
Abstract
There is a long history linking traumatic brain injury (TBI) with the development of dementia. Despite significant reservations, such as recall bias or concluding causality for TBI, a summary of recent research points to several conclusions on the TBI-dementia relationship. 1) Increasing severity of a single moderate-to-severe TBI increases the risk of subsequent Alzheimer’s disease (AD), the most common type of dementia. 2) Repetitive, often subconcussive, mild TBIs increases the risk for chronic traumatic encephalopathy (CTE), a degenerative neuropathology. 3) TBI may be a risk factor for other neurodegenerative disorders that can be associated with dementia. 4) TBI appears to lower the age of onset of TBI-related neurocognitive syndromes, potentially adding “TBI cognitive-behavioral features”. The literature further indicates several specific risk factors for TBI-associated dementia: 5) any blast or blunt physical force to the head as long as there is violent head displacement; 6) decreased cognitive and/or neuronal reserve and the related variable of older age at TBI; and 7) the presence of apolipoprotein E ɛ4 alleles, a genetic risk factor for AD. Finally, there are neuropathological features relating TBI with neurocognitive syndromes: 8) acute TBI results in amyloid pathology and other neurodegenerative proteinopathies; 9) CTE shares features with neurodegenerative dementias; and 10) TBI results in white matter tract and neural network disruptions. Although further research is needed, these ten findings suggest that dose-dependent effects of violent head displacement in vulnerable brains predispose to dementia; among several potential mechanisms is the propagation of abnormal proteins along damaged white matter networks.
INTRODUCTION
Dementia and traumatic brain injury (TBI) are among the most prevalent neurological disorders [1]. Alzheimer’s disease (AD), the most common form of dementia, is a virtual epidemic, increasing in prevalence as populations achieve greater longevity. Likewise, TBI is a major public health problem, occurring in as many as four million people every year in the U.S. [2]. There is a long literature associating TBI and dementia [3, 4], from early descriptions of the “punch-drunk” syndrome and “dementia pugilistica” among boxers to the current burgeoning information on chronic traumatic encephalopathy (CTE) among athletes and others exposed to multiple, repetitive mild TBIs [5]. These developments, along with a series of epidemiological studies, have solidified TBI as a risk factor for neurocognitive syndromes; however, in the process they have led to many questions about how TBI and dementia interrelate [6].
Although this is a complex relationship, the current literature has begun to answer these questions and unravel the relationship between TBI and dementia. A review of the literature suggests a number of tentative conclusions, subject to many reservations such as the potential for recall bias in reporting head injuries and referral bias in research participation [7]. The nature of TBI itself is broad and can range from single severe injuries to multiple small subconcussive TBIs and to concussion, a term often used synonymously with mild TBI. Likewise, the term dementia refers to a decline in two or more areas of cognition that can be associated with a range of pathologies beyond that of AD. Both TBI and dementia each vary in a number of other ways, including symptoms, course, other associated factors, biomarkers, animal modeling, and others. While unable to determine all variables that affect the TBI-dementia relationship, a current review of this evolving literature does point to ten potential emerging trends and preliminary conclusions regarding the complex relationship.
GENERAL TBI-DEMENTIA CLINICAL RELATIONSHIPS
Increasing severity of a single moderate-to-severe TBI increases the risk of subsequent AD, the most common type of dementia
Studies report a relationship between a prior moderate-to-severe TBI and the eventual development of AD [8–13]. Although a number of studies have failed to document the association [13–16], most reports, and three meta-analyses of 11–19 well-characterized studies [3, 17], indicate that, in the aggregate, there is evidence to conclude that a single moderate-to-severe TBI is a risk factor for AD (See Fig. 1) [3, 17–19]. The largest and most recent meta-analysis of the TBI risk for AD versus controls found an overall odds ratio (OR) of 1.4 (95% Confidence Interval [CI]: 1.02–1.90), and an increased risk of TBI for mild cognitive impairment, which is often a precursor to dementia [17]. In the two earlier meta-analysis, males, but not females, were at increased risk of AD from TBI [3, 8], but this gender difference does not persist in other studies [17, 20]. In addition, the limited number of prospective studies show a relationship between moderate-to-severe TBI and AD or other dementia [9–11]. In one of the most important ones, investigators evaluated the risk for dementia among 548 U.S. World War II veterans 50 years after confirmed TBIs, compared to 1,228 with other types of injuries [10]. They found that a record of moderate or worse TBI was significantly associated with AD or other dementia [10].
