family history for AD compared to those without the family history [7].
Conversion from aMCI to AD
The second MRI/VBM study analyzed gray matter loss that occurs in specific brain regions during conversion from aMCI to AD [8]. The study was preceded by data collection from 61 subjects with aMCI who presented for MRI scans on three occasions over a period of time lasting approximately three years. Criteria for aMCI were: memory complaint, impaired memory for their age, almost normal cognitive function, fairly normal activities of daily living, and absence of dementia.
An inclusion criterion for the MRI/VBM analysis was that the patients with aMCI converted to AD around the three-year timeframe. Hence, the study included 33 subjects with aMCI, who all converted to AD at a similar rate and had three MRI scans performed during the course of conversion, which occurred over a median interval of three years. The 33 aMCI subjects were compared with 33 healthy controls [8].
The first of three scans was performed approximately 3 years prior to the diagnosis of AD. The second was about one year before AD diagnosis and the third scan was done at the time of AD diagnosis. At each time point, all 33 scans for each group were combined with VBM software and templates to determine average gray matter volumes for the aMCI/AD group and the control group.
At the time of the first scan, the averaged gray matter loss in the aMCI cohort was primarily confined to the anterior medial and inferior temporal lobes (bilateral entorhinal cortex, anterior hippocampus, and left amygdala) with some involvement of the fusiform gyrus. The posterior hippocampus was relatively unaffected. Gray matter loss was bilateral, affecting the left hemisphere slightly more than the right hemisphere [8].
At the time of the second scan, the averaged gray matter atrophy in the aMCI group was greater in the medial temporal lobes, inferior temporal lobes, and posterior regions of the temporal lobes, now including the entire hippocampus. Parietal lobes also showed atrophy whereas the frontal lobes were relatively spared [8]. These results show that significant gray matter atrophy occurs before AD is diagnosable.
The 33 aMCI subjects were ultimately diagnosed with AD at the time of their third scan. Gray matter atrophy in the converted AD group’s brains was more extensive throughout the medial temporal lobes and the temporoparietal association neocortices than in the scan taken one year earlier. By this time, gray matter of the frontal lobes showed substantial atrophy for the first time, particularly in anterior frontal lobes and superior frontal gyri. Some gray matter loss was also evident in the midbrain [8]. Other authors confirm that subjects who convert from aMCI to AD have significantly less hippocampal and amygdala gray matter volume and less temporal and parietal lobe cortical thickness than non-converters [11].
Cognitive test scores for the aMCI/AD group also changed over the period during which the three scans were obtained: MMSE scores fell from 27 to 25 to 24, decreasing 0.9 points per year; CDR scores rose from 1.0 to 2.0 and 3.5, increasing 0.7 points per year; and Dementia Rating Scale scores decreased from 132 to 125 to 120 or 3 points per year over the span of this period [9]. Other investigators have reported that atrophy of the anterior hippocampus coincides in subjects with a CDR of 0.5 whereas atrophy of the posterior hippocampus is only seen in subjects with a CDR of 2 or 3 [12].
Maps of gray matter atrophy in AD brains
The third example involves MRI/VBM mapping of the spreading loss of gray matter, imaged on two occasions separated by approximately 1.5 years, averaged from 12 patients diagnosed with probable AD compared to 14 healthy elderly controls [9]. Novel brain mapping methods combined the corresponding cortical regions of all subjects to create average maps that could be visualized in high resolution.
The resulting maps differentiated AD from normal aging (controls) and were able to distinguish different phases of AD. The temporal sequence of deficits that occurs as AD spreads across the cortex was visualized and related to cognitive decline with MMSE scores. The serial MRI images of AD brains indicated gray matter loss had spread from limbic and temporal cortices into frontal and occipital brain regions while sparing sensorimotor cortices. Some local rates of gray matter loss were more than 5% per year in AD as opposed to 0.9% per year in controls. The left hemisphere was affected significantly earlier than the right hemisphere. Frontal regions were spared early in the disease but eventually showed more than 15% loss [9].
Relationship between AD clinical progression and neuropathological progression
Results from MRI/VBM studies provide quantitative, dynamic visualization of cortical atrophy in living subjects with aMCI and AD and have the advantage that they can be supplemented by cognitive testing. The MRI/VBM studies confirm that gray matter atrophy occurs in a well-defined sequence with AD progression in a manner that is fundamentally consistent with the temporal sequence previously predicted by studies that relied on postmortem specimens of neuropathological deterioration in AD-affected brain regions. Both approaches have been valuable for analyzing AD progression from one region to another in the aging brain.
SEQUENCING THE BRAIN REGIONS INVOLVED IN AD NEUROPATHOLOGICAL PROGRESSION
Several neuropathological studies, briefly presented here, have identified brain regions specifically involved in AD and attempted to determine either the order in which they are affected in AD progression or the extent of severity to which they are affected [2, 13–17]. Two others examined how many Aβ deposits and NFTs form in brains of older non-demented subjects, relative to their ages [18, 19]. Another identified brainstem regions also involved in AD [20].
These studies originally analyzed the numbers and distributions of both NFTs and neuritic plaques (NPs) in order to determine which AD hallmark is more suited as a biomarker of brain regions involved in AD progression. All studies ultimately used NFT numbers for evaluating the timing and/or severity of AD-affected brain regions. Reasons for this choice are that NFTs develop within cells located in specific laminae within well-defined brain regions [13]. The characteristic distribution pattern of NFTs permits the differentiation of stages in AD progression. In contrast, NPs have a patchy distribution pattern within discrete architectonic units, significant variation from one brain to another, and multimodal formation [13]. Also, unlike NFTs, Aβ correlates poorly with rates of brain atrophy [21].
Braak staging of AD neuropathological progression
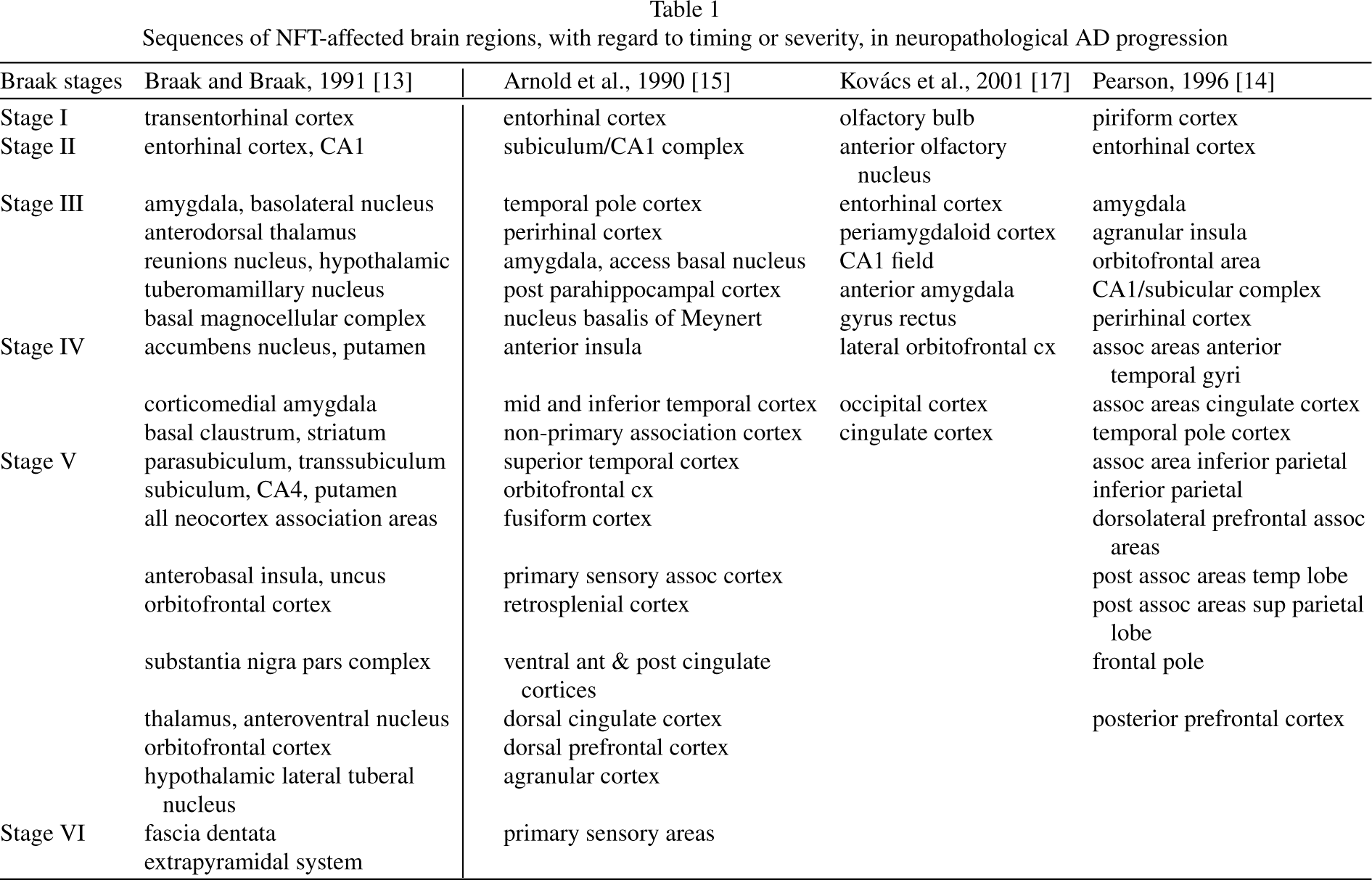
In 1991, Braak and Braak published a study in which they examined 83 human brains with varying degrees of AD involvement, including 21 from elderly demented humans whose brain tissue had sufficient densities of NFTs to confirm their clinical diagnosis of AD [13]. The study concluded that NFT pathology follows a predictable pattern that allows AD-affected brains to be categorized into six stages: Stages I–II or transentorhinal cortex stages are pre-clinical. (The transentorhinal cortex is in the perirhinal cortex where it interfaces with the entorhinal cortex.) [1].; Stages III–IV or limbic stages are characterized clinically by incipient AD; and Stages V–VI or neocortical stages are characterized by overt AD (Table 1). Studies of severe AD are complicated by wide-scale neuron loss and increasing neuropathology in glia and non-pyramidal cells [22]. The Braak hierarchical model of AD neuropathology has been critically tested and validated by Gertz et al. [23].
Sequences of NFT-affected brain regions, with regard to timing or severity, in neuropathological AD progression
Cell-cell connectivity and AD neuropathological progression
Pearson et al. showed that cell–cell connectivity plays an essential role in AD progression [14]. More specifically, they observed that AD neurodegeneration commences in olfactory structures located in the medial temporal lobe and then spreads widely via anatomical connections to more distant cortical regions (Table 1).
Deafferentation of corticocortical fibers in one brain region may result in NFT damage within the region to which those corticocortical fibers normally project [14]. NFTs appear to develop in conjunction with denervation of short feed-forward and/or feed-back corticocortical fibers as well as the ascending or descending paths. The number of pyramidal cells with NFTs in layer V of the middle temporal neocortex is in a 2 : 1 ratio with NFT-containing pyramidal cells in layer III. Moreover, the NFTs of layers III and V are arranged in clusters that are in register with each other. There is close correspondence between the distribution of NFTs in the neocortex and damage in anatomically connected regions [14].
NFT counts indicate AD-affected brain regions
The cells of origin for the perforant pathway, located in layers II and III of the superficial entorhinal cortex, collect incoming information from sensory association cortices, which is relayed to the dentate gyrus and CA1/subiculum zone of the hippocampal formation. These cells contain more NFTs than cells in any other of the 30 cortical regions of the AD brain examined in a study by Arnold et al. [15] (Table 1). They are selectively vulnerable during aging and MCI as well as AD, suggesting they become damaged at a very early stage [24]. Other large pyramidal cells, in layer IV of the entorhinal cortex, are also heavily invested with NFTs, but at a later stage than those in the superficial entorhinal cortex. By endstage AD, all cell layers of the entorhinal cortex are devastated [1].
The hippocampal CA1/subiculum zone is also severely affected by NFT formation whereas other areas of the hippocampal formation are largely spared. The entorhinal cortex/hippocampus/amygdala regions are the most severely affected in AD, on the basis of NFT numbers, whereas the motor and primary sensory cortices are the least affected [15]. Many more NFTs are located in limbic and temporal lobes than in the parietal, frontal, and occipital lobes. Neurons in layers II and IV of the entorhinal cortex have substantial afferent and efferent interconnections with temporal and posterior parietal higher order neocortical association areas [25], the most NFT-damaged regions in the AD neocortex [26]. The laminar distribution of NFTs in all neocortical association areas favors layers III and V with relatively few NFTs appearing in cortical layers II, IV, and VI [14, 15].
NFTs and NPs have different distribution patterns, with NFTs being more selectively distributed than NPs [15]. Most NPs are found in neocortical layers II, III, IV, and V [14]. The selective involvement of NFT damage in neurons of layers III and V of the association cortices is expected to disrupt corticocortical and corticosubcortical connections [14, 15, 27].
Olfactory deterioration in AD neuropathological progression
Kovács and colleagues [16, 17] counted NFTs in olfactory structures to link the olfactory bulb and related olfactory regions to the Braak hierarchical model for AD staging [16], and to explain the well-known observation that olfaction is impaired at an early stage of AD (Table 1). They concluded that: (1) the olfactory bulb is one of the first structures to develop NFTs; (2) this may occur prior to NFT damage in the entorhinal cortex; (3) NFTs are observed in olfactory bulbs at Braak stages 0 or I; and (4) impaired olfaction in early stages of AD is attributable to early NFT damage in the olfactory bulb and anterior olfactory nucleus [16, 17].
NFTs in subcortical nuclei
Pyramidal neurons of neocortical layer V project to the striatum, the nucleus basalis of Meynert, and the brainstem. The nucleus basalis is virtually devoid of tangles in brains from non-demented individuals under age 75 years and shows only a few NFTs in brains of older controls [19]. The nucleus basalis of Meynert is highly variable between AD cases, some being barely affected and others severely affected by NFTs [15].
Certain brainstem nuclei are selectively vulnerable to AD [20] and their deterioration may give rise to additional features of AD. The dorsal raphe complex is severely invested by NFTs in most AD cases. The dorsal raphe complex consists mainly of serotonergic neurons and accounts for a substantial proportion of the major ascending serotonergic projections to the forebrain. Dorsal raphe pathology could account for the significant decrease in serotonergic innervation and serotonin decrease in the CSF of AD patients [20].
The locus coeruleus is also severely affected in AD. Damage occurs in the locus coeruleus at a very early stage of AD, possibly preceding damage in the transentorhinal cortex [28], a finding that should be examined with MRI/VBM. This nucleus has major noradrenergic projections to basal forebrain nuclei and several cortical regions. Pathology in the locus coeruleus may underlie disorders of selective attention and neural control of sleep and wakefulness [20].
NFTs and Aβ deposits: Which occurs first in AD?
A search for NFTs and Aβ in limbic regions from non-demented subjects under age 65 revealed that 22 (58%) of the 38 brains showed AD change in the form of NFTs only [18]. Three (8%) seemed to have only Aβ deposits and 13 (34%) had both NFTs and Aβ deposits. Almost all Aβ deposits observed in brains of these nondemented middle-aged humans were primitive. Classic plaques with cores were seldom observed [18]. These findings [18] are very similar to findings from a study by Kovács et al. [16]. who reported that all 15 AD cases and 13 (87%) of the 15 aged controls they examined exhibited NFTs in the olfactory bulb whereas only 5 (33%) had Aβ deposits and these were diffuse. Compact and neuritic Aβ plaques were not seen [16]. Thus, NFTs appear earlier than Aβ deposits in olfactory and limbic regions, even in controls [16, 18].
The view expressed by Kovács et al. [16] that the olfactory region is affected by AD neuropathology prior to the entorhinal cortex is countered by the argument that AD affects the entorhinal cortex before the olfactory mucosa or olfactory bulb [18]. According to Ulrich, NFTs are sometimes seen in the entorhinal cortex and/or hippocampus while absent from the olfactory bulb. However, it is never the case that NFTs are present in the olfactory bulb while absent from the entorhinal cortex and/or hippocampus [18].
