
Abstract
The 24-residue peptide humanin (HN) has been proposed as a peptide-based inhibitor able to interact directly with amyloid-β (Aβ) oligomers and interfere with the formation and/or biological properties of toxic Aβ species. When administered exogenously, HN, or its synthetic S14G-derivative (HNG), exerted multiple cytoprotective effects, counteracting the Aβ-induced toxicity. Whether these peptides interact directly with Aβ, particularly with the soluble oligomeric assemblies, remains largely unknown. We here investigated the ability of HN and HNG to interact directly with highly aggregating Aβ42, and interfere with the formation and toxicity of its oligomers. Experiments were run in cell-free conditions and in vivo in a transgenic C. elegans strain in which the Aβ toxicity was specifically due to oligomeric species. Thioflavin-T assay indicated that both HN and HNG delay the formation and reduce the final amount of Aβ42 fibrils. In vitro surface plasmon resonance studies indicated that they interact with Aβ42 oligomers favoring the formation of amorphous larger assemblies, observed with turbidity and electron microscopy. In vivo studies indicated that both HN and HNG decrease the relative abundance of A11-positive prefibrillar oligomers as well as OC-positive fibrillar oligomers and had similar protective effects. However, while HN possibly decreased the oligomers by promoting their assembly into larger aggregates, the reduction of oligomers caused by HNG can be ascribed to a marked decrease of the total Aβ levels, likely the consequence of the HNG-induced overexpression of the Aβ-degrading enzyme neprilysin. These findings provide information on the mechanisms underlying the anti-oligomeric effects of HN and HNG and illustrate the role of S14G substitution in regulating the in vivo mechanism of action.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of progressive senile dementia, affecting a large proportion of aged people in developed countries and causing memory impairment, disordered cognitive function and decline in language abilities [1]. Deposition in several brain areas of amyloid plaques mainly composed of amyloid-β (Aβ) peptides, and intra-cellular neurofibrillary tangles of hyperphosphorylated tau are the central neuropathological features of AD [2]. Studies aimed at deciphering the relationship between plaque burden and neuronal death indicate that oligomeric pre-fibrillar assemblies of Aβ, more than fibrils, play a key role in the toxicity and early cognitive impairment in AD [2–5]. Despite efforts to develop disease-modifying molecules, no effective therapy is yet available for AD. Numerous strategies have been designed aimed at preventing or delaying the self-assembly of monomeric Aβ into oligomeric forms, including molecules that interact directly with the misfolded protein, modify the kinetics of fibril formation, and facilitate their re-absorption [6–8].
Peptide-based inhibitors that interact directly with Aβ and interfere with the formation and/or biological properties of Aβ aggregates have been proposed as therapeutic candidates. The 24-residue peptide humanin (HN), first described as a neuroprotective factor identified from the cDNA from a protected lobe of an AD patient’s brain [9], has stimulated interest on account of its potential as an endogenous protective molecule.
HN is a mitochondrial-derived peptide, physiologically expressed in various organs and highly conserved across species, from the nematode Caenorhabditis elegans to humans, which may act as a retrograde signaling molecule to transmit information from the mitochondria to the nucleus and beyond [10]. This peptide is cytoprotective against a variety of stress and disease conditions, including AD, and low circulating HN levels may represent a risk for the insurgence of age-related pathologies [10, 11].
At central level, the neuroprotective action of HN was mediated by different mechanisms, involving the ability to prevent the aggregation and deposition [12, 13] of Aβ and to counteract its neurotoxic effects. HN can also bind to its putative receptor activating the intracellular signaling cascade and inhibiting c-Jun N-terminal kinase [14]. Studies in vitro have shown that HN acts as an anti-oxidant molecule, counteracting the Aβ-induced oxidative stress [15, 16]. It also protects neuronal cells from apoptosis induced by the amyloid-β protein precursor (AβPP) by binding to the apoptosis-regulator protein Bax [14].
Structure-function relationship studies showed that the amino acid residues between the Pro3 and Pro19 of HN were particularly important for its neuro- and cyto-protective activities [9]. On this basis a new synthetic peptide was developed in which Gly was substituted for Ser14 (HNG), proving about 1000 times more effective than HN on neuronal cell death in vitro [14]. When chronically administered to transgenic mice overexpressing both AβPP and mutant human presenilin-1, HNG significantly reduced the neuronal Aβ deposits [17–19]. All these findings, upholding the neuroprotective effect of endogenous HN and HNG, support their use as peptide-based inhibitors.
To clarify the underlying mechanisms better, we applied a multidisciplinary and integrated approach to investigate the ability of HN and HNG to interact directly with Aβ42 and interfere with the formation and toxicity of oligomers. Experiments in cell-free conditions indicated that HN and HNG have similar effects on Aβ aggregation, interacting with preformed toxic soluble oligomeric species. In vivo studies were also done to investigate how HN and HNG counteracted the toxicity specifically due to Aβ oligomeric species. We used transgenic CL4176 C. elegans, in which the inducible expression of human Aβ42 results in the specific accumulation of oligomeric assemblies in the body wall muscle cells which leads to paralysis [20–23]. This strain has been widely employed as a simplified invertebrate prototype of Aβ amyloidogenesis, and is a valuable in vivo animal model to elucidate the pharmacological mechanisms involved in the protective action of some anti-Aβ compounds [20, 25]. HN and HNG similarly reduced soluble oligomers, and protected nematodes against the Aβ toxicity. However, while HN specifically decreased the oligomers by promoting their assembly into larger aggregates, the main in vivo effect of HNG was the reduction of total Aβ levels. These data indicate for the first time the relationship between the amino acid sequence of HN-derived peptides and their mechanisms of action.
MATERIALS AND METHODS
Peptides synthesis
HN (MAPRGFSCLLLLTSEIDLPVKRRA), HNG (MAPRGFSCLLLLTGEIDLPVKRRA) and an unrelated 24-amino acid fragment of the human albumin (rAlb25-48, EAHKSEIAHRFKDLGEQH-FKGLVL), used as negative control, were synthesized by solid-phase chemistry using FMOC-protected amino acid (Flamma, BG, Italy) with an Applied Biosystems 433A peptide synthesizer (Life Technologies, Monza, Italy) on a 0.1 mM scale. The fluorenylmethyloxycarbonyl group was automatically removed using 20% piperidine (Sigma-Aldrich, Saint Louis, MO, USA) in N-methylpyrrolidone solution (Applied Biosystems, Foster City, CA, USA) and amino acids were activated with -O-(benzotriazol-1-yl)- N,N,N′,N′-tetramethyluronium tetrafluoroborate and N,N,-diisopropylethylamine (both from Sigma-Aldrich, St. Louis, MO, USA). After the last coupling cycle of each amino acid a capping step with acetic anhydride (Sigma-Aldrich, St. Louis, MO, USA) was included. Peptides were cleaved from the resin (Rink Amide MBHA, 100–200 mesh, 0.7 mmol/g) with trifluoroacetic acid (TFA): triisopropylsilane solution (95:5 vol/vol), precipitated, and washed with diethyl ether. They were purified by reverse phase HPLC on a semi-preparative C18 column (Vydac, Grace, Columbia, MD, USA) with mobile phases of 0.1% TFA in water (eluent A) and 0.08% TFA in acetonitrile (eluent B), using a linear gradient from 5 up to 100% of eluent B in 60 min. Peaks were collected and characterized by matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry (MS) with a Bruker Reflex III TOF mass spectrometer operating in reflector mode (Bruker, Billerica, MA, USA). The peaks identified on the basis of the mass/charge (m/z) were consistent with the theoretical molecular weight of the peptides, i.e. 2687 Da for HN and 2656 Da for HNG (Supplementary Figure 1). The purity was great than 95%. Peptides were lyophilized and stored at – 20°C until use.