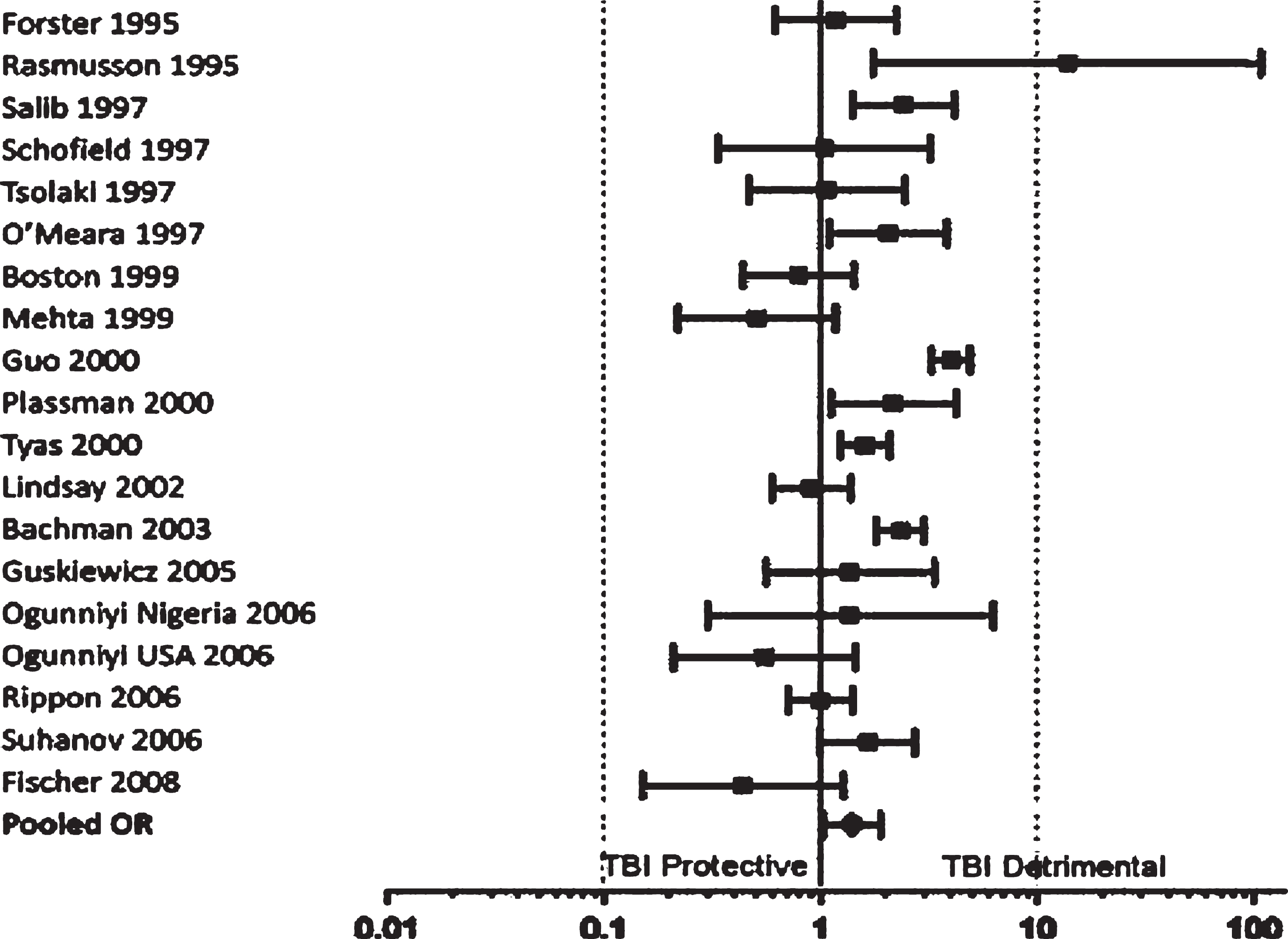
Odds ratios and confidence intervals for Alzheimer’s disease. Reprinted from the Journal of Neurosurgery, Perry et al., 2016 [17], with permission.