The hippocampus, entorhinal cortex, and nucleus basalis of Meynert contain fewer NPs per field than any other region [15]. The entorhinal cortex has one of the lowest NP densities, and most NPs in this region are located in layer III. By the time AD is evident, entorhinal cortical cells of origin for the perforant pathway are heavily invested with NFTs with many in the form of extracellular “ghosts” [25]. Damage to the perforant pathway involves deterioration of its glutamatergic terminals that normally innervate the hippocampal formation. This ultimately results in disconnection of the hippocampal formation from the entorhinal cortex and, hence, the neocortex [29]. A layer of NPs forms in the center of the dentate gyrus molecular layer, precisely where the terminals of the perforant pathway were previously located [29, 30].
The amyloid cascade hypothesis assumes that Ap deposits precede NFTs and cause their formation [31]. Aβ deposition is much more prominent in the neocortex of controls and AD cases than in limbic regions [14, 16]. The average Aβ plaque density in the temporal, insular, and orbital cortices is three-fold greater than in the hippocampal CA1 field. Also, Aβ plaques were observed to have formed in the temporal neocortex prior to neocortical NFTs [19].
However, these findings are insufficient to support the concept that Aβ deposition precedes and causes NFTs. Studies of AD progression have shown that the limbic regions which, on average, exhibit AD neuropathology 25 years earlier than neocortical regions [32] show NFTs in most brains earlier than Aβ deposits [16, 18]. The occurrence of Aβ plaques preceding NFTs in the neocortex raises the possibility that neo-cortical Aβ plaques could form in response to NFT damage from layer IV neurons of the entorhinal cortex and/or from AD-affected hippocampal CA1 neurons that normally project to the neocortex, rather than from neocortical sources. For example, the hippocampus has stronger feed-forward output to the medial temporal gyrus than feedback from the medial temporal gyrus [33]. Thus, NFT-induced damage in the hippocampus could result in Aβ plaques in the medial temporal gyrus. Tau pathology in limbic regions precedes Aβ pathology by at least 27 years [22, 34]. Immunostained tau is visible in brain tissue of many children and young adults despite the absence of evidence for immunoreactive Aβ [22].
Connectivity and disconnectivity in AD
AD-affected brain regions are interconnected with each other [14], causing some investigators to describe AD as a connection syndrome and others as a disconnection syndrome. Olfactory pathway-innervated brain regions include certain neocortical regions. Subcortical neurons that directly project to these neo-cortical regions are also selectively vulnerable in AD. It has been postulated that AD neuropathology results from an agent that crosses synapses, spreading in an anterograde direction between connected corticocortical and corticosubcortical cells of AD-vulnerable brain regions [26]. Spread of an agent from one brain region to another may provoke cell changes that lead to the formation of NFTs in the soma and dendrites of target neurons. For example, hyperphosphorylated tau has been proposed to spread between interconnected brain regions in AD [35–37]. An alternate explanation for the apparent spread of hyperphosphorylated tau in interconnected brain regions is discussed below in the section “Aluminum and AD Progression”.
AD damage may also spread between specific brain regions in a retrograde direction since synaptic terminals have active incorporation of plasma membrane, indiscriminately taking in neuropil substances that can be transported back to the cell soma [38]. This mechanism involves plasma membrane invagination with vesicles budding into the cytoplasm [39]. Small membrane-bound vesicles are transported retrogradely by kinesin along microtubules at rates similar to fast anterograde transport [40]. AD progression could also spread simultaneously in both anterograde and retrograde directions via different sets of fibers [14].
Paradoxically, the neuropathology of AD is also consistent with its being a disconnection syndrome, involving disruption to afferent and efferent connections of NFT-containing corticocortical cells [27]. AD neuropathology probably involves connections at one stage and disconnections at another. For example, cell-cell connectivity could allow the propagation of a neurotoxic agent throughout a sequence of specific brain regions, giving rise to NFTs and cytoskeletal damage that eventually disrupt these connections and result in functional loss.
A consensus on AD neuropathological progression
The Braak hierarchical model for AD progression and other studies described above support the concept that, toward the end of a long prodromal phase, NFT neuropathology is already spreading in a stepwise manner along well-defined corticocortical connections between the entorhinal cortex and other limbic structures. More brain regions become involved as AD progresses. MRI/VBM studies on brains of living AD-affected humans and postmortem brains of humans who died with AD have confirmed this observation and enabled a consensus view on the general sequence of AD progression.
The entorhinal cortex and the transentorhinal cortex are damaged earlier in AD than other limbic regions. Damage in the limbic regions is followed by NFT damage in temporoparietal neocortices with association areas affected earlier and to a greater extent than primary sensory areas. The frontal and occipital cortices then show damage while the sensorimotor cortices are largely spared. Some subcortical regions are also involved in AD but those of the brainstem have been less studied. AD-affected brain regions are all interconnected.
AD preferentially affects certain cell types in AD-vulnerable brain regions. Next we consider possible reasons why certain types of neurons are preferentially affected, both earlier and to a more severe extent than other cell types.
CELLULAR AND TISSUE FEATURES THAT INCREASE CELL SUSCEPTIBILITY TO NFT FORMATION
The large corticortical stellate neurons in layer II and underlying pyramidal cells in the superficial part of layer III of the entorhinal cortex are, as mentioned, the cells of origin for the perforant pathway (Fig. 1) [25]. These particular cells have a high risk for developing NFTs in AD and must have at least one, or more, unique qualities that predispose them to NFT formation.
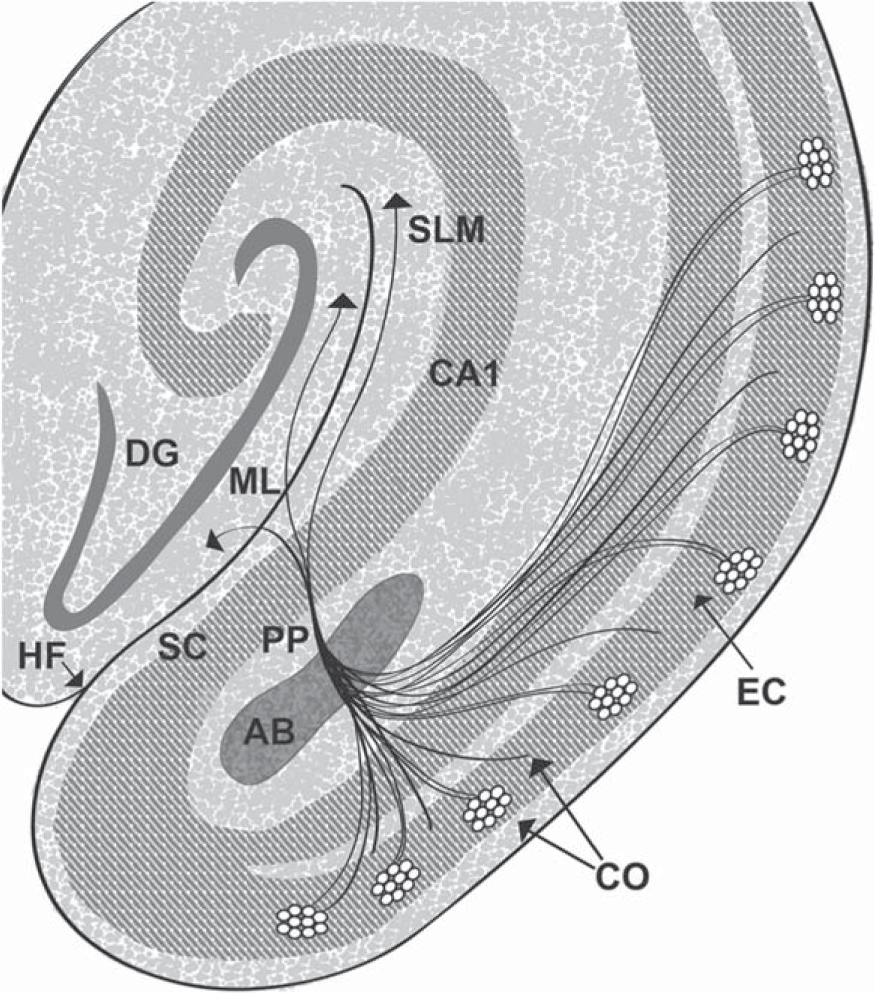
Entorhinal cortex cells of origin for the perforant pathway. Schematic representation of the perforant pathway. The perforant pathway is similar for humans and rats apart from minor variations. The cells of origin (CO) for the perforant pathway reside in layer II (shown as cell islands) and in the superficial part of layer III of the entorhinal cortex (EC). The cells of origin receive information from many cortical regions. Axons of the cells of origin converge in the angular bundle (AB). The axons leave the angular bundle and diverge into fascicles known as the perforant pathway (PP) because the axons perforate through the gray matter of the subicular cortex (SC) to the hippocampal formation (dentate gyrus and hippocampus proper). A contingent of fascicles enters the stratum lacunosum moleculare (SLM) of the hippocampal CA1/subicular zone (CA1). More fascicles cross the hippocampal fissure (HF) to enter the molecular layer (ML) of the dentate gyrus (DG). Reproduced from [41] (Copyright: JR Walton).
Several cellular and tissue features have been proposed to account for this phenomenon.
Firstly, the cell types that develop NFTs are generally complex with a long axon and large surface area [42]. Secondly, they have high energy demands, high densities of transferrin receptors, and elevated iron uptake [43, 44]. Thirdly, NFTs form more readily in cells that are poorly myelinated [45]. Fourthly, a cell may be more susceptible to NFT formation if exceptionally well-vascularized.
Cell size and complexity
The reasoning behind the proposal that complex cells with a large surface area are most prone to NFT development is that these traits would provide increased exposure to toxic agents in the extracellular milieu [42]. This proposal is probably based on observations that most cells that develop NFTs are large corticocortical and corticosubcortical pyramidal or stellate cells. In contrast, small neurons, such as the spiny stellate cells in entorhinal cortex layer IV and inhibitory interneurons seldom, if ever, contain NFTs [46].
The total dendritic length in layer II neurons of the entorhinal cortex and the hippocampal CA1 zone of non-human primates can both reach 8.0 mm, implying these two cell types have comparable complexity [42]. However, the entorhinal cortex is consistently damaged earlier and more severely by NFT formation than the CA1/subiculum zone despite the comparable complexity of corticocortical cells in both brain regions. This led Stranahan and Mattson [42] to conclude that some other cell feature must also be important.
High energy demands, densities of transferrin (Tf) receptors, and iron uptake
A second possibility for increased cell vulnerability to AD could come from particularly high energy demands, high densities of Tf receptors, and the large mitochondrial iron requirement necessary to generate sufficient ATP to meet such high metabolic requirements. Cells with these features may be more vulnerable to NFT formation than cells with relatively low metabolic demands.
Plasma iron circulates in the form of transferrin-bound iron (Tf-iron). Figure 2 illustrates iron uptake and utilization, storage, and exit from a generic cell [47]. These processes are similar in capillary endothelial cells of theblood-brain barrier. The Tf-iron attaches to Tf receptors on the luminal surface of the cerebral capillary endothelium, initiating iron transport across the blood-brain barrier [49]. Upon being released to the cytoplasm of endothelial cells, iron excessive to that required by the endothelial cell is transported across its abluminal surface and into the extracellular matrix by ferroportin. Iron also crosses theblood-brain barrier by Tf-independent mechanisms [50, 51]. Iron exiting from the endothelium binds to extracellular Tf generated by oligodendrocytes, astrocytes, and/or the choroid plexus [52]. The Tf-bound iron is then taken up into neurons via Tf receptors on the neuronal surface [53].
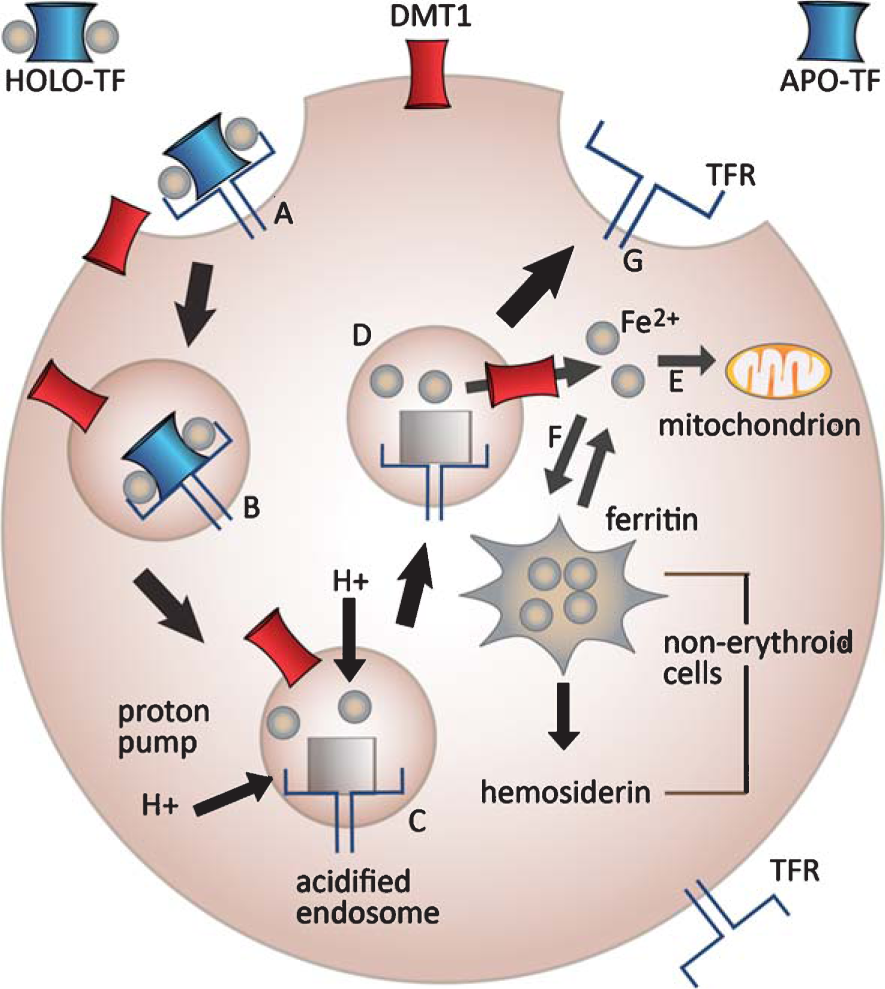
Generic diagram of iron uptake into cells. A) Tf-iron binds to Tf receptors (TFR) at the cell surface. B) The Tf-iron/TfR complexes are internalized in clathrin-coated vesicles. C) ATP-dependent proton pumps (H+) acidify the endosome, releasing iron from the Tf/TFR complex. D) Iron exits the endosome via the divalent metal ion transporter DCT1 [48]. E) The free iron (Fe2+) is either used by the cell for mitochondrial respiration or incorporated into ironcontaining proteins. F) The remainder of iron is either stored in ferritin deposits or transported out of the cell via ferroportin (not shown). G) Meanwhile, the Tf/TFR complex is returned to the cell membrane where the complex dissociates, releasing Tf for re-use. Reproduced from [47] with permission from John Wiley & Sons.
High energy demands
Multipolar stellate cells and pyramidal cells in islands of the superficial entorhinal cortex are especially vulnerable to NFT formation. These cells of origin for the perforant pathway are grouped into functional modules that have particularly heavy metabolic demands during waking hours and paradoxical (REM) sleep [54]. Cytochrome oxidase staining confirms that these entorhinal cortical cells function as cytochrome oxidase-rich modules [54]. Intrinsic electrical properties of the stellate cells, involving oscillations associated with gamma and theta rhythms [55], are highly correlated in all behavioral states and also contribute toward their necessarily high bioenergetic needs [54]. Cells in the perirhinal cortex (including the transentorhinal cortex) also display oscillations in the theta range [56]. Consequently, corticocortical cells in both the entorhinal and perirhinal regions have high glucose utilization and a very high level of mitochondrial respiration [57].