Circular dichroism (CD) analysis
The secondary structure of HN and HNG was determined using a Jasco J-815 CD spectro-polarimeter (Jasco Europe, Cremella, Italy) equipped with a Peltier heating system.
Synthetic Aβ42 oligomers
Depsi-Aβ1-42 peptide was synthesized as previously described [26]. The peptide was precipitated into cold diethylether, washed three times and purified with semi-preparative HPLC (C4 Water Symmetry 19×150 mm) using the following solvent systems: A: 0.1% TFA in H2O and B: 0.08 % TFA in CH3CN. Purity was determined under similar conditions using an analytical C4 Waters Symmetry column (4.6×150 mm) (Supplementary Figure 2A). Finally, the molecular mass of the final product was analyzed with a MALDI-TOF mass spectrometer (Bruker) (Supplementary Figure 2B). The purity was great than 95%. Peptides were lyophilized and stored at – 20°C until use.
The advantages of using depsi-peptide as a starting point to have reproducible seed-free stock solutions of monomeric Aβ1-42 have been previously demonstrated [26–30]: in particular, depsi form has a much lower propensity to spontaneous aggregation, and is 100 times more water soluble (15 mg/mL) than the native Aβ1-42 prepared by the standard synthesis protocol [27], avoiding the need for vortexing. The Aβ1-42 depsi-peptide adopts and retains a monomeric unordered state under acidic conditions and when the solution is stored at – 80°C for several days [29]. The native Aβ1-42 sequence is then easily and irreversibly obtained by a “switching” procedure involving a change in pH [26, 31], which produces Aβ1-42 with an unordered structure that undergoes the usual oligomerization and amyloid fibril formation. In more detail, the depsi-Aβ1-42 peptide was dissolved in 0.02 % TFA solution, pH < 3, to obtain a 500–600 μM solution, and injected into a Fast protein liquid chromatography apparatus (Biologic DuoFlow FPLC system, Biorad) equipped with a Yarra 3000 column (Phenomenex, Torrance, CA). Depsi- Aβ1-42 peptide was eluted with 0.02% TFA solution at a flow rate of 1.0 mL/min and detected by monitoring the UV absorbance at 214 nm. When necessary, the monomer fraction was concentrated to 100 μM using a 3 kDa cut-off filter (YM-3, Millipore). The final concentration of purified depsi-Aβ42 peptide was calculated on the basis of the absorbance, measured at 214 nm using the UV/VIS spectrophotometer (Lambda 19, Perkin Elmer, Italy), and the theoretical molar extinction coefficient ɛ (214 nm)Aβ42 = 76848 M-1 cm-1 [32]. The purified peptide stock solution was stored at – 80°C.
To prepare Aβ42 oligomers, depsi-Aβ1-42 peptide stock solution was dissolved and we applied the “switching” procedure, involving a change in pH, to obtain the Aβ42 monomer [26, 33]. The solution was then immediately diluted to a final concentration of 100 μM in 10 mM phosphate buffered saline (PBS) containing 150 mM NaCl, pH 7.4, and incubated at 25°C or 37°C to to obtain the maximal amount of biological relevant oligomers after 5 h or 2 h, respectively [30, 34].
Thioflavin T (ThT) fluorescence assay
We investigated the effects of HN and HNG on the aggregation kinetics of Aβ42 peptide using an in situ ThT fluorescence assay [35] based on the increase of the fluorescence signal of ThT when bound to amyloid fibrils [36]. Aβ42 (2 μM) in 50 mM sodium phosphate, pH 7.4, containing 20 μM ThT, was incubated with or without 2 μM HN or HNG, or 2 μM rAlb25-48 used as negative control peptide. Samples (3×100 μL) were incubated at 28°C in96-microplate wells (Mikroplate Corning 3651, Corning Incorporated Life Sciences, Acton, MA) and the ThT fluorescence intensity was measured, under non-shaking conditions, every 2.5 min for 15 h using a M200 Infinity plate reader spectrofluorimeter (Tecan F200, Männedorf, Switzerland) at emission wavelength 440 nm and excitation wavelength 495 nm. For control experiments 2 μM HN or HNG were incubated with 20 μM of ThT. The half-times of transition (i.e. the times at which 50% of the maximal ThT signal was reached, t1/2) were obtained by normalizing the ThT traces on the corresponding maximal ThT fluorescence intensity. Growth rates were estimated from the slope at the inflection point of the normalized reaction curves, assuming complete Aβ monomer incorporation intofibrils.
Surface plasmon resonance (SPR)
For SPR we used the ProteOn XPR36 Protein Interaction Array System (Bio-Rad). Anti-Aβ monoclonal antibody 4G8 (Covance, Emeryville, CA) was immobilized by amine coupling chemistry onto the surface of a GLC sensor chip (Bio-Rad) [30]. This antibody, which recognizes amino acid residues 17–24 of Aβ, was here employed as capturing agent for the detection of oligomeric species as already described [30]. Briefly, the surface of the sensor chip was activated for 5 min with a mixture of 0.2 M 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride and 0.05 M sulfo-N-hydroxysuccinimide (Bio-Rad); this was followed by injection of 4G8 (30 μg/mL in sodium acetate, pH 5.0), which flowed for 5 min at 30 μL/min. The remaining activated groups were blocked with a 5 -min injection of 1 M ethanolamine. The amount of 4G8 covalently immobilized was about 5000 Resonance Units (RU, 1 RU = 1 pg/mm2). A reference channel was prepared in parallel using the same activation/deactivation procedure but without adding the antibody.
After rotation of the microfluidic system, aliquots of Aβ42 monomers co-incubated at 25°C for 5 h with or without HN or HNG and Aβ42 oligomers treated for 30 min with HN or HNG were injected over immobilized 4G8 for 2 min at a flow rate of 30 μL/min. Identical experiments were performed also using rAlb25-48 as negative control peptide. HN, HNG and rAlb25-48 (1 μM) were injected onto 4G8 immobilized on the SPR sensor chip to evaluate their possible nonspecific binding.
Dissociation was measured for the next 11 min. According to our previous data, Aβ42 monomers completely dissociate from 4G8 after >10 min in running buffer, at which times the SPR signal is only due to oligomers bound pseudo-irreversibly to the antibody. The running buffer, also used to dilute the samples, was 10 mM PBS containing 150 mM NaCl and 0.005% Tween 20. All these assays were done at 25°C. The sensorgrams (time course of the SPR signal in RU) were normalized to a baseline of 0. The signal on the surfaces immobilizing the antibody was corrected by subtracting the nonspecific response on the reference surface.
Atomic force microscopy (AFM)
Synthetic Aβ42 (100 μM) was incubated at 25°C for 5 h with or without 100 μM HN or HNG, diluted in 10 mM PBS to a final concentration of 10 μM and incubated for 0.5–2 min on a freshly cleaved mica disk. The disk was washed with water and dried under a very gentle nitrogen stream. The sample was then mounted on a Nanoscope V multimode AFM (Veeco/Digital Instruments, Santa Barbara, CA, USA) operating in tapping mode using standard phosphorus-doped silicium probes (Veeco). The scan speed was 1 Hz. Three pictures from distinct regions were taken for every sample.
Transmission electron microscopy (TEM)
Five-μL drops of synthetic Aβ42 (100 μM) alone or incubated with 100 μM HN or HNG for 2 h at 37°C, were diluted in 10 mM PBS to a final concentration of 10 μM and placed to dry at room temperature on a 100 mesh Formvar/carbon-coated copper grid (EMS, Hatfield, PA, USA) for 30 min. Grids were washed three times with H2O, counterstained with 2% uranyl acetate and observed with an Energy Filter Transmission Electron Microscope (EFTEM, ZEISS LIBRA® 120) equipped with YAG scintillator slow-scan CCD camera.