In sum, a single moderate or severe TBI, especially if recent rather than remote, is essentially the major known environmental risk factor for AD [3, 21], and this risk increases with increasing severity of the head trauma. These studies indicate that a when TBI is of moderate severity, there may be a two-fold increased risk of AD or other dementia, and when TBI is severe, there may be a four-fold increasedrisk [10].
Repetitive, often subconcussive, mild TBIs increases the risk for chronic traumatic encephalopathy (CTE), a type of dementia
The vast majority of TBIs are mild (loss of consciousness <30 minutes; post-traumatic amnesia < 24 hours) [2]. Recent studies now show that mild TBI, particularly if repetitive, can also increase the risk of dementia [22–28]. The long-term risk of dementia from repetitive mild TBIs is a crucial question for athletes and among veterans [29, 30], as mild TBIs are common from participation in contact sports and from military deployment [31–33]. A Taiwanese study of one million people with three years follow-up found a three-fold increased risk for dementia in those with a history of mild TBIs [23], and the California statewide health database found an increased risk of dementia from mild TBIs in elderlyindividuals [34].
The repetitive occurrence of mild TBIs appears to be much more of a risk for later dementia from CTE, a neuropathological disorder associated with TBI, than just one prior episode. We have long known of the association of TBI with the “punch drunk syndrome” or dementia pugilistica on neuropathology in boxers, now known at CTE [35]. Clinically, CTE often resembles AD in the delayed deterioration of cognition [36, 37], although one form of CTE manifests primarily with behavioral changes such as depression and suicidality, apathy and aloofness, poor impulse control, and behavioral disinhibition [5, 38]. Recently, there is widespread attention of the risk of CTE to professional football players and other athletes with increasing years of exposure to possible mild and repetitive TBIs [39, 40]. The clinical and neuropathological aspects of CTE have occurred not just in athletes but, despite a scarcity of neuropathological cases, among veterans, who are often prone to repetitive mild TBIs [5, 39–41]. This is further implied from epidemiological data from the VA National Database [24]. Of 188,764 veterans older than 54 years of age and followed for about 9 years, the presence of any TBI, including blast-force TBI, increased their risk of developing dementia by 60% (OR: 1.57; 95% CI: 1.35–1.83) [24].
TBI may even increase the risk for other neurodegenerative disorders that can be associated with dementia
Epidemiological studies report that TBI may increase the risk of developing non-AD neurodegenerative disorders potentially associated with cognitive impairment or dementia. Examining a California statewide administrative health database, investigators assessed the risk of dementia among 51,799 patients 55 years of age or older who had had a TBI during the preceding 5–7 years [34]. They found that moderate-to-severe TBI can increase the risk of different types of dementia with a minimum hazard ratio adjusted for age of 1.26 (95% CI: 1.21–1.32) [34]. In the prospective study of U.S. World War II veterans, there was an increased risk of both AD and non-AD dementias after a single moderate or severe TBI [10]. In addition to AD, specific neurodegenerative conditions linked to TBI include frontotemporal dementia (FTD) and to Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS), two conditions with the potential for associated cognitive decline.
TBI appears to be a risk factor for FTD. A study from the National Alzheimer’s Coordinating Center (NACC) database of autopsy-confirmed cases of FTD found that a history of a remote TBI with five or more minutes of loss of consciousness was more common among 1,016 cases with FTD pathology compared to 2,015 normal controls (OR: 1.67; 95% CI: 1.00–2.78) [42]. In a prior retrospective case-control study of 80 patients with FTD compared to controls, investigators found 19 (23.8%) who had had a history of TBI [43], and in another retrospective case–control study involving 845 veterans, a history of prior TBI was association with a 4.4 times greater risk of developing FTD compared to controls [44]. Furthermore, in a large retrospective cohort study involving 147,510 patients in Taiwan, those with evidence of a prior TBI had a 4.13 times greater risk of developing FTD compared to non-TBI controls [45].