Transferrin receptor density
Neurons with large metabolic demands have high densities of Tf receptors on their surface that allow them to import sufficient iron to meet their high-energy demands [43]. Glial cell membranes also have Tf receptors [58]. Brain regions containing neurons with high densities of Tf receptors show almost identical distribution to those with neurons that have with high numbers of mitochondrial respiratory chains and high cytochrome oxidase levels [59]. Cytochrome oxidase, the terminal enzyme of the electron transport chain, serves as a marker of metabolic activity in neurons [44].
The entorhinal cortex, hippocampus, amygdala, temporal and parietal cortex, locus coeruleus, and dorsal raphe nucleus all contain relatively high densities of Tf receptors compared to other brain regions [43, 44, 59–61]. However, high Tf receptor density on its own is insufficient to explain selective neuronal vulnerability to AD since other cell groups that have high Tf receptor densities, such as the olives and trigeminal nucleus, fail to show evidence of selective neurodegeneration in AD [60]. This suggests that high Tf receptor density in cells is more relevant to AD vulnerability if those cells are interconnected with cells in other AD-vulnerable brain regions.
Iron regulatory proteins (IRPs)
Iron is an essential metal in cell metabolism for ATP production, DNA polymerases and repair, synthesis of heme proteins, and catecholamine neurotransmitters. Each mitochondrial respiratory chain requires up to 40 iron atoms [61]. Intracellular iron metabolism is controlled by iron regulatory proteins 1 and 2 (IRP1, IRP2), that act as iron sensors, as described in Fig. 3 [62, 63]. Normally, when cells are iron-deficient, synthesis of the iron-storage protein ferritin is repressed (Fig. 3A) whereas Tf receptor synthesis and iron uptake resume (Fig. 3B). When the cells have enough iron, ferritin synthesis is activated (Fig. 3C) and Tf receptor synthesis shuts down, reducing iron uptake (Fig. 3D). This process is cyclical in healthy cells, ensuring that cells have sufficient, but not excessive, free iron.
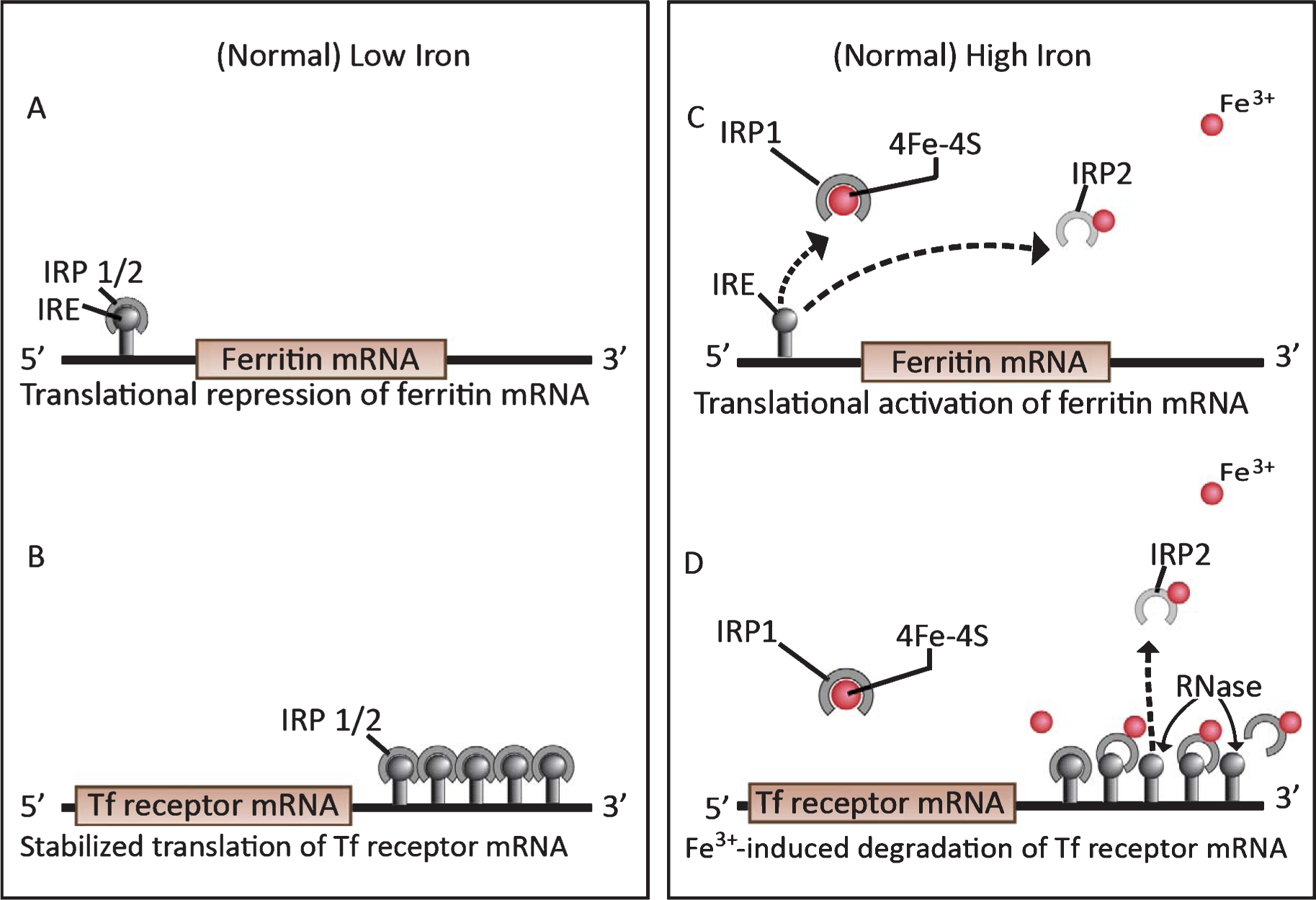
Iron regulatory proteins (IRP) 1 and 2 under low and high iron conditions in neurons. Ferritin mRNA activity (A&C) is reciprocal with Tf receptor mRNA activity (B and D, respectively). A) When intracellular iron is deficient, IRPs 1 and 2 bind with high affinity to the iron regulatory element (IRE) in the 5′ UTR of ferritin mRNA, repressing its translation. B) IRP binding to the five IREs in the 3′ UTR of Tf receptor mRNA stabilize its translation by protecting the mRNA from RNase degradation. This allows the continuation of Tf receptor synthesis and iron uptake until the iron supply is again replete. C) The small spheres indicate excess ferric iron ions (Fe3+). When the iron supply is replete, Fe3+ binds to a Fe3+ -binding site on the IRP2, oxidizes the IRP2 and causes it to lose its affinity for the IRE. IRP detachment from the 5′ end of ferritin mRNA activates ferritin translation. The dotted line indicates a deteriorating IRP2 that has detached from its IRE and signaled for degradation by the ubiquinol-proteosomal pathway (61–63). The higher Fe3+ level changes the conformation of IRP1 from an RNA-binding protein to an inactive form that binds an iron-sulfur cluster (4Fe-4S). D) As in C, Fe3+ binds to iron-binding sites on the IRP2 s, oxidizing them, and causing IRP2 detachment from the IREs and signaling for IRP2 degradation [62–64]. This exposes the Tf receptor mRNA to degradation by intracellular RNases. Consequently, iron uptake ceases. Reproduced from [61] with permission from Elsevier.
Iron metabolism becomes disregulated in the most vulnerable regions of AD brains. The disregulation occurs as a result of IRP2 stabilization which in turn maintains Tf receptor mRNA in a stabilized state [65]. IRP2 stabilization makes the cells behave as though they are in a permanent state of iron deficiency. This stabilized state occurs because, for some reason, IRP2 does not degrade as normally happens when iron levels are adequate. Consequently, cells in these vulnerable regions continue to synthesize Tf receptors and import more iron than the cell ferritin deposits can store.
In AD, iron levels become significantly more elevated in the hippocampus, amygdala, temporal cortex, and parietal cortex compared to control brains [66]. This excess iron contributes to increasing oxidative damage in large corticocortical cells of the AD-vulnerable brain regions. Excess iron imported into these neurons participates in NFT formation by aggregating hyperphosphorylated tau and binding to NFTs [67].
Scanty myelination increases cell susceptibility to NFT formation
Poorly myelinated cells are highly prone to NFT formation [68, 69]. Primary sensory areas of the neocortex are very well myelinated but the density of myelination decreases with increasing distance from primary sensory areas [68–70]. Thus, neocortical association areas are less well myelinated, particularly in the higher order association areas. Limbic tissues including the entorhinal cortex, hippocampal formation, and amygdala are in the phylogenetically-oldest allocortex and these are the most poorly myelinated and most NFT-prone regions of the AD brain [45].
Increased vulnerability is conferred on the poorly myelinated entorhinal cortical neurons by their huge demands for energy generation. A high degree of axonal myelination on neurons in primary sensory areas increases the velocity of their signal conduction while reducing cellular energy expenditures about 5000 times [71]. Conversely, poorly myelinated neurons, such as those of the entorhinal cortex, have leaky axons and must generate vastly (∼5,000-fold) more energy to compensate for the significant wastage. Poorly myelinated pyramidal cells are much more susceptible to oxidative damage. Consequently, those cells accumulate lipofuscin pigment and exhibit early evidence of cellular aging [45].
Poor myelination in the olfactory limbic system is likely to be one reason why the entorhinal cortex, hippocampus, amygdala, and olfactory bulb are generally more vulnerable to NFT pathology than neocortical regions. However, poor myelination on its own fails to explain why the entorhinal cortex is more vulnerable to NFT formation at an earlier time than other limbic structures.
Redundant vascularization may increase cell susceptibility to NFTformation
The final feature considered here that may increase a cell’s susceptibility to NFTs pertains to vascular redundancy. Neuronal firing increases blood flow by evoking the redistribution of patent capillaries [72]. Hence, neurons with high firing activity increase blood flow. Conversely, neurons damaged and unable to fire have a decreased vascular supply.
Blood flow to a brain region is also dependent on capillary density. This may explain why stellate and pyramidal cells in the superficial entorhinal cortex are selectively more vulnerable to NFT pathology than cells in other limbic regions. The entorhinal cortex of the guinea pig brain is supplied by caudal and rostral posterior arteries, with a very broad overlap between their distal territories, and also by the middle cerebral artery [73]. Such redundancy is potentially protective against hypoxic events. The redundant vascular input to the entorhinal cortex also increases its exposure to blood-borne toxicants and inflammatory factors. In contrast, the ventral part of the hippocampal formation is exclusively supplied by the rostral posterior cerebral artery [73]. This may explain hippocampal susceptibility to transient global ischemia [74].
The human entorhinal cortex receives its vascular input from both the posterior and middle cerebral arteries and has abundant anastomotic links [75]. Satellite arterioles, among the dense reticular matrix of axons that surround islands of multipolar stellate cells and pyramidal cells in layer II, give rise to a dense mesh-work of capillaries that invade these islands (Fig. 4) [1].
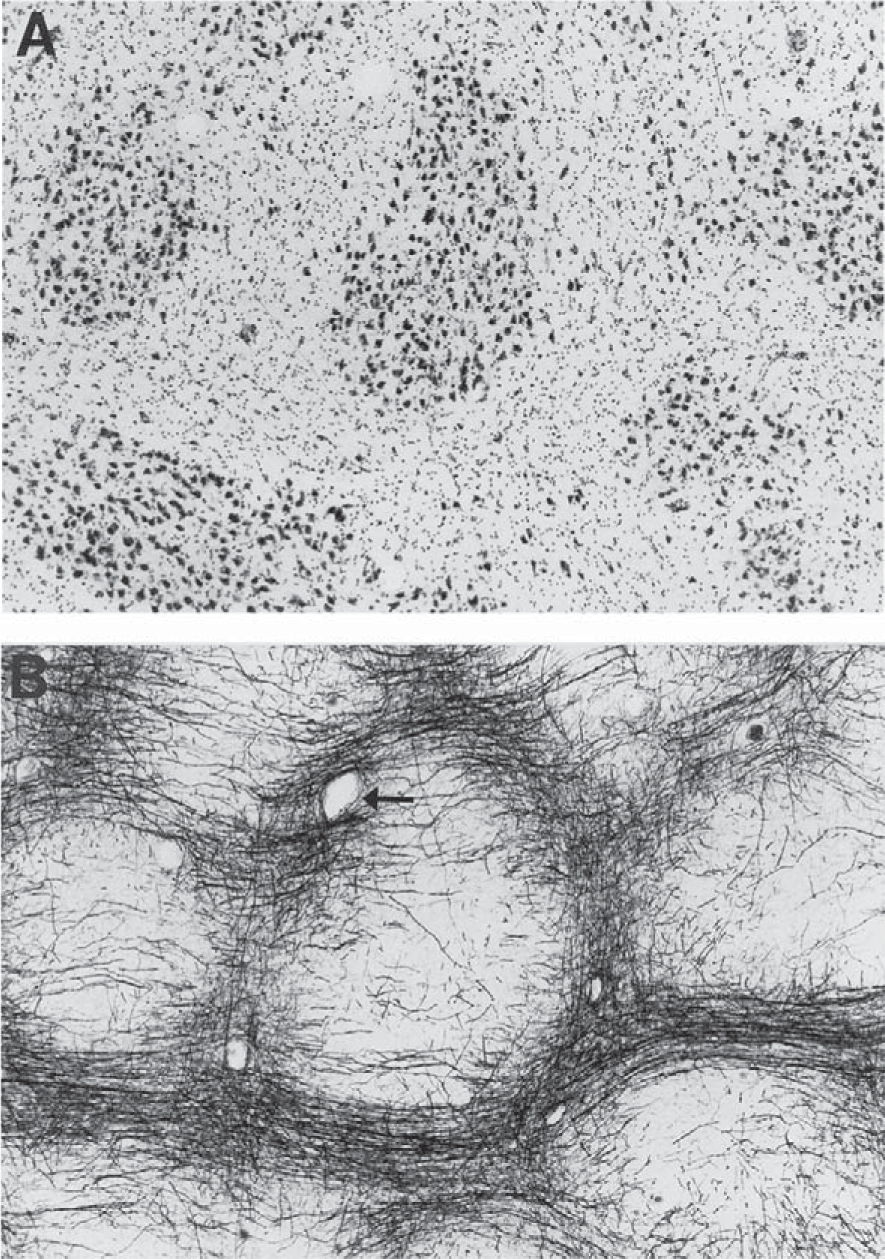
Vascularization of cell islands in the human entorhinal cortex. A) This tangential section through layer II of the human entorhinal cortex is stained with cresyl violet. Note the islands of large neurons. B) An adjacent section to that shown in A, stained with the Gallyas method, shows the complex axonal matrix that surrounds the cell islands. Note the satellite arterioles (e.g., arrow) among the surrounding axonal fibers. The arterioles that surround the cell islands branch and give rise to a dense capillary meshwork that heavily invests the cell islands. Reproduced from [1] with permission from John Wiley & Sons.
Capillaries of different brain regions have a relatively uniform density of Tf receptors on their surface [76]. However, a higher capillary density should provide more endothelial cells and more Tf receptors that can, in principle, move larger amounts of iron and other blood-borne agents across the blood-brain barrier into the extracellular matrix of well-vascularized neurons.
LARGE CELL SIZE, HIGH ENERGY AND IRON DEMANDS, SCANTY MYELINATION, AND REDUNDANT VASCULA RIZA TION ARE INSUFFICIENT TO CAUSE NFT FORMATION
Here we focus on the large corticocortical cells of the superficial entorhinal cortex that become profoundly affected at a relatively early stage of AD [77], to assess whether they have any single feature, or combination of the cell and tissue features described above, that might account for their susceptibility for early NFT formation. Stellate and pyramidal cells of the superficial entorhinal cortex are poorly myelinated cells, extremely active, and have a high density of Tf receptors on their surfaces and high iron uptake. The superficial entorhinal cortex has a redundant vascular supply and its cell islands are surrounded by a dense capillary network.
It is likely that each of the proposed features contributes to increased susceptibility for NFT formation in these cells of origin for the perforant pathway. However, these features are also characteristic of their counterpart cells that are functioning well in healthy young and non-demented aged brains. Hence, a trigger that disrupts normal cell metabolism in these AD-vulnerable cells is also necessary.