Turbidity assay
Changes in Aβ42 polymerization was assessed by performing turbidity assay [34]. Aβ42 (100 μM) in 50 mM sodium phosphate (pH 7.4) was incubated with or without 100 μM HN or HNG at 37°C for 2 h and the absorbance at 400 nm was measured using a M200 Infinity plate reader spectrofluorimeter (Tecan M200, Männedorf, Switzerland). Control experiments were performed in parallel by incubating buffer solution alone or containing 100 μM HN or HNG. These values were used to correct the absorbance value of corresponding solution containing Aβ42.
C. elegans studies
The transgenic CL4176 strain (smg-1(cc546ts)I; dvIs27[pAF29 (myo-3/Aβ1–42/let UTR)+pRF4 (rol-6(su1006)]) expressing human Aβ42 in the body-wall muscle [37] and the control CL802 strain (smg-1(cc546) I; rol-6(su1006) II) were obtained from the Caenorhabditis Genetic Center (CGC, USA) and propagated on solid Nematode Growth Medium (NGM) seeded with OP50 E. coli (CGC, USA) for food [20]. Age-synchronized worms were obtained by transferring nematodes to fresh NGM plates to reach maturity at three days of age and lay eggs overnight. Isolated hatchlings from the synchronized eggs were placed at 16°C on fresh NGM plates (35×10 mm culture plates, 100 worms/plate) seeded with OP50 E. coli. Aβ42 expression was induced by putting worms at 24°C, 60 h after plating. Worms were treated 12 h after the temperature rise (at L3 larval stage) with 0–100 μM of HN, HNG (100 μL/plate), or 100 μM tetracycline hydrochloride (Fluka, Switzerland) as positive control [20]. All compounds were freshly dissolved in water before use. Control worms received vehicle alone (Vehicle). Paralysis of the nematodes was evaluated after 24 h, at the L4 larval stage, by scoring worms that did not move or only moved their head when gently touched with a platinum loop.
Aβ expression
Transgenic CL4176 and CL802 worms, fed vehicle, HN or HNG as described above, were collected with M9 buffer, centrifuged at 1100×g for 4 min and washed twice to eliminate the bacteria. Worms were suspended in lysis buffer (5.0 mM NaCl, 5.0 mM EDTA, 1.0 mM dithiothreitol and protease inhibitor mixture in 25 mM Tris/HCl buffer, pH 7.5) and homogenized using a TeSeE homogenizer (Bio-Rad) with acid-washed glass beads (Sigma) [20]. For dot-blot analysis, equal amounts of protein (20 μg) were spotted onto nitrocellulose membranes (Millipore). To evaluate the total Aβ level, the membranes were incubated with an anti-Aβ mouse monoclonal antibody, clone WO2 (1:1000, Millipore), recognizing amino acid residues 4–10 of Aβ (1:1000, Millipore) [21, 24]. The membranes were incubated with two different anti-conformational antibodies: the rabbit polyclonal A11 antibody recognizing prefibrillar oligomers (1:1000 dilution, Biosource, USA) [2] and the rabbit polyclonal OC antibody recognizing anti-amyloid fibrils as well as Aβ fibrillar oligomers (1:1000 dilution, Chemicon) [38, 39]. To minimize background staining due to non-specific membrane-binding of the antibody, the membranes were saturated for 1 h at room temperature by incubation with 10 mM PBS, pH 7.4 containing 0.1% (v/v) Tween 20, 5% (w/v) low-fat dry milk powder and 2% (w/v) bovine serum albumin. Peroxidase-conjugated anti-mouse IgG (1:20.000, Sigma) and peroxidase-conjugated anti-rabbit IgG (1:20.000, Sigma) were used as secondary antibodies. A 0.1% Ponceau Red solution (Sigma Aldrich) was used to stain the blotted membranes for total protein visualization.
We identified the Aβ species in lysates from transgenic C. elegans strains by immunoblotting using 15% Tris-glycine gel, and by western blotting [20]. After heating samples in sample buffer containing 5% β-mercaptoethanol (1:1 vol/vol, Bio-Rad), 50 μg of the total protein were loaded in each lane of the gel. The membranes were blocked with 10 mM Tris-HCl solution, pH 7.5, containing 100 mM NaCl and 0.1% (vol/vol) Tween 20, and incubated overnight with the monoclonal anti-Aβ antibody 6E10 reactive to amino acid residue 1–16 (1:1000, Covance) or mouse monoclonal anti-actin antibody (Clone C4 MAB1501, 1:1000, Chemicon International). Peroxidase-conjugated anti-mouse IgG (1:10.000, Sigma) was used as the secondary antibody. The mean volumes of dot-blot immunoreactive spots and of Ponceau-dyed spots were analyzed using Quantity One 1-D Analysis Software (Bio-Rad). The data were expressed as the mean volume of the immunoreactive spot/volume of total Ponceau-dyed proteins in the spot±SD.
Neprilysin expression
The level of the neprilysin was evaluated by dot-blotting analysis performed on lysates of transgenic CL4176 worms, fed for 20 h vehicle, 50 μM HN or HNG. Equal amounts of protein (20 μg) were spotted onto nitrocellulose membranes (Millipore) and the membranes were incubated with the rabbit polyclonal anti-neprilysin antibody (Neutral Endopeptidase, Nep, 1:1000, Millipore). To minimize background staining due to non-specific membrane-binding of the antibody, the membranes were saturated for 1 h at room temperature by incubation with 10 mM PBS, pH 7.4 containing 0.1% (v/v) Tween 20, 5% (w/v) low-fat dry milk powder and 2% (w/v) bovine serum albumin. Peroxidase-conjugated anti-rabbit IgG (1:20.000, Sigma) were used as secondary antibodies. A 0.1% Ponceau Red solution (Sigma Aldrich) was used to stain the blotted membranes for total protein visualization. The mean volumes of dot-blot immunoreactive spots and of Ponceau-dyed spots were analyzed using Quantity One 1-D Analysis Software (Bio-Rad). The data were expressed as the mean volume of the immunoreactive spot/volume of total Ponceau-dyed proteins in the spot±SD.
Statistical analysis
The effects of vehicle, HN and HNG on cultured worms were compared by an independent Student’s t-test or one-way ANOVA followed by the Bonferroni post hoc test. Inhibition curves were fitted according to the logistic equation (sigmoid curve) (GraphPad Prism 6.07 for windows), to obtain IC50 with their 95% confidence intervals (CI). Extra sum-of-squares F-test included in the GraphPad Prism software was used to check the statistical significance of the differences between IC50. p < 0.05 was considered significant.
RESULTS
Circular dichroism studies were done first to investigate the secondary structure of HN and HNG. Peptides were dissolved in water at the final concentration of 40 μM, put into a 0.1 mm path-length quartz cuvette and the spectra were obtained at 25°C by scanning the far-UV region between 190–260 nm at 20 nm/min with a response time of 0.5 s. After averaging six accumulations, the spectrum of the buffer was subtracted, the mean residue molar ellipticity calculated and the spectrum smoothed using a regularization method [40]. The spectra of both peptides were superimposable with strong negative dichroism around 200 nm (Fig. 1), indicating that the structure is mainly random, which is in line with published data [41, 42] and suggests the absence of any β-sheet containing aggregates, which occur at higher peptide concentrations [43]. The absence of β-sheet was also confirmed by the lack of any ThT signal upon incubation of 2 μM HN or HNG (data notshown).

Circular dichroism (CD) spectra of HN and HNG. Polypeptides were dissolved at the final concentration of 40 μM in water. CD spectra are the average of six scans. Data are mean residue molar ellipticities (MRE). The two spectra gave a strong negative band around 200 nm, indicative of a low degree of ordering in the monomer.