Studies have also reported TBI as a risk factor for PD. In a case-control study of 196 persons with PD in the Rochester, Olmsted County Epidemiology Project [46], there was a greater history of prior TBI among the PD cases than among matched controls (OR: 4.3; 95% CI: 1.2–15.2), and this was most significant among those who had more severe TBI or were previously hospitalized with a TBI. In a large retrospective cohort study of inpatient or emergency room data of patients presenting with TBI, there was an increased risk of PD in the subsequent 5–7 years of 44% (adjusted hazard’s ratio of 1.44; 95% CI: 1.31–1.58), and the risk was greater with increased severity and frequency of the TBI [47]. An interesting case-control study of 93 twins reported that prior TBI increased the risk for PD (OR: 3.8; 95% CI: 1.3–11) [48]. The risk of PD after a history of TBI was also increased in the prospective study of U.S. veterans from World War II [10]. Additionally, one meta-analysis of 22 case studies found an increased risk of PD from TBI of 57% [49], and a second meta-analysis of 15 studies found an increased association of TBI with PD (OR: 1.45; 95% CI: 1.18–1.78) [17].
After reports of an unusually increased risk of ALS after TBI among football (soccer) players in Italy [50, 51], investigators examined this association in other populations. In one case-control study of 109 patients with ALS compared to matched controls, investigators found an increased risk for ALS among those with a history of two or more TBIs (OR: 3.1, 95% CI: 1.2–8.1) or if a single TBI had occurred in the prior 10 years (OR: 3.2; 95% CI: 1.0–10.2) [52]. In another case-control study of 77 patients with ALS compared to 185 controls, those with a TBI between age 30–40 had an OR for ALS of 14.2 (CI 1.04–194.42) [53]. Among veterans, including 241 with ALS and 597 controls, those with TBIs in the prior 15 years had an adjusted OR of 2.33 (95% CI: 1.18–4.61) [54], and this association was strongest among those with had an APOE ɛ4 allele. One meta-analysis reported a moderately increased risk of ALS with prior TBIs (OR: 1.7; 95% CI: 1.3, 2.2) [52], but a subsequent meta-analysis of four studies did not quite find an elevated risk (OR = 2.07; 95% CI = 0.94–4.56) [17]. In contrast, some case-control studies did not find an association between TBI resulting in hospitalization and subsequent ALS, including in the Oxford Linkage study and among 4,004 ALS patients from the Swedish Patient Register [55, 56]. Furthermore, one meta-analysis of 12 studies did not find that a single prior TBI was a risk factor for ALS [57]. More epidemiological studies with neuropathological correlation would help resolve some of these discrepancies.
TBI appears to lower the age of onset of dementia, potentially adding “TBI cognitive-behavioral features”
There is evidence that TBI may lower the age of onset of any dementia or permit crossing of the threshold into dementia at an earlier age [20, 58–60]. There is a particularly high prevalence of early-onset dementias among populations that have a high incidence of TBIs, such as U.S. and other veterans [58, 61]. Among Swedish young men who had median follow-up of 33 years, those with a history of prior TBI had an increased risk for early-onset dementia of 50% (OR: 1.5; 95% CI: 1.1–2.0), which increased with the severity and frequency of TBIs [22]. Moreover, in a meta-analysis, the relative risk for AD was 1.63 with a TBI 10 or more years prior to onset, but increased to 5.33 with a TBI within 10 years, suggesting that more recent TBIs accelerate the emergence of AD [3].
In a large study, investigators used the NACC Uniform Dataset (UDS) to compare the prevalence of a history of TBI between AD patients with an early age of onset (<65 years; n = 1,429) versus those with a later age of onset (>65 years; n = 4,292) [20]. This study also examined the prevalence of TBI in a cohort of 115 early-onset AD patients attending a university subspecialty clinic. Despite potential recall bias, the prevalence of a history of TBI among the early-onset AD patients from both cohorts, was 13.5–13.9%, compared to 7.8% for the NACC late-onset cohort (p < 0.001). The NACC early-onset AD patients with a history of TBI, compared to those without, were more likely to have disinhibition and irritability, clinical signs of frontal lobe dysfunction often associated with “TBI cognitive-behavioral features” (see Fig. 2) [20].