AGENTS PROPOSED AS THE ENVIRONMENTAL COMPONENT OF AD
The main candidates for the environmental component of AD are prions (atypical slow viruses), conventional viruses, and metal neurotoxicants. These candidates are reviewed to determine which best fits the profile of an environmental trigger that slowly accumulates in the brain over decades, specifically in affected neurons of AD-vulnerable brain regions, to reach toxic levels in old age and produce AD neuropathology without rapidly lysing the affected neurons.
A microorganism proposed as the environmental cause of AD should fulfill Koch’s postulates. These are: (1) the microorganism must be present in every case of the disease under study; (2) the microorganism should be capable of being isolated and grown in pure culture; (3) the microorganism must, when inoculated into susceptible animals, cause the disease under study; (4) The microorganism must be recovered from the inoculated animals and identified.
Prions
Human prion diseases are infectious, transmissible, and hereditable, between species as well as within species unlike AD. The brain regions that are affected relatively early in prion diseases include the pulvinar, ventrolateral, and mediodorsal thalamic nuclei, the putamen, and caudate nucleus [78]. The hippocampal formation is relatively spared. Damage in these brain regions is inconsistent with AD progression. Prion protein deposits, vacuoles, and rapid cell death further distinguish prion disease neuropathology from AD neuropathology [79].
A large study conducted at the National Institutes of Health carried out extensive testing of brain samples from 52 autopsy-confirmed AD patients [80], transplanting them into the brains of 61 non-human primates to learn whether a transmissible agent is involved in AD. Only two animals developed a spongiform encephalopathy. A further 17 cases were tested for more than 50 months without any effect. The investigators re-tested samples from the same brain tissue that had previously produced positive results for encephalopathy. Positive findings were elusive upon re-testing. This led the authors to conclude that an infectious agent is not involved in AD causality [80]. Subsequent testing of 36 additional AD brain samples confirmed these conclusions [81].
These AD results contrast with those from another NIH study of prion disease transmissibility. Transplanted brain samples into non-human primates showed transmission rates of 100% for eight subjects with iatrogenic Creutzfeldt-Jakob disease, 90% for 234 patients with sporadic Creutzfeldt-Jakob disease, 68% for 36 cases with familial Creutzfeldt-Jacob disease, and 95% for 18 (biopsied) patients with kuru [81]. These samples were injected into 45, 1167, 197, and 45 primates, respectively. As for AD, animals that died from any cause were given neuropathological examinations and designated as positive if they showed typical spongiform change in their brain.
Common viruses
Common viruses have also been considered as possible environmental causes of AD. Herpes simplex virus 1 (HSV1) can persist in a latent form for many years without showing overt disease after primary infection. It can reactivate from the latent state and cause cold sores and mouth, throat, face, and eye problems, despite the presence of circulating antibodies. HSV1 is one of many micro-organisms that stimulate NF
HSV/HSV1 immunoreactivity has been reported in AD and normal brain tissue [85–87] and HSV DNA was detected in 3 of 4 brain smears [88]. However, larger DNA hybridization studies have been unable to confirm the specific presence of HSV DNA in AD brain tissue [89–91]. HSV antibody titers of AD patients are similar to those of controls [92–94.
HSV1 is also neurotrophic. HSV1 can produce Herpes simplex encephalitis (HSE) and may be the only microorganism that specifically attacks the stellate and pyramidal cells of origin for the perforant pathway, producing an encephalitis with AD-like impairment of short-term memory [95, 96]. Then again, HSE is a rare condition that affects children and adolescents as well as older people, with 50% of HSE cases being under age 50 [97]. Unlike sporadic AD, HSE shows no preference for the elderly. Abundant immunoreactivity was observed in temporal lobe regions of an encephalitis case. Large numbers of glia and neurons showed evidence of active infection. Similar HSV antigenicity is not observed in any temporal lobe region of AD cases or controls [98]. Approximately 70% of untreated HSE cases undergo rapid death [97] and 2.5% regain normal brain function. Relapses of infections can occur within weeks or months [99]. These traits are uncharacteristic of AD.
Proponents for HSV1 causality of AD describe a scenario in which HSV1 enters the brain in old age [84]. This late and rapid progression is inconsistent with the slow progression of AD, in which brain changes gradually give rise to MCI and then to overt AD [100].
In summary, large AD transplantation studies carried out at the NIH failed to show evidence of transmissibility [80, 81]. Also, serum antibody titers measured in AD patients and non-demented controls for HSV, and other common viruses (Measles virus, Adenovirus, Coxiella burnettii, Cytomegalovirus, Influenza A, Influenza B, Chlamydia Group B (psittacosis virus), and Mycoplasma pneumoniae) have all failed to show statistically significant difference [93, 94]. Overall, the results indicate that, in the case of AD, Koch’s postulates have yet to be fulfilled by any infectious agent.
Toxic metals
Metal neurotoxicants have been suggested as environmental candidates for AD etiopathogenesis. Lead neurotoxicity preferentially affects children up to 4 years of age. One survivor of lead encephalopathy, who died at age 42 years with severe mental deterioration, had brain lead levels ten times higher than those of AD patients [101]. Nevertheless, the evidence for lead involvement in AD is scant.
Blood mercury levels were found to be twice as high in AD patients as in controls [102]. Most evidence relating to brain mercury levels in AD and human controls indicates that mercury does not progressively accumulate in human brains with increasing age as does aluminum [103, 104]. Aluminum best matches the profile for the AD environmental candidate.
Old age is commonly regarded as a major risk factor for AD. AD’s long prodromal phase suggests that AD involves the slow and prolonged accumulation of a toxic agent in the brain over decades of time [26, 83]. If aluminum is indeed causal to AD, its prolonged accumulation is likely to be life-long, with aluminum exposure beginning at, or even prior to, birth [105–107.
ACUTE, SUB-ACUTE, AND CHRONIC FORMS OF ALUMINUM NEUROTOXICITY
There are three main forms of aluminum toxicity (acute, sub-acute, and chronic). All three forms share some similarity and yet are significantly different from each other. They are similar is that they all cause some form of brain disease. The three forms differ in the type of subjects they affect. The rates at which these conditions occur and the aluminum concentrations involved are distinctly different.
Acute aluminum neurotoxicity
Acute aluminum neurotoxicity occurs when a large amount of aluminum (as much as 500 μg/l or more) enters the circulation. Acute aluminum neurotoxicity can affect humans with normal kidney function as well as those with chronic renal failure, resulting in an encephalopathy that typically involves grand mal seizures, culminating in coma and death within days or several weeks [108, 109.
Sub-acute aluminum neurotoxicity
Sub-acute aluminum neurotoxicity involves intermediate aluminum levels in blood or CSF over several years. An example is dialysis encephalopathy syndrome (DES) or dialysis dementia, a progressive encephalopathy that particularly affects dialysis patients with chronic renal insufficiency routinely exposed to high levels of circulating aluminum [110]. Dialysis patients have impaired ability to excrete the aluminum they acquire, mainly from aluminum-containing products such as phosphate binders, antacids, and/or contaminant aluminum in dialysis water. DES onset is insidious, often involving problems with speech. Seven to nine months after symptoms first appear, the patient becomes totally mute, unable to perform purposeful movements and soon dies.
DES epidemics have previously occurred in dialysis patients [110–112]. Removal of aluminum from the dialysis water and dialysis equipment, monitoring of plasma aluminum levels, and interventional treatments with aluminum-chelating agents, usually desferriox-amine, have prevented many adult deaths from this cause [113]. In general, DES and dialysis patients tend to be younger than most sporadic AD patients.
In a study by Harrington et al. [114], the mean brain aluminum content was 7.35 μg/g in dialysis patients compared to 1.95 μg/g in controls. Brain aluminum content correlated with plasma aluminum content (r = 0.772, p = 0.008) and with terminally truncated tau protein in white matter (r = 0.753, p = 0.001). Eight of 15 brains from dialysis patients exhibited Aβ immunoreactivity. Electron microscopy revealed twisted filaments, indistinguishable from those in NFTs of AD brains, in two brains of dialysis patients with the highest levels of hyperphosphorylated tau in their gray matter [114]. The observed frequency for these AD-type changes exceeded the expected frequency for AD changes in this group (38–68 years; 0–1%, p < 0.0001), corresponding to the expected frequency for AD only above age 80.
Five of the six brains from dialysis patients with high aluminum showed depletion of normal tau accompanied by accumulation of hyperphosphorylated tau protein [114]. This phenomenon of progressive normal tau conversion to hyperphosphorylated tau also occurs in AD [115]. The tau-related changes reported in brains of these dialysis patients are comparable to those that occur in AD brains at an earlier stage and that eventually lead to NFT formation.
Some people feel that brains of dialysis patients with high plasma aluminum levels would have abundant NFTs if aluminum is involved in NFT formation in AD patients. However, given that chronic renal failure and AD are two distinctly different disease entities that develop at vastly different rates, their neuropathology can also be expected to differ. DES patients have very high brain aluminum concentrations (e.g., 25 μg/g brain tissue (dry weight)). This may result in aluminum precipitates, forming deposits in lysosomes of cells instead of reacting with hyperphosphorylated tau to form NFTs as aluminum does when present in plasma and brain concentrations 2–3 times higher than normal values.
NFT paucity in brains of dialysis patients and DES patients is also likely to relate to: (1) the age difference at which most patients develop chronic renal failure or sporadic AD; (2) the sub-acute aluminum exposure period over which plasma levels in chronic renal failure patients are significantly elevated (amounting to a few years rather than four or more decades); and (3) the extremely slow, decades-long time over which human NFTs are estimated to form [22]. Dialysis patients may die of their condition prior to the time required for significant NFT formation unless treated by kidney transplantation.
NFTs also form very slowly in brains of non-human primates, even after aluminum injection directly into the brain [116]. In contrast, aluminum-induced NFTs develop in brains of rabbits and cats within 48 and 76 hours, respectively, after intracerebral aluminum injection [117, 118], showing species difference in the timing of NFT formation.
Chronic aluminum neurotoxicity
Finally, there is chronic aluminum neurotoxicity. Considerable evidence supports the possibility that AD is a form of chronic aluminum neurotoxicity that occurs in humans.
ALUMINUM EXPOSURE AND ABSORPTION OF INGESTED ALUMINUM
The main source of aluminum exposure for most humans is from oral ingestion of aluminum additives used to enhance various aspects of commercially-prepared foods and beverages, including alum-treated drinking water [119]. In water treatment parlance, alum generally refers to aluminum sulfate (Al3(SO4)3), a relatively soluble form of aluminum used in the clarification of some bottled waters and many urban drinking water supplies.
Aluminum compounds have versatile properties and serve many useful functions as additives: anti-caking agents in salt, coffee whitener, pancake mix, and other powdered foods; emulsifiers and melting agents in cheeses; clarifying agents in water, puddings, and other processed foods where precipitates may form; pickling agents; meat binders for sausages and luncheon meats; hardening agents for candied fruits; gravy and sauce thickeners; rising agents in baking powder, self-rising flour, and baked goods; dough strengtheners; buffering and neutralizing agents; and mordants that bind dyes to confectioneries to make them colorful.
Aluminum absorption occurs across all parts of the intestine including the colon [120]. A major reason why aluminum accumulates so slowly in the brain is because most ingested aluminum is blocked from absorption into the systemic circulation by a mucus layer that lines the gastrointestinal tract [121].
Small amounts of Al26, a radioactive tracer for natural aluminum (Al27), are detectable by accelerator mass spectrometry in the brains of rats within weeks after ingesting an equivalent amount of Al26 to the aluminum contained in a single glass of alum-treated drinking water [122–124]. Al26 is not found in nature so its presence in the brain after 26Al treatment is unambiguous. Non-dietary sources of aluminum further contribute to the aluminum burden of the brain and skeletal tissues. Trans-dermal aluminum absorption occurs from topical applications (mainly aluminum-based deodorants and sunscreens) [125, 126]. Some injected vaccines include aluminum as an adjuvant to boost the body’s immune response [127, 128]. Simulated vaccination in mice produces an aluminum peak in their brains 2–3 days post-injection [129]. Aluminum is a component of some pharmaceuticals (such as aluminum antacids and buffered aspirins) [130] and certain medical treatments such as irrigation of the bladder [131]. Aluminum is also contained in a bone cement used in surgery [132]. Additional information about human exposure to aluminum is available elsewhere [133, 134.
More aluminum enters than leaves the brain, resulting in a net accumulation of aluminum in the brain over time. Aluminum is the only common neurotoxicant known to accumulate in the brain with increasing age, even in non-demented humans albeit at lower rates [135–138]. In 2007, the UN/WHO expert committee on food additives reduced the provisional weekly tolerable level for aluminum from 7 mg Al/kg body-weight (bw)/week to 1 mg Al/kg bw/week [139]. Many humans routinely consume considerably more aluminum than this recommended limit [133].
METHODS THAT ALLOW POSTMORTEM ALUMINUM DETECTION IN BRAINS OF HUMANS AND EXPERIMENTAL ANIMALS
Aluminum can be visualized postmortem in hippocampal and cerebral neurons of the AD brain [140] with at least three different staining techniques: the morin fluorescent stain [141], Walton bright field/fluorescent stain [142], a modified protocol for the Walton stain [143], and an immunostain against protein-bound aluminum [144, 145.
A variety of instrumentation is also available for aluminum detection or measurement. These include graphite furnace atomic absorption spectrometry, inductively-coupled plasma atomic emission spectrometry, laser microprobe mass analysis, scanning/transmission electron microscopy and transmission electron microscopy with energy-dispersive X-ray analysis, secondary ion mass spectrometry (SIMS), and accelerator mass spectrometry used in conjunction with the 26Al radioisotope.
Aluminum is a light element present in the brain in small quantities. Some instrumental techniques, including nuclear microscopy, X-ray fluorescence, scanning electron microscopy with energy-dispersive X-ray analysis (except in NFTs, where aluminum is more concentrated), and flame atomic emission methods are too insensitive to measure trace levels of aluminum in biological samples over the 1 to 7 parts aluminum per million (ppm) range contained in AD-vulnerable brain regions. Also, certain other elements, mainly magnesium and phosphorus, can interfere with aluminum detection unless appropriate measures are taken to prevent this problem. Aluminum measurements require clean-room conditions.
THE DILEMMA IN PERFORMING EPIDEMIOLOGICAL STUDIES THAT ASSESS THE EFFECTS OF CHRONIC HUMAN EXPOSURE TO A NEUROTOXIN
The dilemma is that a properly-designed epidemiological study capable of providing the highest level of evidence that AD is a human form of chronic aluminum neurotoxicity would be unethical to perform. It would also be impossible to obtain cohorts of human subjects who could comply with a prospective, decades-long, randomized controlled trial involving assignment to treatment groups that consume either high, medium, or low concentrations of aluminum in their food and water, over several decades, to learn whether chronic aluminum ingestion results in AD. In lieu of any existing human study of this nature, the author applied a comparable protocol to an animal population.
Rodents that mimic human dietary aluminum exposure
Chronic aluminum neurotoxicity is best studied using human-equivalent aluminum exposure in short-lived laboratory animals over most of their lifespan [41, 143, 146]. A progressive dementia-like condition develops in a dose-dependent manner in rats that consume soluble aluminum in their drinking water for most of their adult life, in total amounts equivalent to those consumed by humans living in contemporary westernized societies from their foods, beverages, and aluminum additives [133, 134]. This approach mimics the aluminum dietary levels consumed by humans as well as the prolonged duration of human exposure to dietary aluminum. As it turned out, this animal model developed cognitive deterioration in old age after a long prodromal period and proved to be a faithful translational rat model for AD [41, 143.
Aluminum treatment was delayed until the rats were at age 12 months (commencing their middle age) to allow time for normal brain development. All animals in this longitudinal study were exposed to the same levels of aluminum in the air they breathed and in the measured food aliquots they received. The amounts of water the rats consumed ad libitum were also measured. The only treatment difference between the three rat groups was the amount of aluminum contained in their drinking water [146]. This level of controlled dietary aluminum exposure is more accurate than could be achieved with a human population.