This allowed us to investigate the effects of HN and HNG on the Aβ aggregation kinetics in cell-free system using a ThT assay. A 24-amino acid fragment of the rat albumin (rAlb25-48) has been employed as negative control. Equimolar concentrations of HN or HNG, added to 2 μM Aβ42 solution, similarly delayed fibril formation and halved the maximal ThT signal (Fig. 2A and Table 1) suggesting their ability to reduce the amount of Aβ fibrils and/or change their structure. This effect was specific for HN and HNG since 2 μM rAlb25-48 did not modify the aggregation kinetics of Aβ42 peptide (Fig. 2 and Table 1). To further quantify these effects, the ThT traces were normalized (Fig. 2B) and the half time of transition t1/2 (i.e., when half the maximum ThT signal is reached), and the growth rate of fibril formation were calculated. HN and its derivative similarly and significantly increased t1/2 and reduced the fibril growth rate (Table 1).

Effects of HN and its derivative on the kinetics of Aβ42 fibril formation. A) Two-μM Aβ42 were dissolved in 50 mM sodium phosphate, pH 7.4, with or without 2 μM HN, HNG or rAlb25-48 and 20 μM thioflavin T (ThT). The time course of changes in ThT fluorescence intensity are shown, as arbitrary units (A.U.). The results of three independent experiments are shown. B) Data in A) after normalization on the maximal ThT signal. Data are the mean±SD, n = 3. **p < 0.0001 versus Aβ42 alone according to one-way ANOVA and Bonferroni’s post hoc test.
Effect of HN and HNG on the Aβ42 aggregation
Two-μM Aβ42 were dissolved in 50 mM sodium phosphate, pH 7.4, with or without 2 μM HN, HNG or rAlb25-48 and 20 μM thioflavin T (ThT). Data are the mean±SD, n = 3. **p < 0.0001 versus Aβ42 alone according to one-way ANOVA and Bonferroni’s post hoc test.
We investigated the ability of HN and HNG to interfere with Aβ42 oligomerization with an SPR-based immunoassay, developed to specifically recognize transient and biologically relevant oligomers [30]. Oligomers bind to chip-immobilized 4G8 antibody in a pseudo-irreversible manner (very slow dissociation, probably due to multivalent binding), while monomers bind but rapidly dissociate, and higher-order aggregates show no SPR signal [30].
This assay is useful to test potential anti-oligomeric compounds [30]. In initial studies, we added the peptide to a freshly prepared Aβ42 solution and recorded the oligomer-dependent SPR signal after 5 h incubation (Fig. 3A) [30]. Both HN and HNG inhibited this signal (Fig. 3B) and the effect was dose-dependent (Fig. 3C). IC50 were 6.9 μM (5.5–8.5 CI) for HN and 10.2 μM (8.8–12.0 CI) for HNG. This inhibitory effect could be ascribed only to the interaction of HN and its derivative HNG with Aβ42, since the two peptides did not bind immobilized 4G8 (Supplementary Figure 3).

Effects of HN and HNG on Aβ42 oligomers detected by a surface plasmon resonance-based immunoassay. A) In the first experimental protocol synthetic monomeric Aβ42 (100 μM) was incubated at 25°C for 5 h with or without different concentrations of HN or HNG (0.3–100 μM), diluted 100-fold then flowed onto 4G8 immobilized on the SPR sensor chip. B) Representative sensorgrams obtained by flowing Aβ42 solutions with or without 100 μM HN or HNG, for 2 min (bars). The signal at the end of the dissociation phase (i.e. at 780 s)is indicative of the oligomer-dependent SPR signal. C) Dose-response effects of HN and HNG on the Aβ42 oligomer-dependent SPR signal. Data are percentages of the maximum SPR signal in the absence of inhibitors. D) In the second experimental protocol synthetic monomeric Aβ42 (100 μM) were pre-incubated at 25°C for 5 h to form oligomers, HN or HNG (0.3–100 μM) were then added, and after 30 min the solutions were diluted 100-fold and flowed onto 4G8 immobilized on the SPR sensor chip. E) Representative sensorgrams obtained by flowing Aβ42 solution with or without 100 μM HN or HNG for 2 min (bars). F) Dose-response effects of HN and HNG on the Aβ42 oligomer-dependent SPR signal. Data are percentages of the maximum SPR signal in the absence of inhibitors.
Similar SPR experiments were performed with the control peptide rAlb25-48. To this end, freshly prepared Aβ42 was incubated with the control peptide, and after 5 hours of incubation, the solution was injected over immobilized 4G8 (Supplementary Figure 4). Data showed that rAlb25-48 did not have any significant effect on Aβ42 oligomers formation, thus confirming the specificity of HN and HNG in their anti-oligomeric activities.
Since this inhibition could be due to an effect either on oligomer formation or on oligomer binding to immobilized 4G8, we ran additional studies in which HN and HNG were added to preformed oligomers (Fig. 3D). In this case too, HN and HNG reduced the oligomer-dependent SPR binding signal (Fig. 3E), in a dose-dependent manner, with IC50 14.5 μM (95% CI 11.1–18.9) for HN and 11.9 μM (9.3–15.2) for HNG (Fig. 3F). HN was twice as active when added at the beginning of the aggregation than when added to preformed oligomers (6.9 versus 14.5 μM, p < 0.01, extra sum-of-squares F-test) suggesting that it either i) interferes with oligomer formation and/or ii) both act on preformed oligomers (in this case preventing their binding to immobilized 4G8). In contrast HNG gave similar IC50s, indicating interactions with preformed oligomers only. Hypothetically this effect may be due to the ability of HN and HNG to shield hydrophobic residues responsible for the interaction of oligomers with immobilized 4G8. Otherwise, HN and HNG might favor the oligomer’s aggregation into larger assemblies, which are not sensed by the SPR assay.
To test this latter possibility, AFM, TEM and turbidity analysis were done. Incubation of 100 μM Aβ42 at 37°C for 2 h resulted in the appearance of short scattered protofibrils (95–155 nm long) (Fig. 4A, B). HN co-incubated with Aβ42 induced assembly of the short protofibrils into dense agglomerates which could be seen with TEM (Fig. 4D) but not AFM (Fig. 4C). This might be due to the different electrostatic properties of the agglomerates and oligomers, which did not allow their immobilization on the negatively charged mica surface [30] (Fig. 4C). Results were similar when Aβ42 was incubated with HNG (Fig. 4E, F).

Atomic force microscopy (AFM) and transmission electron microscopy (TEM) analysis. Synthetic Aβ42 (100 μM) was incubated at 37°C for 2 h with or without 100 μM HN or HNG. Samples were then diluted ten times with 10 mM phosphate buffer, pH 7.4. Representative (A, C, E) AFM and TEM (B, D, F) images of Aβ42 alone (A-B) and Aβ42 co-incubated with HN (C-D) or HNG (E-F). Scale bar: 2 μm×2 μm scan for AFM images and 500 nm for TEM images.
Aggregation of Aβ42 can also be assessed by turbidity at 400 nm. As shown in Fig. 5 the absorbance of 100 μM Aβ42 incubated for 2 h at 37°C increased in the presence of 100 μM HN and HNG. These findings confirmed the presence of larger aggregates as suggested by TEM analysis.