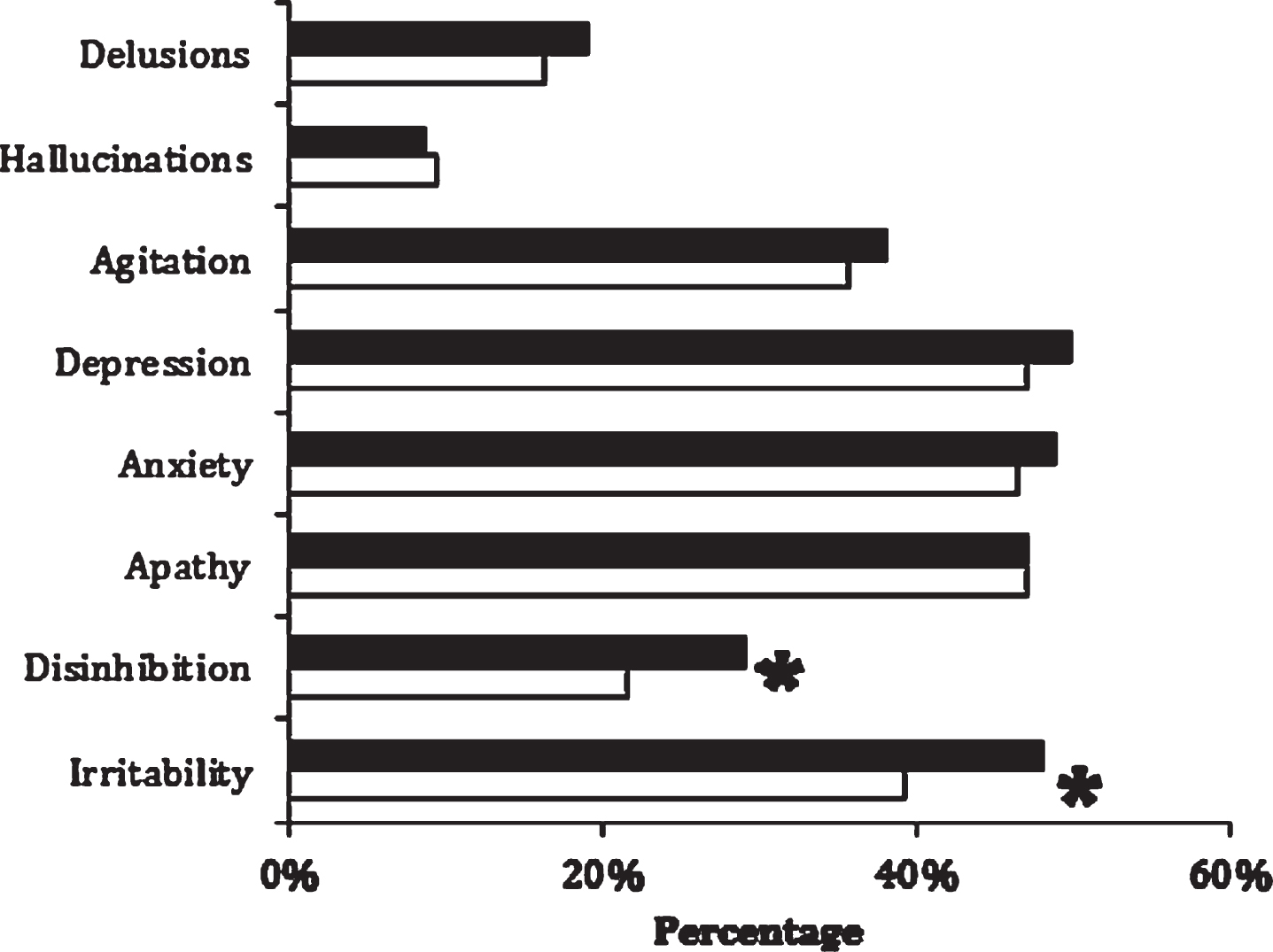
Prevalence of neuropsychiatric inventory symptoms among early-onset Alzheimer’s disease patients with (n = 178) and without (n = 1,234) a history of traumatic brain injury. *Significance to p = 0.01 level for disinhibition (29.0% versus 21.6%; χ2 = 6.19) and for irritability (48.1% versus 39.3%; χ2 = 6.40). Reprinted from the Journal of Alzheimer’s Disease, Mendez et al., 2015 [20], with permission from IOS Press.