Training and weekly testing on a task used for memory assessment
The rats were trained to perform a rewarded continuous alternation T-maze task commonly used for memory assessment [147]. Rats that died before 28 months of age, or were unable to perform all ten choices of the T-maze task within 5 minutes, were excluded from the study. The study rats were tested on the T-maze task each week from age 9 months to the end of their natural lifespan. They performed the maze task with 70%–100% accuracy upon entering middle age (at 12 months).
All rats that consumed aluminum at the low end of the human dietary aluminum range performed the task in old age as well as, or better than, they had performed in middle age. Twenty percent of the rats that consumed aluminum at the intermediate dose level and seventy percent of those that consumed aluminum at the high dose level, that is, at the high end of the human total dietary aluminum range, obtained significantly lower mean T-maze performance scores in their old age (Fig. 5) than in their middle age and developed progressive cognitive deterioration from around ages 27–28 months. Their mean lifespan was 30 months of age.
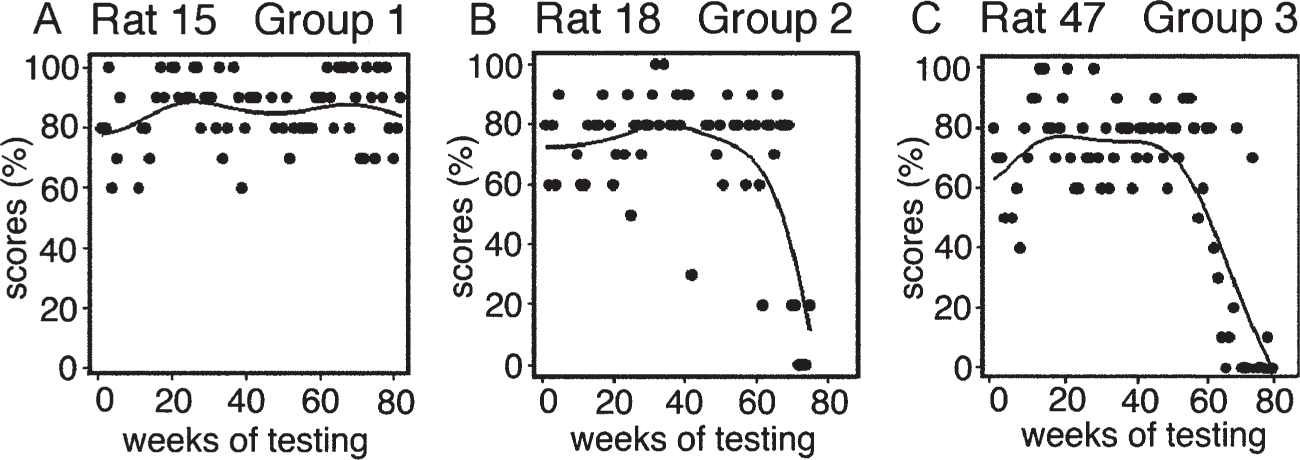
Line and scatter plots, with Loess smoothing, exemplify overall T-maze performance scores of rats from the three aluminum treatment groups. Each dot recognizes one weekly test score for choice accuracy out of 100%. A) Scores of a rat that chronically consumed a low level of aluminum in the human dietary aluminum range and remained cognitively-intact. B) Scores of a rat that exhibited cognitive deterioration after chronically consuming aluminum at an intermediate level. C) Scores of a rat with cognitive deterioration that chronically consumed aluminum equivalent to an amount at the high end of the human total dietary aluminum range. Reproduced from [145] with permission from Elsevier.
Testing continued until a terminal condition became evident. The longest-living animals were tested on more than 100 occasions. The rats that developed cognitive deterioration in old age showed no evidence of improvement with continued testing, indicating that their condition was irreversible. These rats displayed abnormal neurological signs and exhibited novel behaviors such as confusion, attentional deficit, perseverative activity and urinary incontinence while in the T-maze. The rats that developed cognitive deterioration in old age did so in an aluminum dose-dependent manner. Almost all rats in the study had normal kidney and liver functions in old age [146].
Rats that developed chronic aluminum neurotoxicity/cognitive deterioration generally did so at a mean age of age 28 months, after 16 months of chronic aluminum exposure at human relevant levels. Wistar rats are estimated to age approximately 35 times faster than humans [148] so 28 month old rats would approximate the age of 82-year old humans. The 16 month exposure period is almost equivalent to a human exposed to dietary aluminum for 47 years.
The amounts of aluminum the animals consumed correlated positively with their serum aluminum levels. Animals in the highest aluminum dose group had a mean serum aluminum level that was twice as high as that of the low aluminum controls yet some rats in the highest aluminum group had serum aluminum levels that were six times higher than the low aluminum controls. Two rats in the intermediate group had higher serum aluminum levels than other members of their treatment group. Those two rats were the only ones in their group to develop cognitive deterioration in old age, indicating they absorbed more aluminum than the others that consumed the same aluminum dose level. This may reflect some difference in their genetic constitution that enhances aluminum absorption [146].
AD-equivalent neuropathology in the animal model for AD
Neuropathological examination of sections processed with Walton’s stain for aluminum [143] involved cell counts performed with image analysis software. These revealed that the superficial entorhinal cortex in brains from rats with cognitive deterioration had a significantly larger proportion (60%) of pyramidal and stellate cells at stage IV aluminum accumulation compared to 23% at this stage in low aluminum controls [143]. These are the cells of origin for the perforant pathway (Fig. 6).
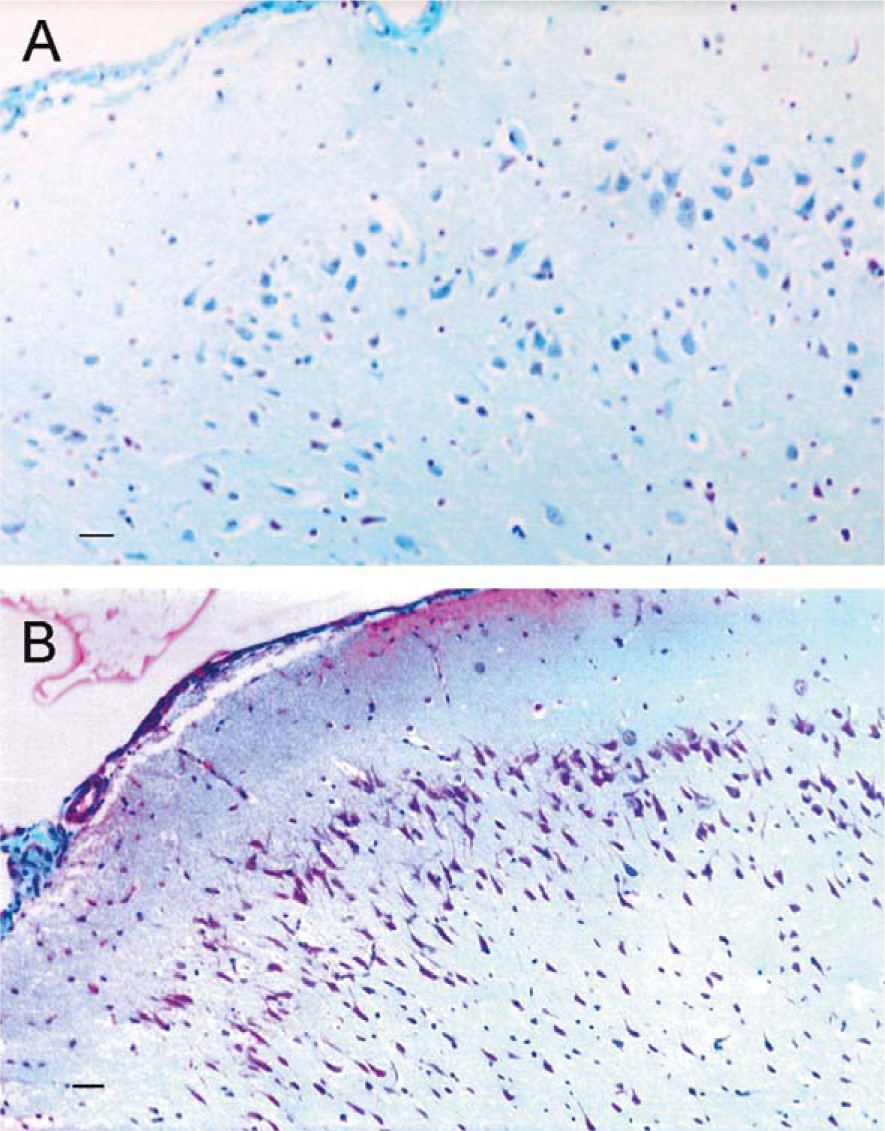
Staining for aluminum in the entorhinal cortex of a cognitively-intact rat (A) and a rat with cognitive deterioration (B). (A) Stellate and pyramidal cells of origin for the perforant pathway from a low aluminum control stain blue, lacking the magenta staining characteristic of aluminum with the Walton stain. Aluminum staining in this section primarily stains glial cells and erythrocytes. (B) In contrast, stellate and pyramidal cells of origin for the perforant pathway in the entorhinal cortex, from a rat with cognitive deterioration, stain magenta to purple, indicating high-stage aluminum accumulation. Reproduced from [149] with permission from the Royal Society of Chemistry.
Also, high-stage nuclear aluminum accumulation affected a much larger proportion of the superficial entorhinal cortical cells than of cells in any other brain region examined. Cells in the hippocampal subiculum/CA1 zone tended to exhibit high stage aluminum accumulation in the form of discrete cell bands or lesions.
The temporal association cortex of rats with cognitive deterioration also had a significantly larger proportion (40%) of neurons with high-stage aluminum accumulation than in the same brain region of controls (13%) [143]. The temporal association cortex of cognitively-deteriorated rats also exhibited many more pyramidal cells with high stage aluminum accumulation than the primary sensory cortex. High-stage aluminum in the nucleus of the rats with cognitive deterioration preferentially affected the amygdala, olfactory bulb, piriform cortex, frontal cortex, nucleus basalis of Meynert, dorsal raphe nucleus, locus coeruleus, and other brain regions vulnerable to NFT damage in AD [146].
The brain regions most affected in this aluminum-inducible translational rat model are homologous to those most affected in humans with AD [13, 146]. Cognitive deterioration in these rats involves destruction of the perforant pathway by a mechanism shared by humans with AD [1, 29, 41]. In an earlier study, aluminum was directly injected into the brains of rabbits and NFT damage was observed in regions of the rabbit brains homologous to those where NFTs form in AD brains [150]. Other studies reported that dialysis patients exhibit aluminum deposition in the same brain regions that are vulnerable to NFTs in AD [43, 44, 151]. Thus, several models have shown that aluminum preferentially accumulates in brain regions that are particularly susceptible to damage in AD.
AD PATIENTS ABSORB ALUMINUM EFFICIENTLY AND HAVE HIGHER PLASMA AND BRAIN ALUMINUM LEVELS THAN AGE-MATCHED NON-DEMENTED CONTROLS
Aluminum absorption and plasma aluminum levels in AD
AD patients absorb about 64% more aluminum than age-matched controls from a standardized dietary aluminum dose [152]. Consequently, AD patients have higher plasma/serum aluminum levels than controls [153–159].
Down syndrome is commonly regarded as a model for accelerated AD. Down syndrome patients have even greater aluminum absorption than AD patients from a standardized aluminum dose. Down syndrome patients absorb approximately 6-fold more aluminum than age-matched controls from a standardized dietary aluminum dose and 4-fold more from a standardized pharmacological aluminum dose [160].
The reason(s) for increased aluminum absorption in AD patients and even more so in Down syndrome patients is unknown. The fact that Down syndrome patients absorb much more aluminum than age-matched controls from a standardized dose suggests that the absorption difference could be attributable to an intestinal protein encoded by a gene on chromosome 21. AβPP is on chromosome 21 and recent evidence indicates that AβPP-deficient mice demonstrate impaired intestinal absorption [161]. AβPP and Aβ are both expressed in enterocytes [162], the absorptive epithelial cells of the small intestine, so they are candidates for elevating aluminum absorption. Aluminum has previously been shown to upregulate expression of the AβPP gene [83] and stimulate AβPP mRNA [163] expression in neural cells so aluminum may have the same effects on the AβPP gene and mRNA expression in intestinal cells. AβPP and/or Aβ have yet to be tested to learn whether either or both are capable of enhancing aluminum absorption and, if so, the conditions under which this occurs. Regardless, higher levels of aluminum absorption give rise to higher brain aluminum levels [146].
AD and brain aluminum
Aluminum has been regarded as a candidate cause of AD since 1973 when it was first shown that gray matter from brains of AD patients contains more aluminum than that of non-demented age-matched controls [135]. This finding has been confirmed by at least 11 reports from different researchers working in at least seven different countries using a variety of analytical techniques [104, 135, 143, 144, 164–170].
Aluminum concentrates in cell bodies localized in gray matter. When AD patients were initially reported to have higher brain aluminum levels than non-demented controls, the authors emphasized the importance of careful dissection to obtain 10–20 mg samples of gray matter for measurements of aluminum concentration. Samples larger than 20 mg are likely to give erroneous results by diluting the sample with white matter that has lower aluminum content.
Two early studies were unable to confirm higher aluminum values in AD brains than in controls [136, 137]. The study by Markesbery et al [137]. utilized tissue samples ranging from 100 to 250 mg (dry weight). Ganrot [171] described methodological flaws in both studies. Markesbery’s group [137] subsequently revisited this issue, using conditions specified in the original report (20 mg (dry weight) sample sizes and more sensitive instrumentation) and then reported significantly higher aluminum values in biopsy samples of AD hippocampus, inferior parietal lobule, superior and middle temporal gyri than in controls [166]. Brain sections used as non-demented elderly controls in the McDermott study [136] were re-examined and found to contain abundant NFTs, indicating that their classification as controls was incorrect [172].
Aluminum values in gray matter of brains from aged, non-demented humans typically range from about 1 to 2.5 μg Al/g brain tissue (dry weight) [135, 168]. Aluminum values in gray matter of AD brain are 2- or 3-fold higher, generally around 3 to 7 μg Al/g brain tissue [135, 168]. Approximately 20% of samples measured in AD-affected brain regions have aluminum values greater than 5.5 μg Al/g brain tissue [104, 135]. Importantly, toxic effects occur in cats at cerebral aluminum concentrations between 4 and 6 μg Al/g brain tissue [173]. Also, the LD50 (50% lethality dose) for brain aluminum in rabbits is already evident at 5.5 μg Al/g brain tissue [172]. Aluminum combined with food acids, such as aluminum maltol (approved as a food additive in some countries), are much more toxic than poorly soluble inorganic aluminum compounds [172].
AβPP/tau triple-transgenic (3xTg-AD) [174] mice are reported to have higher aluminum levels in their brains than controls, even without aluminum supplementation [175]. It would be interesting to know if other untreated AβPP-transgenic animal models also have elevated aluminum levels in their brains which would imply more efficient aluminum absorption.
Down syndrome patients are reported to exhibit brain aluminum values as high as those in AD brains but at earlier ages [135]. Down syndrome patients generally show AD neuropathology by early middle age [176] and have a significantly higher rate of AD-type dementia than age-matched controls [177].
PHYSICAL PROPERTIES OF ALUMINUM ACCOUNT FOR ITS NEUROTOXICITY
The aluminum ion (abbreviated Al3+) has a high charge (+3) and a very small ionic radius (0.51 Å) in its preferred coordination state with six water molecules. Al3+ is smaller than magnesium (Mg2+) and slightly smaller than the ferric ion (Fe3+) [178]. The size of Al3+ allows it to effectively compete in cells with the essential metals Mg2+ and Fe3+.
Mg2+ regulates more than 300 enzymes [179]. Nanomolar amounts of Al3+ successfully compete with mM amounts of Mg2+, substituting for Mg2+ in the active sites of some key regulatory enzymes, including protein kinase C, and in ATP and GTP co-factors. In this way, Al3+ alters, and impairs, the activities of key regulatory proteins and cellular signal-ingpathways [180, 181 (review)]. Al3+ also substitutes for Fe3+ in Tf and on IRP2 [182, 183.