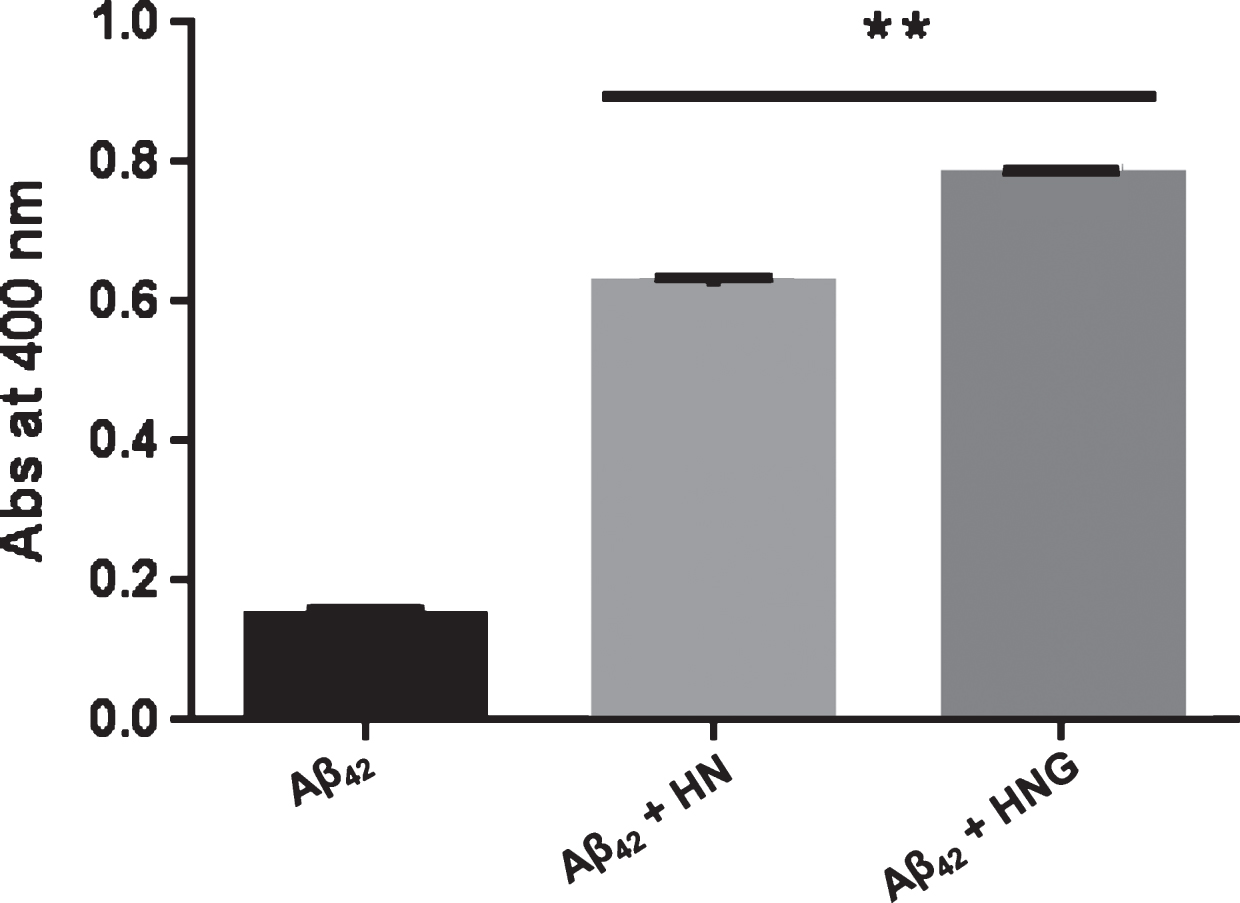
Turbidity analysis. Synthetic Aβ42 (100 μM) in 50 mM phosphate buffer (pH 7.4) was incubated at 37°C for 2 h with or without 100 μM HN or HNG. Turbidity was determined by measuring the absorbance of samples at 400 nm. Data are the mean±SD, n = 3. **p < 0.0001 versus Aβ42 alone according to one-way ANOVA and Bonferroni’s post hoc test.
We further investigated the ability of HN and its derivative to interfere with Aβ42 oligomers in vivo using C. elegans as the animal model of Aβ-induced toxicity. We employed the transgenic CL4176 strain, in which the paralysis phenotype is specifically caused by the deposition of oligomeric Aβ42 in the body muscle cells and no fibrillar amyloid aggregates are formed [20, 37]. After synchronization of the eggs, worms were placed at 16°C on NGM plates seeded with OP50 E. coli for 60 h and Aβ42 expression was induced by raising the temperature to 24°C. The worms were given increasing concentrations of HN and HNG (5–250 μM) 12 h after the temperature rise and their paralysis was scored 24 h later. Both peptides protected CL4176 worms from paralysis in a dose-dependent manner, with similar IC50 (31.9±1.5 μM for HN and 32.6±1.9 μM for HNG, mean±SE) (Fig. 6).
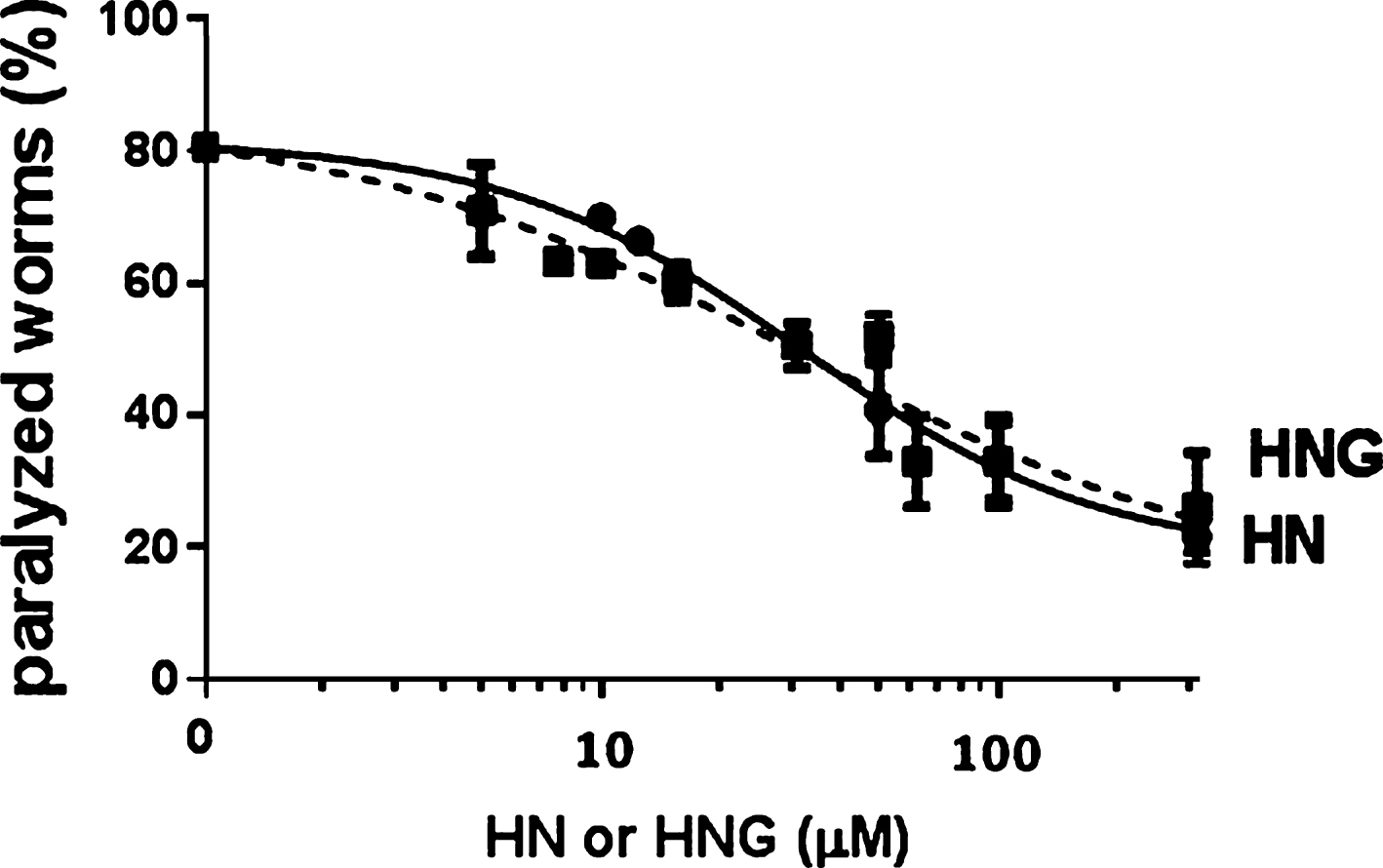
Dose-response effects of HN and HNG on Aβ42-induced paralysis in transgenic C. elegans. Egg-synchronized CL4176 worms were placed at 16°C on fresh NGM plates seeded with OP50 E. coli and the temperature was raised 60 h after plating. Twelve h later, worms were fed HN or HNG (5–250 μM) and the number of paralyzed worms was scored after 24 h. Data are percentages±SE of paralyzed worms compared to vehicle-treated ones (100 worms/group, three independent assays).