TBI not only lowers that age of onset of dementia, but also alters the clinical manifestations. Frontal injury from TBI may cause patients to have executive deficits, and orbitofrontal and anterior temporal injury may manifest as disturbances in social awareness and impulse/emotional control. In addition, dementia patients with a history of TBI have had better memory and verbal fluency but moredepression and neurological symptoms in gait and motor speed [62]. This “TBI cognitive-behavioral profile” includes disproportionately impaired cognitive tests of attention, processing speed, and executive functions, compared to memory and language functions, as well as a greater psychiatric history (depression, apathy, irritability, poor impulse control and social-emotional disturbances, post-traumatic stress disorder, and substance abuse) [63–65].
SPECIFIC RISK FACTORS FOR TBI-ASSOCIATED NEUROCOGNITIVE DECLINE AND DEMENTIA
Any blast or blunt physical force to the head as long as there is violent head displacement
Most data are on blunt-force or impact TBI, but recent studies on blast-force TBI have implications for the relationship of TBI to neurocognitive decline and dementia. The majority of mild TBIs from recent combat deployments have been due to exposure to explosive devices such as mortar blasts or improvised explosive devices [66], and this blast-force mild TBI has become the “signature” injury of recent U.S. military engagements [67]. In addition to persistent post-concussive symptoms [32, 68–70], blast-force mild TBI may be associated with post-traumatic cognitive impairment [71, 72], and there may be late cognitive, behavioral, and neuroimaging changes [63, 65]. As indicated in the VA National Database study, blast-force TBI may increase the risk for the eventual development of dementia [24]. This cognitive sequalae from blast-force TBI may resemble that from blunt-force TBI [73].
The clinical, as well as neuropathological, aspects of CTE have occurred in those exposed to blast-forces [39, 74]. A case series found CTE in postmortem brains from U.S. veterans exposed to blast-force as well as blunt-force TBI [72, 74]. Investigators have replicated this evidence of blast-force CTE among mice exposed to blast winds of 330 miles/hour [72], and studies on blast impact on mice show that, although it is a different brain force than from brain impact, the brain pathology resembles that of CTE and of blunt-force trauma [72]. Furthermore, L.E. Goldstein and colleagues indicate that there may be an absence of the neuropathology of CTE when the animal’s head is kept stationary during experimental head trauma, hence equating the risks from both blast and blunt TBI with violent head displacement [72, 75]. Other studies indicate that most TBI has in common a sliding of the brain relative to the skull with rotation and deformation stress at anchoringpoints [76].
Decreased cognitive and/or neuronal reserve and the related variable of older age at TBI.
Prior mild TBIs may permit the development of AD not by the direct promotion of AD neuropathology but by lowering neuronal or cognitive reserve. The concept of “reserve” refers to resiliency in the face of brain damage, with “brain reserve” suggesting neuronal number and vulnerability to pathology and its clinical expression and “cognitive reserve” suggesting neuroplasticity, connectivity, and the capability for alternate, cognitive strategies. Common indicators of cognitive reserve include level of education, occupational attainment, and baseline intelligence. Epidemiological studies show that the lower the education and occupational ranking, the greater the risk for AD and other dementias [77, 78]. The attainment of high occupational achievement appears to delay the onset of cognitive and behavioral decline and the clinical aspects of CTE [79]. Conversely, a lower intelligence quotient before penetrating TBI among Vietnam veterans may be associated with greater cognitive decline on 30-year follow-up [80].
Neuronal and cognitive reserve decline with age, suggesting that age at the time of the TBI is a risk factor. The older the age at the time of TBI, the more likely the person is to develop dementia, as well as to develop amyloid-β plaques, a hallmark pathology of AD [81]. In another study, mild TBIs increased the risk of developing dementia by 20% if they occurred at age 65 or older, but not at a youngerage [34].