Al3+ competes with Mg2+, Fe3+, ferrous iron (Fe2+), and calcium (Ca2+) for cell ligands, particularly phosphates. Al3+ also competes with Ca2+ ions for their sites on membranes and in calcium channels [184–186].
The high charge:size ratio of Al3+ causes it to dissociate from ligands very slowly, approximately 108 times more slowly than Ca2+ and 104 times more slowly than Mg2+ [178]. The slow ligand exchange rates of Al3+ prevent Al3+ from successfully participating in enzyme reactions as they require rapid dissociation. Hence, Al3+ disrupts cell metabolism, largely by interfering with the activity of key regulatory enzymes and cell signaling processes [180, 182].
Another aspect of Al3+ toxicity is its pro-oxidant status. Al3+ produces oxidative stress on its own and synergistically with copper and iron [187–189]. These neurotoxic features of aluminum severely incapacitate neuronal function rather than killing cells outright.
ALUMINUM INVOLVEMENT IN DEMENTIAS OTHER THAN AD
High aluminum levels in dialysate water have elevated brain aluminum levels in dialysis patients and caused multiple cases of DES or dialysis dementia [111, 190, 191]. Aluminum levels in the brains of untreated DES patients have exceeded 25 μg/g in brain tissue with death occurring within months to several years [191].
Aluminum has also been implicated in other dementias; namely, Balint’s disease from occupational aluminum exposure [192], amyotrophic lateral sclerosis/parkinsonism dementia complex of Guam [193, 194], and Parkinson’s disease with AD-type dementia [195]. Furthermore, aluminum is the acknowledged cause of cognitive impairment in hundreds of Canadian miners who inhaled McIntyre powder over years in a prophylactic attempt to reduce their chance of developing lung silicosis. The number of miners who became cognitively impaired correlated in a time-dependent manner with their length of exposure to McIntyre powder; i.e., from 0.5 to 10 years, 10–20 years or more than 20 years [196]. McIntyre powder consists of poorly soluble aluminum hydroxide and pulverized (metallic) aluminum.
EVIDENCE FOR ALUMINUM INVOLVEMENT IN AD PATHOGENESIS
The studies that follow, including some based on transgenic animal models and others on wild-type animal models, replicate what appear to be the most important neuropathological aspects of AD.
NFT formation
Protein phosphatase 2A activity and mRNA levels are diminished in AD, resulting in hyperphosphorylation of cytoskeletal proteins
AD brains show depressed activity of protein phosphatase 2A (PP2A), the main enzyme in mammalian brain that removes phosphates from phosphorylated tau [197], and abnormally low mRNA levels for PP2A [198]. The low PP2A activity in AD brains results in hyperphosphorylation of cytoskeletal proteins (in particular, hyperphosphorylated tau and hyperphos-phorylated neurofilament protein) in pyramidal cells and on NFTs [199–206]. Inhibited PP2A activity resulting from a PP2A mutation in transgenic mice also correlates inversely with increased levels of hyper-phosphorylated protein [207].
In AD brains, intracellular NFTs at an early stage of NFT formation are reported to stain more intensely for hyperphosphorylated neurofilament protein and less for hyperphosphorylated tau. At later stages, the NFTs stain more intensely for hyperphosphorylated tau and less for hyperphosphorylated neurofilament protein [199]. Some authors [200] have suggested that phos-phorylated neurofilament protein participates in the formation of human NFTs by serving as a scaffold for tau deposition since hyperphosphorylated neurofilament immunoreactivity appears in patterns (linear skeins, flame-shaped, or intraneuronal globose skeins) very similar to those of silver-stained NFTs. Ultra-structural immunocytochemical studies have shown that antigenic determinants in the 200-kDa neurofilament protein, Bodian’s silver-binding sites in the carboxyl-terminal tailpieces of neurofilament subunits and microtubule-associated proteins, especially tau, are all integral components of the filaments that constitute the NFTs of AD brains [201]. These authors note that NFT filaments comprise sequences from both tau and neurofilament proteins in a hyperphosphorylated form.
Several studies, for example, that by Ksiezak-Reding et al. [208], have reported that immunostaining for hyperphosphorylated neurofilaments in NFTs is artifactual, resulting from antibodies that are cross-reactive for both neurofilament and tau proteins. Vickers et al [202]. analyzed the report by Ksiezak-Reding and colleagues, noting that those authors had used inappropriately small dilutions to demonstrate cross-reactivity between neurofilament and tau proteins. Vickers et al. observed that faint cross-reactivity to tau was just visible in an immunoblot assay with the SM1310 neurofilament antibody if its concentration was higher than that normally used. However, cross-reactivity for native tau was absent when all three of their antibodies for hyperphos-phorylated neurofilaments (SM1310, SM 132, and M14) were used at appropriate dilutions. Also, Vickers et al. used a double-labeled immunostaining technique that revealed NFTs are immunoreactive both for hyperphosphorylated tau and for hyperphosphorylated neurofilament protein [202].
Down syndrome patients likewise exhibit a dramatic decrease in PP2A levels which correlates inversely with elevated levels of hyperphosphorylated tau [209] and hyperphosphorylated neurofilament protein [210].
Aluminum inhibits PP2A activity in vitro and in vivo, resulting in hyperphosphorylation of cytoskeletal proteins
Aluminum inhibition of PP2A activity results in the in vitro formation of hyperphosphorylated tau, the key protein of mature NFTs [211]. Aluminum-inhibited PP2A activity also results in the accumulation of hyper-phosphorylated tau in brains of wild-type rats exposed to human-equivalent levels of aluminum for most of their adult life [146, 212], suggesting that aluminum-induced inhibition of PP2A activity may also explain the increases of hyperphosphorylated tau that occur in brains of aluminum-exposed renal dialysis patients [114], Down syndrome patients [209], and AD patients [115].
Immunostaining evidence indicates that relatively small amounts of aluminum are sufficient to inhibit PP2A activity in neurons. Evidence for this is contained in serial sections of CA1 pyramidal cells from the brains of wild-type rats chronically exposed to aluminum at human-equivalent levels [212]. Immunos-tained sections show CA1 cells with abundant amounts of hyperphosphorylated tau. CA1 cells in adjacent sections stained for aluminum are mostly at intermediate stages (II-III) of aluminum accumulation [212]. Small amounts of hyperphosphorylated tau are just visible in some neurons equivalent to those with stage I aluminum accumulation.
Aluminum inhibition of PP2A activity leads to the accumulation of hyperphosphorylated cytoskele-tal proteins in neurons of humans, rats, rabbits and in vitro experimental systems [114, 115, 211–219]. Nascent intracellular tangles, recently induced by aluminum in rabbit brain, initially exhibit phosphorylated and non-phosphorylated neurofilament protein [215, 216]. Over the next few days, the rabbit tangles change, gradually developing epitopes of hyperphosphorylated tau, AβPP, Aβ, ai-antichymotrypsin, and ubiquitin-protein conjugates [219]. Hence, NFTs that are rapidly induced in certain animal brains by intracerebral aluminum injection evolve to resemble human NFTs [199] more than commonly acknowledged.
A consequence of tau hyperphosphorylation induced by aluminum in neurons is that the excess phosphate interferes with tau’s ability to bind to microtubules. This increases the level of soluble hyperphosphorylated tau in the cytoplasm [220]. The cell continues to produce more normal tau, which soon becomes hyperphosphorylated, contributing to the increasing accumulation of hyperphosphorylated tau. Aluminum causes hyperphosphorylated tau to aggregate into granules of increasing size while sparing normal tau [218].
Around this stage, caspase truncates hyperphospho-rylated tau [221] in AD cells. Aluminum activates caspase 3 [222, 223], the same type of caspase involved in tau truncation [221, 224]. Since aluminum activates caspase 3, inhibits PP2A activity [211, 212], thereby promoting the accumulation of hyperphosphorylated tau, and is present in AD cells where NFTs form, aluminum may also participate in caspase-induced truncation of hyperphosphorylated core tau in these cells.
Aluminum induces the formation of NFTs from hyperphosphorylated tau in brains of humans, particularly those with AD
At the pre-tangle stage, aluminum avidly binds to phosphates on the hyperphosphorylated tau, causing this form of tau to aggregate, precipitate, and form granules of increasing size in AD hippocampal and cortical neurons [218, 225] (Fig. 7A, B).
By the late pre-tangle stage, the aggregated granules have coalesced to form a homogeneous, confluent cytoplasmic mass that stains for both aluminum and hyperphosphorylated tau [225] (Fig. 7C, D)
At an early tangle stage, nascent filaments can be seen within the otherwise homogeneous aluminum/hyperphosphorylated tau complex (Fig. 7E, F).
As more filaments precipitate within the complex, they grow and form NFTs that continue to stain for both aluminum and hyperphosphorylated tau [225] (Fig. 7G, I).
These NFTs are the markers used by neuropathologists to identify AD-affected brain regions involved in AD progression. The aluminum component of NFTs has been confirmed by several different types of instruments [169, 170] as well as staining techniques. Some iron is also found in NFTs [67], suggesting that NFTs are a cell-protective response, involving sequestration of potentially-toxic free metal ions. Aluminum binding to growing NFTs in the neuronal cytoplasm may be a cellular mechanism for slowing aluminum uptake into the nucleus where aluminum avidly binds to nucleic acids and chromatin [225].
NFT formation in AD-affected hippocampus of human brain. A, B) Pre-tangle pyramidal cells stained for hyperphosphorylated tau show (A) small and (B) larger (fused) granules (arrows). C, D) Cytoplasmic pools form from granule fusion and stain for (C) aluminum and (D) hyperphosphorylated tau. Arrows mark the thinner margins of the cytoplasmic pools that consist of an Al/hyperphosphorylated tau complex. E, F) Thin filamentous structures just visible in the cytoplasmic pools, stained for (E) aluminum and (F) hyperphosphorylated tau, represent nascent filaments that develop into NFTs. G, H) Mature NFTs continue to stain for (G) aluminum and (H) hyperphosphorylated tau. I) A large NFT stained to show aluminum (pale purple filaments) has consumed the cytoplasmic pool so individual filaments are more clearly seen (arrow). MB = 7 μm for A–H; 3 μm for I. Reproduced from [225] with permission from IOS Press.
Only a portion of the large CA1 neurons in AD-affected human brains, and aluminum-exposed rabbits, form NFTs [225, 226], indicating that the conditions for NFT formation are rather specific. For species reasons, NFTs are absent from virtually all aged rat neurons, even in those that contain abundant amounts of aluminum and hyperphosphorylated tau [212]. Instead, aged rat hippocampal and cortical neurons show various stages of nuclear aluminum accumulation as do many hippocampal and cortical neurons of aged humans (Fig. 8) and aluminum-exposed rabbits [140, 143, 226].
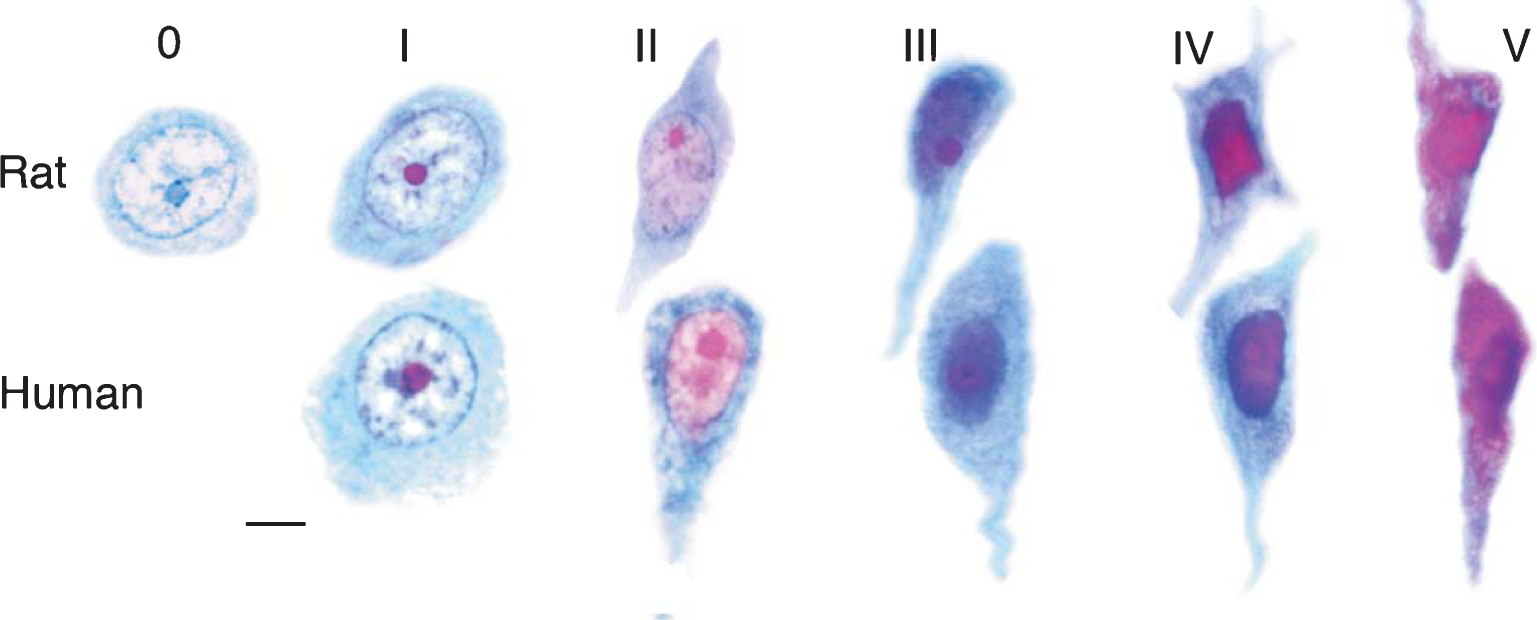
Stages of aluminum accumulation in rat and human hippocampal pyramidal neurons. Stage 1: All sections of hippocampal pyramidal neurons from older human brain, that contain a visible nucleolus, exhibit at least stage I aluminum accumulation (staining only the nucleolus). Stage I neurons have a normal shape. Stages II and III: The nucleoplasm progressively deepens in hue, appearing pink at stage II and purple at stage III. The nucleus and overall cell shape show subtle shrinkage and dendritic changes. Stage IV: Aluminum in the nucleus appears bright magenta. The cell is obviously shrunken and its neurites appear tortuous and retracted. At stage V, aluminum is distributed throughout the nucleus and cytoplasm. These cells are deformed but appear to be viable, exhibiting neither necrosis nor apoptosis. Magnification bar = 5 μm. Reproduced from [143] with permission from Elsevier.
It would be rare for any single animal model to replicate all aspects of a complex disease like AD. For example, AβPP- and tau-transgenic mice are useful models for understanding amyloidogenesis and tauopathies, respectively, in that they focus on replicating a specific AD hallmark rather than AD progression.
The aging aluminum-based rat model is valuable in that it replicates key aspects of AD clinical and neuropathological progression. That is, aluminum specifically produces severe neuropathological damage to the cells of origin for the perforant pathway and hippocampal pyramidal cells in the rats that developed cognitive deterioration. Damage to these cells and brain regions in humans with AD isolates the hippocampus from the neocortex and impairs memory function [25, 29, 227].
Hippocampal isolation can logically explain the cognitive deterioration that occurred in a substantial proportion of the rats that chronically consumed aluminum at the high end of the human total dietary aluminum range. The rats with cognitive deterioration had severely impaired memory function and they displayed other AD-type behaviors and neurological signs [41, 143, 146]. Furthermore, the sequence of damage in their brains parallels that which occurs with AD progression. The superficial entorhinal cortex was most damaged, followed by other limbic regions and neocor-tical association areas—in that order—and, to a much lesser extent, motor and primary sensory areas. The animal model replicates cell damage in the sequence of brain regions involved in AD progression.
As for AD hallmarks, the aging aluminum-based rat model for AD shows incipient changes in Aβ and NFT formation. These changes seldom if ever develop into mature plaques and tangles, clearly a result of species differences. However, aluminum participation in the formation of Aβ and Aβ plaques is described below [228] and this paper has already addressed aluminum participation in human NFTs [225]. The aluminum-induced rat model shows neuropathology that can account for dendritic/axonal dieback, synapse loss, and cortical atrophy, including that which appears on MRI scans analyzed for AD progression [143].