To clarify the mechanisms of the in vivo protective activity of HN and HNG we investigated their effects on Aβ expression/degradation and oligomerization. Dot blotting and western blotting experiments were carried out with lysates from transgenic CL4176 worms fed 50 μM HN or HNG for 24 h. Lysates from CL802 nematodes, which did not express Aβ, were used as negative controls [23]. At this dose HN reduced the worms’ paralysis by 27% (80.8±2.1 of paralyzed worms for control and 53.9±6.3% for HN-fed worms, p < 0.01, one-way ANOVA) and HNG by 30% (50.8±1.4, p < 0.01 versus control, one-way ANOVA). The same concentration of polypeptides had no effects in transgenic CL802 control worms (data not shown).
To investigate the effect of HN and HNG on Aβ oligomers, A11 and OC conformation-selective antibodies recognizing different classes of quaternary structure were used [39]. Results from dot-blot analysis indicated that HN and HNG similarly reduced the A11- immunoreactive signal by about 80% and OC-reactive oligomers by 20% (Fig. 7C-F). Worms treated with HNG, but not HN, also showed a 58% reduction of the total Aβ levels (Fig. 7A, B). In line with the dot blotting analysis, western blot analysis indicated an overall reduction of Aβ levels in the lysates of worms fed HNG accompanied by a significant reduction of the 20 kDa low-molecular weight and >48 kDa high-molecular weight oligomers bands, and 4 kDa band (Figs. 7 and 8).
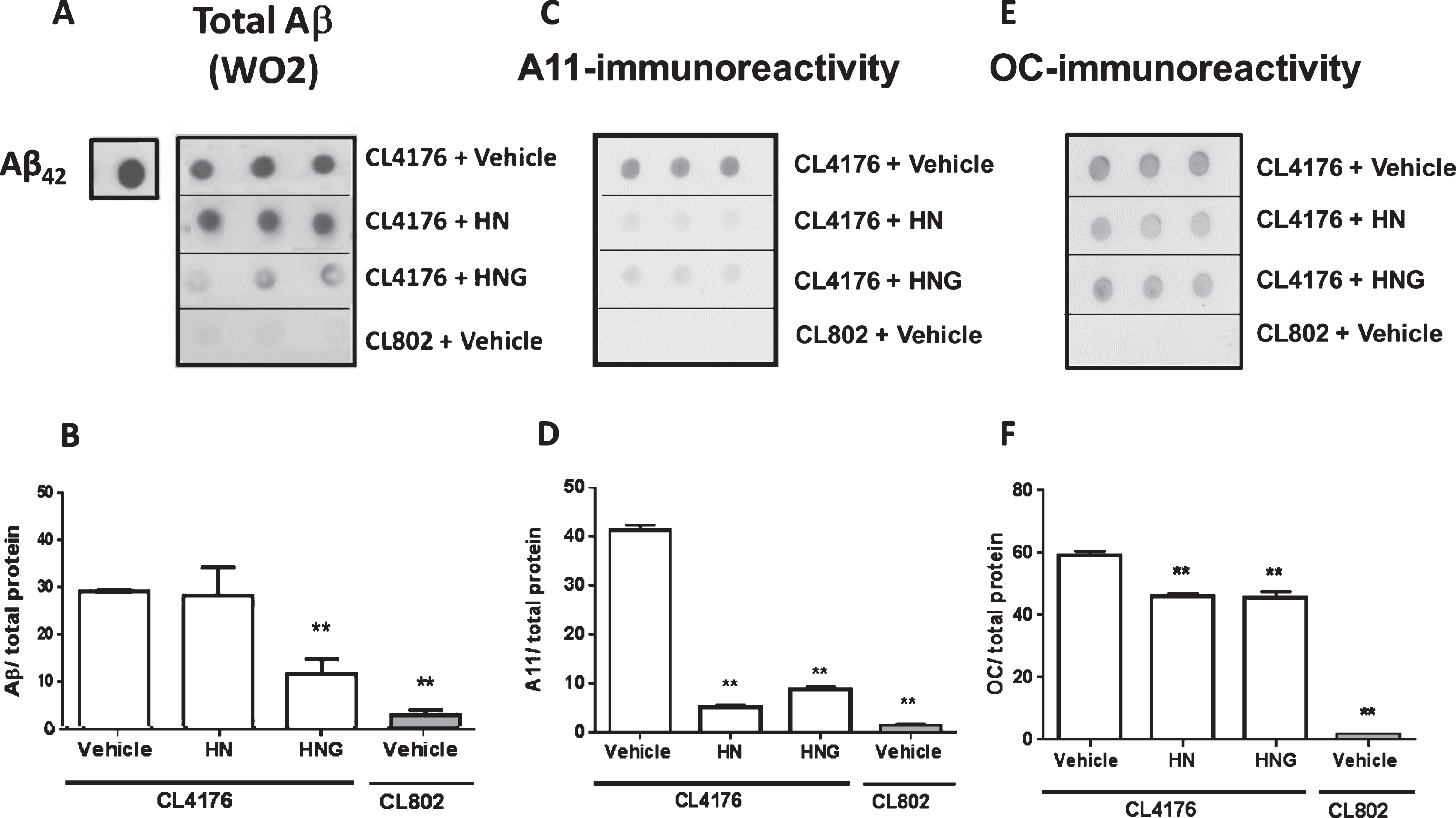
Effects of HN and HNG on total and oligomeric Aβ: dot-blot analysis. Representative dot blot of A) total Aβ (WO2), C) A-11 immunoreactive oligomers and E) OC immunoreactive assemblies in CL4176 transgenic worms fed vehicle or 50 μM HN or HNG for 24 h. CL802 transgenic worms were used as negative control. Equal amounts of protein from worm lysates (20 μg) were spotted in triplicate. Synthetic Aβ42 (0.1 μg) was spotted as positive control. Total proteins on the blotted membranes were stained using 0.1% Ponceau Red solution and were used to normalize the immuno-specific signal for protein loading. The mean volume of the B) WO2 Aβ-immunoreactive, D) A11 oligomer-immunoreactive and F) OC immunoreactive spots were determined using Quantity One 1-D software. Immunoreactivity from three different independent samples, analyzed in triplicate, was expressed as the mean volume of the immunoreactive band/ total proteins±SE. **p < 0.001 versus CL4176 worms fed vehicle (one-way ANOVA).