High stage aluminum accumulation in cells and its consequences
High stage aluminum accumulation refers to intense staining for aluminum in the cell nucleus (Stage IV) or throughout the nucleus and cytoplasm (stage V) (Fig. 8) [143]. The available evidence indicates that aged corticocortical cells with high stage aluminum accumulation, whether in the form of NFTs or nuclear aluminum, are drastically different cells from their healthier counterparts that exhibit low-stage aluminum accumulation, both with respect to structure and function.
An electrophysiological consequence of Al-induced NFT formation in neurons that normally discharge at high spontaneous frequencies is that they become electrically inactive [173]. Human neurons with NFTs have low cytochrome oxidase activity [229]. Low concentrations of aluminum inhibit mitochondrial cytochrome c oxidase activity. Low cytochrome c oxidase activity indicates a low metabolic rate, suggesting these cells are no longer capable of neuronal function [230, 231]. Dysfunctional human cells with NFTs are able to survive in this diminished condition in brain tissue for decades [232]. Dysfunctional cells with high stage nuclear aluminum accumulation likewise persist in animal brains for a prolonged time [143].
Human and/or rat pyramidal cells that exhibit NFTs or high stage nuclear aluminum accumulation consistently show microtubule depletion (Fig. 9) [143, 233, 234]. Aluminum-induced microtubule depletion leads to: shrinkage and change in cell shape [140, 143]; impaired axonal transport [163, 235]; dendritic/axonal dieback [143, 236, 237]; and significant loss of synapse density [143, 238, 239]. Dieback results in shrinkage of the cell’s dendritic tree (Fig. 10) that normally accounts for 95% of cell volume [240]. Widespread dieback leads to significant cortical atrophy [241] that becomes increasingly extensive as AD/chronic aluminum neurotoxicity spreads more widely, in a chain reaction that involves increasing numbers of brain regions.
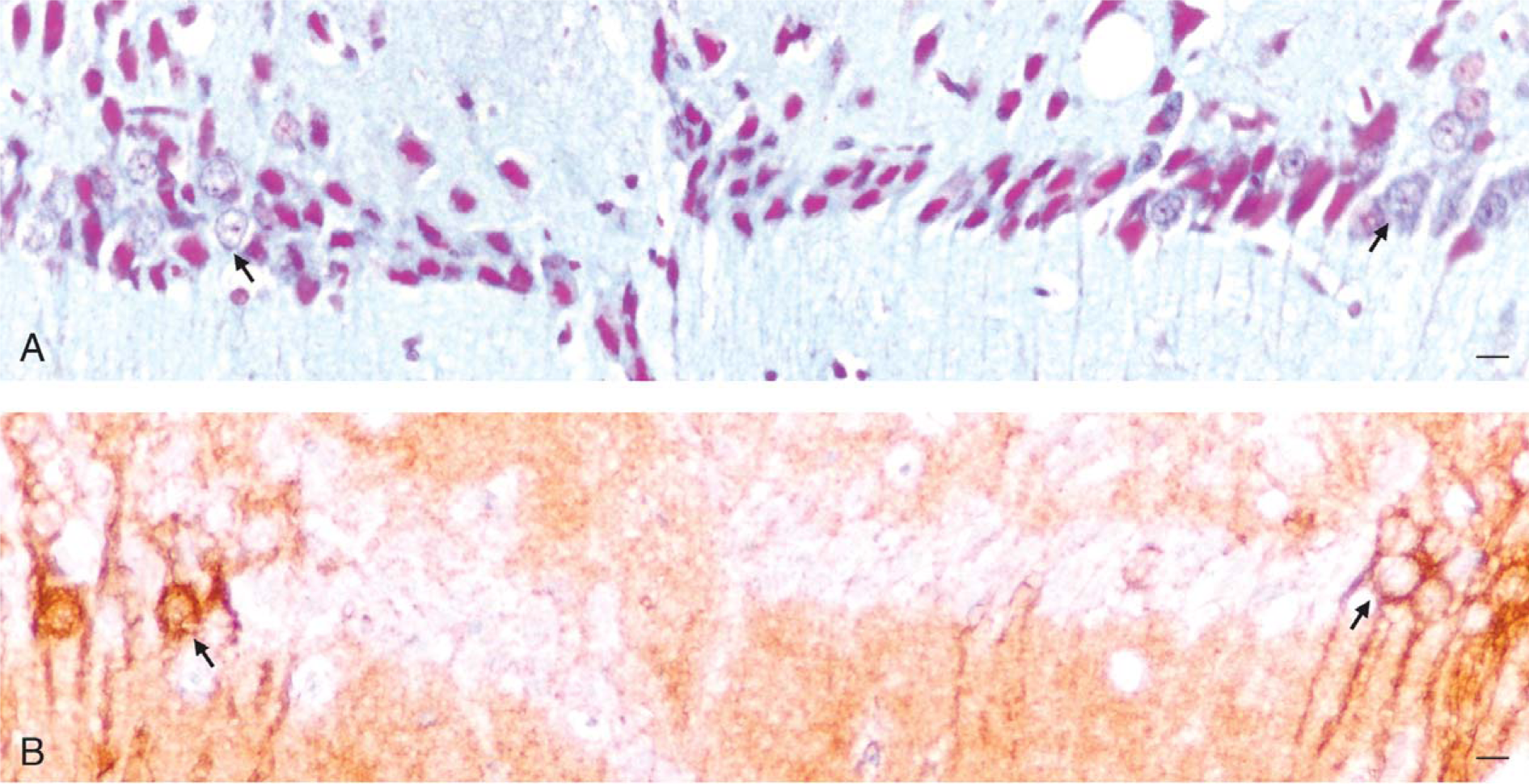
High stage aluminum accumulation correlates with microtubule depletion in rat cortical and hippocampal cells. A) Stage IV pyramidal cells stain magenta for nuclear aluminum within a hippocampal CA1 lesion of an aged rat with cognitive deterioration. Pyramidal cells with a more normal appearance (arrow) are seen at the margins of the lesion. B) An adjacent section immunostained for acetylated-α-tubulin shows that cells within the same lesion fail to immunostain for microtubules whereas microtubules can be clearly seen in the more normal cells along the margins of the lesion (arrow). Magnification bars = 10 μm. Reproduced from [143] with permission from Elsevier.

Camera lucida drawings of AD Golgi-stained pyramidal cells illustrating the process of dendritic/axonal dieback (left to right). Redrawn from [241] with permission from Elsevier.
Aluminum involvement in granulovacuolar degeneration (GVD)
Simchowicz, one of Alzheimer’s students, reported that hippocampal cells of AD tissue show GVD [242]. This pathological feature consists of clusters of intracytoplasmic vacuoles, measuring up to 5 μ in diameter, that each contain a single dense granule, 0.5 to 1.5 μ across. GVD occurs in most AD patients, affecting 9-66% of hippocampal pyramidal cells [243]. Granules in the vacuoles of human hippocampal neurons stain for aluminum [140] and exhibit an aluminum peak when examined by energy dispersive X-ray microanalysis spectroscopy [244]. GVD granules also stain for altered proteins, mainly caspase-cleaved AβPP and hyperphosphorylated tau. GVD formation is experimentally induced in hippocampal cells of rats chronically exposed to dietary aluminum at human-equivalent levels [146, 212] or by repeated intraperitoneal injections of aluminum [245]. Aluminum appears to be the only agent reported to experimentally induce GVD. GVD also occurs together with NFTs in amyotrophic lateral sclerosis/parkinsonism dementia of Guam [193, 246], another neurodegenerative disease that involves chronic aluminum neurotoxicity.
Aluminum involvement in Aβ metabolism
Aβ deposits develop under multiple conditions: as part of the degenerative process in regions of brain where terminals are deteriorating [29]; in association with some blood vessels [247]; and in response to oxidative stress [228]. Aluminum is present in large corticocortical neurons of AD patients [140] and can interact with Aβ at various stages of its formation in experiments. Hence, aluminum may interact with Aβ in the same way in elderly human brains.
Aluminum upregulation expression of the AβPP gene, its mRNA and protein
Aluminum upregulates gene expression for AβPP and other stress-response proteins in human neural cells [83]. AβPP mRNA and AβPP protein expression are increased in the brains of rats chronically exposed to aluminum levels equivalent to those consumed by humans [163]. Aluminum induces axonopathy in hippocampal and cortical regions of rat and rabbit brains in which AβPP-immunostained axons show constrictions and varicosities, indicating impaired axonal transport [163, 235].
Aluminum alters AβPP cleavage to form Aβ instead of sAβPPα
AβPP is cleaved by α-secretase at a site that results in the formation of sAβPPα which is neurotrophic, providing that phosphorylation by protein kinase C (PKC) has occurred [249, 250]. This cleavage precludes the formation of Aβ. However, aluminum, at a concentration of only 2 × 10–8 mol/L, inhibits 90% of PKC activity [251]. Thus, a small amount of intraneuronal aluminum has the potential to redirect AβPP cleavage from its non-amyloidogenic pathway (forming sAβPPα) to its amyloidogenic pathway (forming Aβ). This can explain why exposure of cultured rat neurons to 10–50 μΜ aluminum for three weeks results in a pronounced increase in soluble Aβ in addition to tau accumulation and neurite degeneration [252].
Aluminum interactions with Aβ
Aluminum forms complexes with Aβ in vitro and stabilizes Aβ oligomers [253]. Aluminum also converts soluble human Aβ42 with random-coil structure to a fibrillar precipitate with β-pleated structure [254, 255] and induces human Aβ fibrils to aggregate [248]. Aβ fibrillization is a species-specific effect that occurs in animal species that have the same amino acid sequence for Aβ as in humans. The Aβ sequence of rats and mice differs from the Aβ sequence for humans by three amino acids (Fig. 11) [256]. This species-specific difference is apparently sufficient to block soluble Aβ from fibrillizing and forming Aβ plaques in mouse and rat brains [235, 256].
The Αβ portion of the rat ΑβΡΡ sequence. The protein sequence is shown in the standard one-letter code below the nucleotide sequence. The rat equivalent of the human Αβ polypeptide is located between amino acid residues 598–640 and is shown in gray. Also bold/highlighted are the three positions (601, 606, and 609) where the rat and human sequences differ. The human amino acid residues are indicated below the rat amino acid residues at these positions. The highlighted residues indicate the presumed transmembrane region. Reproduced from [256] with permission from the Nature Group.
Aluminum accelerates and enhances Aβ plaque formation in a transgenic mouse model for amyloidogenesis
Mice have been genetically-engineered to overexpress human AβPP and to form human Aβ. This allows Aβ plaques to form in brains of relatively young transgenic mice. Human AβPP-transgenic mice, fed a diet supplemented by aluminum for one year, exhibited significantly higher levels of plasma Aβ as well as soluble and fibrillar forms of Aβ in their brains [228]. Aβ plaque formation occurred earlier and in appreciably larger amounts in brains of aluminum-exposed transgenic mice than in brains of a transgenic cohort without aluminum supplementation. Thus, aluminum stimulates the formation of Aβ, both in oligomeric and plaque forms, in the brains of AβPP-transgenic mice that express human AβPP and the human Aβ peptide. An implication of this study is that the presence of aluminum in AD brains could likewise stimulate the formation of human Aβ.
Congophilic amyloid angiopathy (CAA), a common accompaniment to AD, involves Aβ deposits in cortical blood vessels. Aβ also deposits in the walls of cortical vessels of mice that have drunk water containing alum for at least one year, serving as a model for CAA in humans [257]. The first neuropathological examination of a person overexposed to alum from the Camelford water pollution incident, who died of an unspecified neurological condition, was determined to have a rare form of sporadic early-onset CAA in leptomeningeal and cerebral cortical vessels. High concentrations of aluminum were found in severely-affected cortical regions of this patient’s brain [258].
Aluminum in Aβ plaque cores
A recent study by Yumoto et al [259]. using transmission electron microscopy with an ultrasensitive spectrometric technique reported that aluminum is detectable in the cores of Aβ plaques. The present author used histological processes to find aluminum in AD neurons and NFTs. It is clear that ultrasensitive highly sophisticated precision techniques such as that of Yumoto need to be brought to bear to determine the presence of minute amounts of aluminum in plaques and the significance of that presence.
A different group, using a prototype nuclear microscope, reported 15 years earlier that aluminum is absent from Aβ plaques [260]. A paper published by the latter authors on the same subject in the following year [261] contained discrepancies; for example, reporting their nuclear microscope had a resolution of 50 ppm compared to the 15 ppm resolution reported in the previous year. The nuclear microscope has yet to be validated for its ability to detect aluminum in histological tissue sections. Regardless, Kawahara et al. report that aluminum concentrations lower than 15 ppm are sufficient to aggregate Aβ [248].
Aluminum induces the formation of a presenilin-2 variant that increases Aβ42 and disrupts signaling in the hippocampal CA1 field of sporadic AD brains
The AD research community has long focused on a search for mutations in three main candidate genes: those for AβPP, presenilin-1, and presenilin-2. The presumed defect would enhance Aβ42 levels in the brain and distinguish sporadic AD cases from nondemented controls. A presenilin-2 gene product that satisfies these requirements was identified by a group of researchers around the turn of the 21st century [262, 263].
This gene product comes from an inducible, alternatively-spliced variant of the presenilin-2 gene designated PS2 V rather than from a mutant gene. The exon 5 sequence is omitted during transcription of the PS2 gene [262]. The shortened mRNA that encodes PS2 V is truncated at its N-terminus. PS2 V is reported to be present in the brains of all sporadic AD cases examined by the authors and absent from 90% of brains from non-demented controls [263]. PS2 V increases Aβ42 production by affecting AβPP cleavage [262]. PS2 V also interferes with AβPP maturation and disrupts the signaling pathway for the unfolded protein response, particularly in the hippocampal CA1 field where PS2 V is associated with dendritic dieback [263].
PS2 V expression is inducible in cultured cells by hypoxia and can be blocked by antioxidants. This suggested to the authors that the PS2 V isoform could be induced by a metal that causes oxidative damage. Neuroblastoma cells were exposed to copper chloride, copper sulfate, zinc chloride, ferrous chloride, ferric chloride, aluminum chloride, and aluminum maltolate to learn whether they might be involved in PS2 V formation. Aluminum (both as the chloride and maltolate salts) is the only metal that consistently induces the PS2 V isoform and does so at a low (25 μΜ) aluminum concentration (Al) [264].
Inflammation in an aluminum-based animal model for AD
Aluminum salts contained in drinking water, at amounts to which humans are routinely exposed, were found to increase cerebral levels of glial activation and inflammatory cytokines as well as increasing the level of AβPP within brains of mice [265]. AD is known to be a mild inflammatory condition associated with elevating basal levels of inflammation markers.
ALUMINUM AND THE FEATURES PROPOSED TO INCREASE CELL SUSCEPTIBILITY TO NFT FORMATION
Aluminum, large cell size, and complexity
Histological stains and SIMS analyses for aluminum in brain tissue from AD cases and animal models for AD reveal that aluminum preferentially accumulates in large corticocortical cells of the entorhinal cortex, hippocampal CA1 field, subiculum, amygdala, and neocortical pyramidal cells with long corticocortical projections in layers III and V [41, 140, 143, 146, 150, 151].
Aluminum and high levels of metabolic activity, transferrin receptors, and iron uptake
Aluminum metabolism is closely intertwined with iron metabolism. The close physical resemblance between Al3+ and Fe3+ ions (charge density, ionic size, and favored coordination number) allows Al3+ to mimic Fe3+ in plasma and cells. Cellular proteins involved in iron transport and iron uptake into cells are somewhat promiscuous in that they fail to discriminate between Fe3+ and Al3+]. This may explain why approximately 90% of plasma aluminum binds to unoccupied iron-binding sites on Tf, circulating as Tf-aluminum, with the remainder binding to albumin and low molecular weight species such as citrate [266, 267].