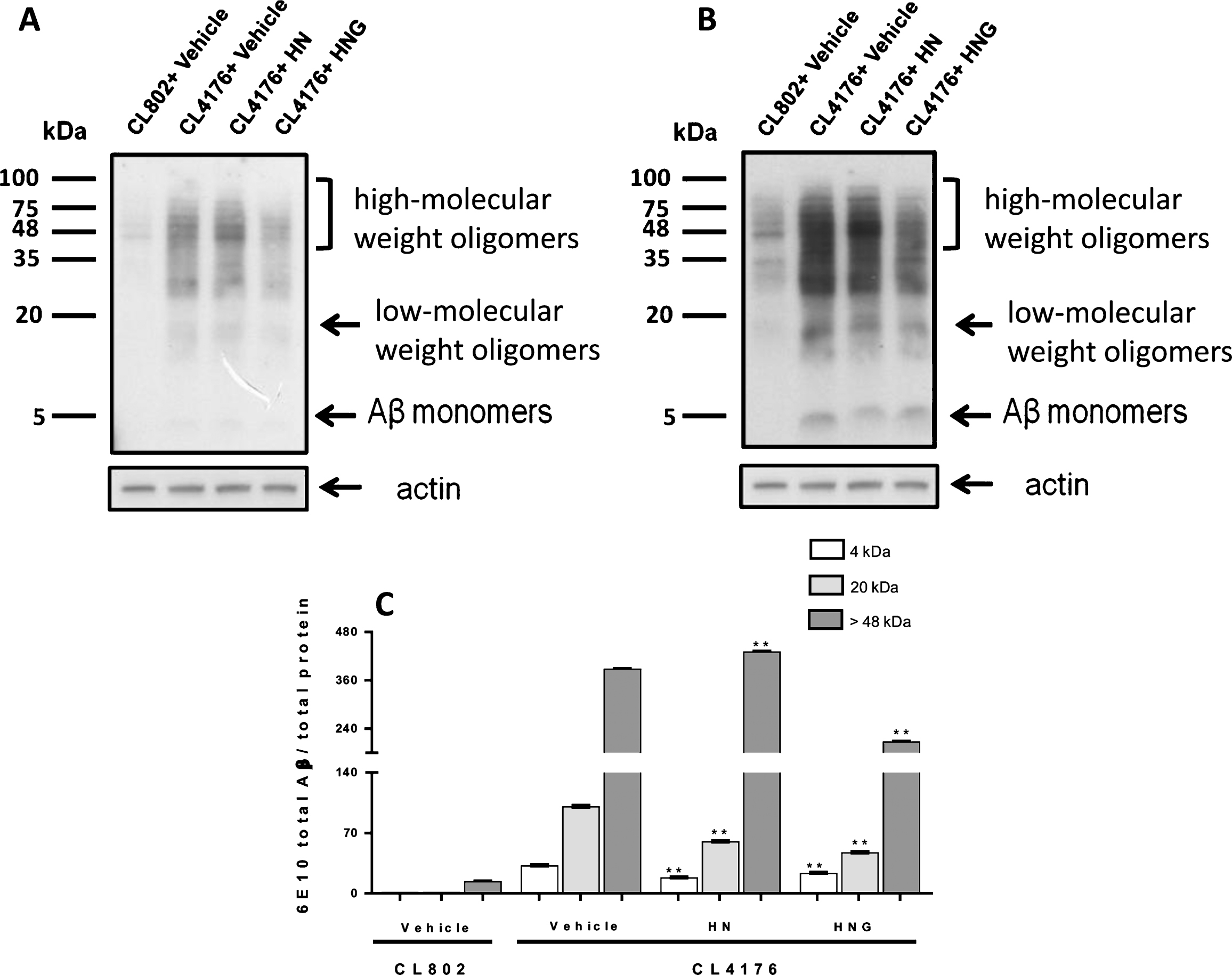
Effects of HN and HNG on total and oligomeric Aβ: western blot analysis. Representative western blot of Aβ species. CL4176 worms fed vehicle or 50 μM HN or HNG for 24 h. CL802 transgenic worms were used as negative control. Equal amounts of protein (50 μg) were loaded on each gel lane and immunoblotted with anti-Aβ antibody (6E10) or actin. Arrows indicate the Aβ monomers (4kDa), low-molecular weight oligomers (∼20 kDa) and high-molecular weight oligomers (from 48 up to 100 kDa). A) Image obtained 3 minutes after membrane exposure, which was the optimal time for visualization of the 20 kDa and >48 kDa bands, corresponding to Aβ low-molecular weight oligomers and high-molecular weight oligomers, respectively. B) Image obtained 8 minutes after membrane exposure which represented the optimal time for visualization of the Aβ monomers at 4 kDa. C) Immunoreactivity of 6E10 for the quantification of the monomeric 4 kDa band was done on the image obtained 8 minutes after membrane exposure. The low-molecular weight oligomers (∼20 kDa) and high-molecular weight oligomers (>48 kDa) bands were quantified on the membrane exposed for 3 min. Data are the mean volume of the immunoreactive band/ total proteins±SD obtained from three different independent samples, analyzed in triplicate. **p < 0.01 versus CL4176 worms fed vehicle (one-way ANOVA).
Western blot analysis indicated that HN significantly reduced the signal of the band at ∼20 kDa, corresponding to Aβ low-molecular weight oligomeric species, and concomitantly boosted the signal of the bands equal or higher of 48 kDa (Fig. 8). These effects were accompanied by a significant reduction of the level of 4 kDa monomeric Aβ (Fig. 8). Moreover, due to the specific ability of CL4176 C. elegans strain to produce only oligomeric Aβ42 [20], the effect of HN was not accompanied by the formation of fibrillar amyloid aggregates. These findings suggest that HN counteracted Aβ-induced toxicity by thus promoting their assembly into larger, not fibrillar, assemblies.
The ability of HNG to lower Aβ levels in vivo has already been reported [18], and was ascribed to the ability of this compound to activate Aβ-degrading enzymes, particularly the neutral neuropeptidease neprilysin [18]. The structure and function of these enzymes are well conserved from mammals to C. elegans [44]. To investigate whether the reduction of total Aβ in CL4176 worms fed HNG was associated to neuropeptidase activation, the level of expression of neprilysin was evaluated. Results from dot-blot analysis indicated that HNG significantly increased the neprilysin immunoreactive signal from 6.12±0.3 neprylisin/μg protein to 24.02±2.9 neprylisin/μg protein whereas HN did not modify it (9.27±2.0 neprylisin/μg protein) (Fig. 9). These data suggest that the HNG-induced reduction of the total Aβ level in worms is likely the consequence of the HNG-induced overexpression of the Aβ-degrading enzyme.