Aluminum utilizes endothelial Tf receptors, evolved for iron uptake, to cross the blood-brain barrier [49, 268] and neuronal Tf receptors for entering large complex neurons [53]. Aluminum has at least three modes for uptake into neurons: (1) trans-synaptic transport between interconnected neurons; (2) Tf-mediated endocytosis; and (3) mechanisms involved in Tf-independent iron uptake (Tf-IU) [269]. Aluminum preferentially deposits in large cells with high metabolic demands, high densities of Tf receptors on their plasma membrane surface, and high iron uptake to meet those metabolic demands [50].
Aluminum accumulation in neurons leads to iron disregulation
Iron homeostasis becomes disregulated in AD brains as a result of IRP2 stabilization in large neurons (Fig. 12A). Micromolar amounts of aluminum (4 to 20 μΜ Al) successfully compete with Fe3+ for iron-binding sites on IRP2. This stabilizes IRP2 and prevents its degradation. According to Yamanaka et al. [183], the affinity of Al3+ for these iron-binding sites is much higher than for other metals, including iron. Aluminum-induced stabilization of IRP2 prevents IRP2 oxidation and degradation in the ubiquitin-proteosomal pathway (Fig. 12B). This allows IRP2 to stay bound to the mRNA for Tf receptors which in turn allows Tf receptor production to continue and ferritin synthesis to remain suppressed as though the affected cells were permanently iron-deficient [62, 63]. Aluminum-induced stabilization of IRP2 is consistent with the IRP2 stabilization that occurs in AD [65] and may account for this phenomenon. In contrast, IRP1 is unaltered by aluminum exposure or AD [65, 183].
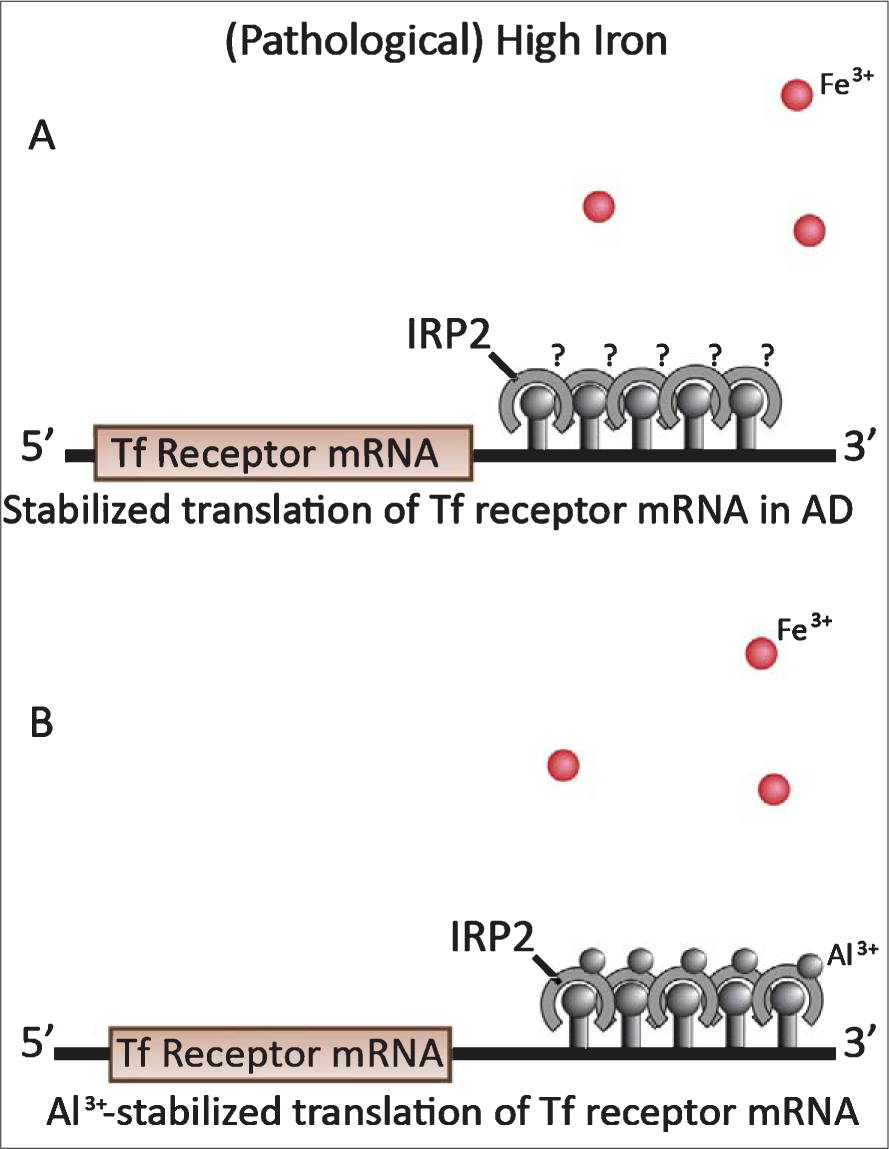
Stabilized translation of Tf receptor mRNA under pathological circumstances. A) Fe3+ ion oxidation of IRP2 does not occur in AD-affected neurons despite high iron levels. This results in stabilized translation of Tf receptor mRNA and continuation of iron and aluminum uptake. B) Aluminum ions bind to Fe3+ ion-binding sites on IRP2 s of aluminum-exposed neurons, preventing Fe3+ ions from oxidizing the IRP2 s and signaling for IRP2 destruction. This results in the stabilized translation of Tf receptor mRNA and continuation of iron and aluminum uptake. In AD and aluminum exposed cells, IRP1 behaves in its normal manner (see Fig. 3C and D).
In vitro and in vivo evidence both attest to aluminum’s effects on iron metabolism. Intraneuronal aluminum accumulation enhances iron uptake and expression of hyperphosphorylated tau in neuroblastoma cells [270]. A kinetic radiolabelled Tf-Fe binding assay showed the number of surface Tf receptors per cell was higher in cells cultured in the presence of aluminum than in those without aluminum exposure [271]. This increase in Tf receptor numbers probably reflects aluminum-induced stabilization of IRP2.
Rats given a series of thrice weekly intraperitoneal injections of aluminum gluconate over 8 weeks (“aluminum-loading”) showed dramatically increased brain aluminum levels, ranging between 14 to 25 μg/g (wet weight) in AD-vulnerable brain regions. The high aluminum levels were paralleled by increased iron levels in the same regions, including 4- to 9-fold iron increases in the temporal, frontal, and parietal cortices and a 4-fold iron increase in the hippocampus of aluminum-exposed rats compared to iron levels in untreated controls [272]. These results suggest that the subtle rise in brain iron stores that occurs for some time with increasing age [103, 273] maybe secondary to the increase that occurs in brain aluminum levels over time [135–138].
Aluminum, iron, and oxidative stress
Aluminum produces oxidative damage on its own and synergistically with iron. Intracellular aluminum forms an aluminum-superoxide complex that produces oxidative damage [187, 274] and aluminum also enhances iron-initiated oxidative damage [187, 275]. Brain cells have a limited ability to react to oxidative stress, particularly in view of their relatively low levels of glutathione and glutathione peroxidase.
Aluminum-loading for eight weeks significantly increases levels of lipid peroxidase while decreasing superoxide dismutase and glutathione peroxidase levels even further. Iron elevation on its own is unable to produce consistent change in any of the cytopro-tective enzymes (superoxide dismutase, glutathione reductase, glutathione peroxidase, andcatalase) [272].
Aluminum stabilizes intracellular ferrous iron by preventing its oxidation to ferric iron [275, 276]. Ferrous iron reacts with hydrogen peroxide, driving the highly toxic Fenton reaction that generates reactive hydroxyl radicals that increase oxidative stress. Aluminum stabilization of the ferrous iron is an important mechanism for enhancing iron-induced free radical production and oxidative stress [276].
Aluminum and myelination
Myelin sheaths provide insulation for nerve fibers, allowing impulses to transmit quickly and efficiently along the length of the axon. A poorly-myelinated neuron with a leaky axon needs to generate vastly more energy to compensate for its axonal wastage. The entry of aluminum and excess iron into the poorly-myelinated neurons of the entorhinal cortex, hippocampus, and other limbic regions could render these neurons susceptible to significant peroxidative stress and its oxidative consequences—oxidized fats and proteins, leading to the formation of reactive aldehydes and carbonyls, cross-linking of proteins, nucleic acids, and proteins with nucleic acids [277, 278].
Aluminum and vascularization
Tf receptor densities on capillary endothelium are 6 to 10 times greater than those on neurons [279]. The vascular plexus that surrounds stellate and pyramidal cell islands of the superficial entorhinal cortex invests the islands with dense capillary networks [1]. In principle, larger amounts of aluminum should be removable from the plasma by these dense capillary networks and transferred into the extracellular matrix surrounding the cells of the superficial entorhinal cortex than in regions where capillaries are less abundant. Increased amounts of extracellular aluminum would then be available to plasma membrane Tf receptors for uptake into neurons. This may be a reason why aluminum preferentially accumulates in the entorhinal cortex early in AD and why these cells are damaged to a greater extent than cells of other limbic regions.
ALUMINUM AND AD PROGRESSION
Aluminum can spread from one brain region to another via interconnected corticocortical cells. Gelfoam® impregnated with 15% aluminum lactate or 5% aluminum chloride was implanted into the nasal cavity of rabbits [280]. One month later, high aluminum levels and neuropathological changes were observed in the olfactory bulb, pyriform cortex, hippocampus, and cerebral cortex. This study demonstrates that soluble forms of aluminum can enter the processes of olfactory neurons in the nasal mucosa and move across pyramidal cell synapses in both retrograde and anterograde directions [26].
Aluminum spread to different brain regions via interconnected corticocortical cells, together with aluminum-inhibited PP2A activity which results in hyperphosphorylated tau, can logically account for the “spread of hyperphosphorylated tau” reported to occur between interconnected corticocortical cells [35–37]. It would be prudent to stain for aluminum, in investigations into the spread of hyperphosphorylated tau, to ensure aluminum absence before assuming that hyper-phosphorylated tau is acting on its own. Aluminum spread between interconnected cells could also contribute towards the spread of NFTs and microtubule depletion in AD-affected brain regions [14, 143].
SUGGESTED SCENARIO FOR ALUMINUM INVOLVEMENT IN AD PROGRESSION
Aluminum ingestion and absorption is an ongoing process in contemporary society as is aluminum uptake into the brain [133]. Dietary aluminum can thus fuel the spread of aluminum over time into an increasing number of brain regions. Minute amounts of aluminum progressively enter neurons via Tf receptors, under normal control of the IRPs, throughout the long prodromal phase.
Intraneuronal aluminum spreads between stellate and pyramidal corticocortical cells of the superficial entorhinal cortex, within islands and between islands, and from these entorhinal cortical cells into connecting corticocortical cells of the hippocampal CA1/subiculum zone, olfactory bulb, neocortex, amygdala, and other brain regions with which the entorhinal cortex communicates. The amount of aluminum provided by projections from the entorhinal cortex to interconnected corticocortical cells in other brain regions could be proportional to their connections.
More than midway through the prodromal period, aluminum accumulation in entorhinal cortical stellate and pyramidal cells begins to reach neurotoxic levels. At this point, intracellular aluminum levels are sufficiently high to compete with essential metals, to inhibit PP2A activity, and disrupt iron metabolism. Cellular IRP2 levels become stabilized and their iron metabolism is disregulated, showing an increased density of Tf receptors on their surface. The disregulated entorhinal cortical cells continue to import aluminum and iron from the neuropil, exporting these metals directly into connecting cells without obvious restraints on this process.
Neurons require tau protein for microtubule assembly and microtubule stabilization. The activity of tau protein depends on alternating phosphorylation and dephosphorylation reactions. Kinases continue to phosphorylate tau but PP2A activity is now too low in these cells to remove phosphate groups. This situation leads to cytoplasmic accumulation of hyper-phosphorylated cytoskeletal proteins; in particular, hyperphosphorylated tau [281–283].
Hyperphosphorylated tau is unable to bind to microtubules so microtubule assembly cannot proceed and existing microtubules break down. Consequently, cells that develop hyperphosphorylated tau, including those in which NFTs form, become microtubule-depleted [233, 234]. Microtubule depletion results in dendritic/axonal dieback, de-efferentation, and deaf-ferentation of the superficial entorhinal cortical cells. Dieback of cell processes, and neuron death in regions where abundant NFTs form, appear in MRI scans as gray matter atrophy.
Meanwhile, aluminum aggregates the soluble hyperphosphorylated tau that occurs in some cells [218], forming granular precipitates which fuse to form larger granules. Eventually, the large granules also fuse to form cytoplasmic pools of the aluminum/hyperphosphorylated tau complex out of which filaments and NFTs precipitate. The NFTs mature, slowly growing as they acquire more aluminum and more hyperphosphorylated tau (Fig. 8G-I). Very large NFTs eventually denucleate their host cells which consequently die and leave the NFTs behind [140].
Such destruction of the entorhinal cortical cells of origin for the perforant pathway, which bi-directionally interconnects the hippocampal formation with the rest of the cortex, results in isolation of the hippocampus, ultimately leading to the memory defects and confusion that herald the onset of overt AD [25, 29, 227].
The disease continues to spread to corticocortical cells in other brain regions, exhibiting the same types of pathology and functional loss as in regions affected at an earlier stage. In time, the transformation of neurons into dysfunctional cells becomes extensive in some AD-affected regions. Microtubule-depleted corticocortical cells in tissues with slower aluminum accumulation and more slowly-growing NFTs, can survive for decades in the dysfunctional state [232]. Microtubule-depleted cells are unable to contribute to neural function. These cells are unable to sustain their high cytochrome oxidase activity [44, 229]. Most of their chromatin is in the heterochromatin form (compacted DNA with reduced transcription) [278]. As each of the regions involved in AD progression becomes affected by these changes, their specialized functions diminish to the extent that their large neurons are damaged.
The scenario presented here is consistent with the atrophy of gray matter that occurs over time with AD progression as described by the Braak hierarchical model, MRI/VBM clinical trials and other neuropathological studies.
CONCLUSIONS
This paper describes studies that have endeavored to define the sequence of interconnected brain regions affected in AD, either by MRI/VBM or neuropathological techniques. There is a consensus among these investigators that AD spreads in a stepwise manner along well-defined corticocortical connections from the entorhinal cortex to other limbic regions in the medial temporal lobe, followed by the temporoparietal association cortices, and then frontal and occipital cortices, and via corticosubcortical connections to the nucleus basalis of Meynert and to susceptible brainstem nuclei.
From the major proposed candidates as the environmental component for AD, aluminum provides the best fit. Aluminum preferentially accumulates in the cells of origin for the perforant pathway of the entorhinal cortex and other large corticocortical cells prone to NFT formation in specific brain regions of AD patients and in rats with aluminum-induced cognitive deterioration.
The main reason for aluminum deposition in AD-affected brain regions is that AD-affected brain regions have large neurons with high metabolic activity and high iron requirements. Aluminum ions are very similar in size and charge to ferric iron ions. The problem is that endothelial cells of the blood-brain barrier and these large neurons are unable to distinguish between the essential iron ions and non-essential toxic aluminum ions that they take in via their transferrin receptors. Eventually, the concentration of intraneuronal aluminum ions can be sufficient to disrupt iron regulation, further increasing iron and aluminum uptake. Aluminum participates in NFT formation in AD-affected neurons of the hippocampus, entorhinal cortex and other NFT-affected brain regions.
Other factors that may help to explain why the entorhinal cortex is one of the earliest- and most severely-affected brain regions in AD are: (1) the entorhinal cortex is poorly myelinated; and (2) the entorhinal cortex has abundant and redundant vascularization that could increase its exposure to plasma aluminum to a greater extent than in other less well-vascularized limbic regions.
This article also describes aluminum contributions to AD neuropathology: to AβPP and Aβ metabolism; NFT formation and growth; and to GVD. Given that aluminum can spread along the olfactory pathway from neuron to neuron, a scenario has been constructed to show how aluminum could spread between interconnected cells that make up the AD progression sequence and, in principle, spread more widely as AD progresses throughout the brain.