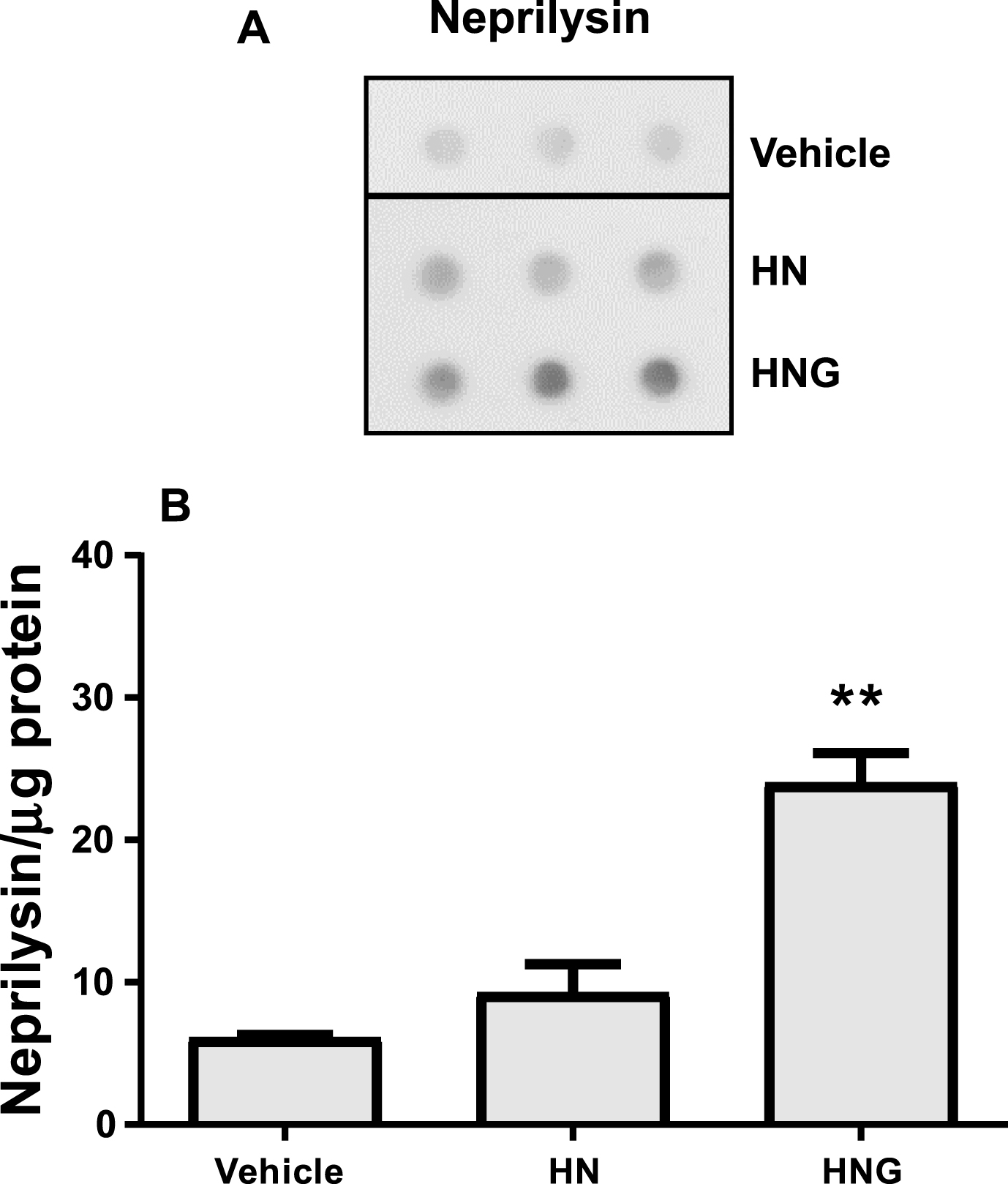
Effects of HN and HNG on neprilysin level. A) Representative dot-blot of neprilysin in CL4176 transgenic worms fed vehicle, 50 μM HN or HNG for 20 h. Equal amounts of protein from worm lysates (20 μg) were spotted in triplicate. Total proteins on the blotted membranes were stained using 0.1% Ponceau Red solution and were used to normalize the immuno-specific signal for protein loading. B) The mean volume of the neprilysin-immunoreactive spots were determined using Quantity One 1-D software. Immunoreactivity obtained from three different independent samples, analyzed in triplicate, was expressed as the mean volume of the immunoreactive band/μg proteins±SD. **p < 0.001 versus CL4176 worms fed vehicle (one-way ANOVA and Bonferroni’s post hoc analysis).
DISCUSSION
The protective effects of HN and HNG against Aβ-induced toxicity have been widely investigated in cells and animal models [13, 42]. Studies performed in cell free conditions showed that HN and HNG similarly interact with Aβ1-40, affecting its folding, [12, 13]. Our present in vitro studies confirm and extend the above observations, demonstrating that HN and HNG directly interact with Aβ42 oligomers, interfering with their toxic activities.
There is increasing evidence of the key role of Aβ soluble oligomers in the pathogenic process of AD, suggesting that small molecules, inhibiting Aβ self-oligomerization or interacting with it might be useful as future pharmacological treatments. We here examined for the first time the potential anti-oligomeric activity of HN and HNG. Quantitative results from SPR experiments indicated that HN and HNG bind to formed soluble Aβ oligomeric intermediates. Qualitative analysis using AFM and TEM, andquantitative data from turbidity assay showed that both polypeptides, similarly to tetracyclines, antibiotics with known anti-aggregating and anti-oligomeric activities [45], promoted the further aggregation of oligomers into larger assemblies with amorphous structures [45]. Similar amorphous structures have been already observed by Zou P et al. after incubation of Aβ1-40 with HNG [13, 46]. This mechanism is presumably responsible for the effects of HN and HNG in hindering the formation of Aβ42 fibrils and the generation of amyloid structuresin vitro. HN and HNG showed similar potencies, indicating that the substitution of Gly for the Ser14 in the HN sequence was probably not important for the interaction with Aβ oligomers.
These observations were confirmed in vivo in experiments in which HN and HNG counteracted the toxicity caused by Aβ42 oligomers in CL4176 transgenic C. elegans. Transgenic worms expressing human proteins have been widely used to elucidate the molecular pathways underlying proteinmisfolding diseases [47, 48]. Moreover, they offer the unique opportunity to test the effect of potential therapeutic compounds on whole animals avoiding ethical problems and at the speed and cost of the in vitro cell-based assays. However, it is important to underlie that C. elegans is a simple multicellular organism avoid of a circulatory and central nervous system and that the findings obtained applying this model, although scientifically relevant, need to be validated in vertebrate animals and translation to human pathology requires caution.
HN and HNG showed similar protective effects against Aβ toxicity, with comparable IC50, and we suggest that this is due to their ability to reduce the amount of toxic oligomers in vivo. Notably, whereas HN specifically reduced the formation of low-molecular weight oligomers favoring their assembly into larger assemblies, the overall decrease of low- and high-molecular weight oligomers caused by HNG is mainly due to the decrease of total Aβ levels. The ability of HNG to lower Aβ levels in vivo, without affecting APP production and secretase activities, has already been reported [18], and was ascribed to the HNG-induced activation of Aβ-degrading enzymes, particularly the neutral neuropeptidease neprilysin [18]. The structure and function of these enzymes are well conserved from mammals to C. elegans [44] allowing us the evaluation of neprilysin involvement in our model. We observed that, similarly to rodents, the reduction of total Aβ in CL4176 worms fed HNG was associated to the increase of neprilysin level.
The two polypeptides showed similar protective effects, with IC50 comparable to those of other compounds which also had anti-oligomeric activity inC. elegans, such as tetracyclines. The pre-clinical and clinical anti-amyloidogenic activity of tetracyclines was studied and confirmed by independent research groups [20, 49–52].
In conclusion, both HN and HNG interact directly with Aβ42 in vitro and interfere with the formation and toxicity of its oligomers. Moreover, they similarly protect transgenic C. elegans against the toxicity specifically induced by Aβ oligomers deposition. Here we show for the first time that HN counteracts the formation of oligomers in vivo by promoting their assembly into larger aggregates, whereas the reduction of oligomers caused by HNG can be ascribed to its ability to reduce the overall Aβ levels.
enlargethispage *2ptIn addition to AD, Aβ amyloid misfolding and deposition is a key feature for the development of inclusion body myositis (sIBM). In this progressive skeletal musculoskeletal pathology, frequently found in older subjects, Aβ deposits accumulated in skeletal muscle cells causing functional neuromuscular dysfunctions [53, 54]. Although sIBM involves an inflammatory process, it does not respond to anti-inflammatory drugs thus remaining an untreatable disease. An effective therapeutic strategy may be represented by the targeting of Aβ. Transgenic C. elegans strains overexpressing human Aβ in muscle cells, like CL4176 strain, have been widely used as good experimental models also for studies aimed at developing new therapeutic approaches for sIBM [53, 54]. In fact, although different tissues are affected in AD and IBM, the functional neuromuscular synaptic dysfunction caused by Aβ amyloidogenic assemblies is a common feature of the two diseases [21, 54]. The findings obtained indicate that HN and HNG may be relevant for the design of future beneficial pharmacological strategies for sIBM, too.
Footnotes
ACKNOWLEDGMENTS
This study was partially supported by Banca Intesa Sanpaolo (2015-2016) and Fondazione Sacchetti (2016). C. elegans and OP50 E. coli were provided by the Caenorhabditis Genetic Center, which is funded by the NIH Office Research Infrastructure Programs (P40 OD010440). We thank Flamma Spa, Bergamo, Italy who kindly provided FMOC amino acids for peptide synthesis.