The presence of apolipoprotein E ɛ4 alleles, a genetic risk factor for AD
Investigators have proposed that APOE ɛ4 alleles may be synergistic or additive with TBI in increasing the risk for developing AD [82, 83]. The presence of one or two APOE ɛ4 alleles is the major known susceptibility gene for the development of AD, increasing the risk by 3 or 12 fold, respectively [84], and shifting the age of onset of dementia to a younger age [85]. APOE ɛ4 impairs the efficiency of proteolytic breakdown and clearance of Aβ [84, 87], and this may link TBI to the development of AD [85, 89]. Several studies indicate that TBI particularly increases risk for AD if the patient has an APOE ɛ4 allele [82, 83]. After an acute TBI, those with an APOE ɛ4 allele have a greater frequency of deposition of Aβ at autopsy than those without an APOE ɛ4 allele, especially if they are homozygous for the gene [87]. In addition to AD, among boxers and football players, the presence of an APOE ɛ4 allele may also increase the likelihood of dementia pugilistica or CTE on neuropathology [90, 91]. But not all studies show a TBI-APOE association. In one case-control study of 230 AD cases and 390 controls with at least one APOE ɛ4 allele, there was no significant variation in the TBI-AD risk by APOE ɛ4 genotype [92].
After a TBI, the major effect of APOE ɛ4 has been poor post-traumatic outcomes [93]. In a series of studies of brain injury, including a meta-analysis of 14 prospective TBI studies, there were poorer neurological outcomes if the patient had an APOE ɛ4 allele than if they did not have an APOE ɛ4 allele [93–97]. Among boxers with 12 or more professional bouts, those with an APOE ɛ4 allele had worse cognitive-behavioral outcomes than those without an APOE ɛ4 allele [90], and among 53 professional football players, those with an APOE ɛ4 allele had worse cognitive test scores than those without an APOE ɛ4 allele [91]. Moreover, in one study, the presence of an APOE ɛ4 allele increased the risk of unfavorable outcomes at six months or more after a TBI [93, 98], and other studies showed that this poorer outcome was particularly evident among younger patients or children [99, 100]. The reason of an association of APOE ɛ4 with poor post-traumatic outcomes is unknown but may relate to impaired mechanisms of repair.
NEUROPATHOLOGICAL FEATURES RELATING TBI TO DEMENTIA
Acute TBI results in amyloid pathology and neurodegenerative proteinopathies
Acute TBI results in multiple neuropathological processes that may predispose to dementia, including inflammation, microglial activation, and oxidative stress [101, 102], but, as evidenced in CTE, one of the most notable mechanisms is a predisposition to abnormal proteins associated with neurodegeneration [103]. A single moderate-to-severe TBI may increase both Aβ and hyperphosphorylated tau deposition, the abnormal protein components that are the substrates of the neuritic plaques and neurofibrillary tangles of AD [104, 105].
After an acute TBI, there has been strong evidence for increased Aβ production and accumulation, both in humans and mice (see Fig. 3) [103, 106–112]. Although not established, this post-traumatic Aβ may be in a prefibrillar, non-aggregated, oligomeric form and may also be detected in the cerebrospinal fluid (CSF) up to 72 hours after acute TBI [113]. Among 152 TBI brains from people ranging from 8 weeks to 85 years old and from 4 hours to 2.5 years after TBI, 20% of those <50 years of age and 60% of those 51–60 had Aβ deposition, compared to none of the controls [81]. Investigators have discovered non-plaque Aβ intracellularly in 80% of temporal cortex samples surgically excised from 18 patients 2–16 hours after a TBI [114]. Additional CSF findings after an acute TBI not only include alterations Aβ, but also in tau, S100b, glial fibrillary acidic protein, neuron-specific enolase, neurofilament light protein, ubiquitin C-terminal hydrolase, and others [115], but the post-TBI levels of all these proteins usually return to normal and have not proven to predict later neurocognitive decline. Genetic polymorphisms are associated with Aβ degradation and clearance and may predispose a subset of post-TBI patients to greater Aβ accumulation and aggregation [110, 117]. In mouse models, the increased production of Aβ after an acute TBI can be decreased by preventing amyloid precursor protein secretase enzymes and the prevention of Aβ accumulation [118, 119].
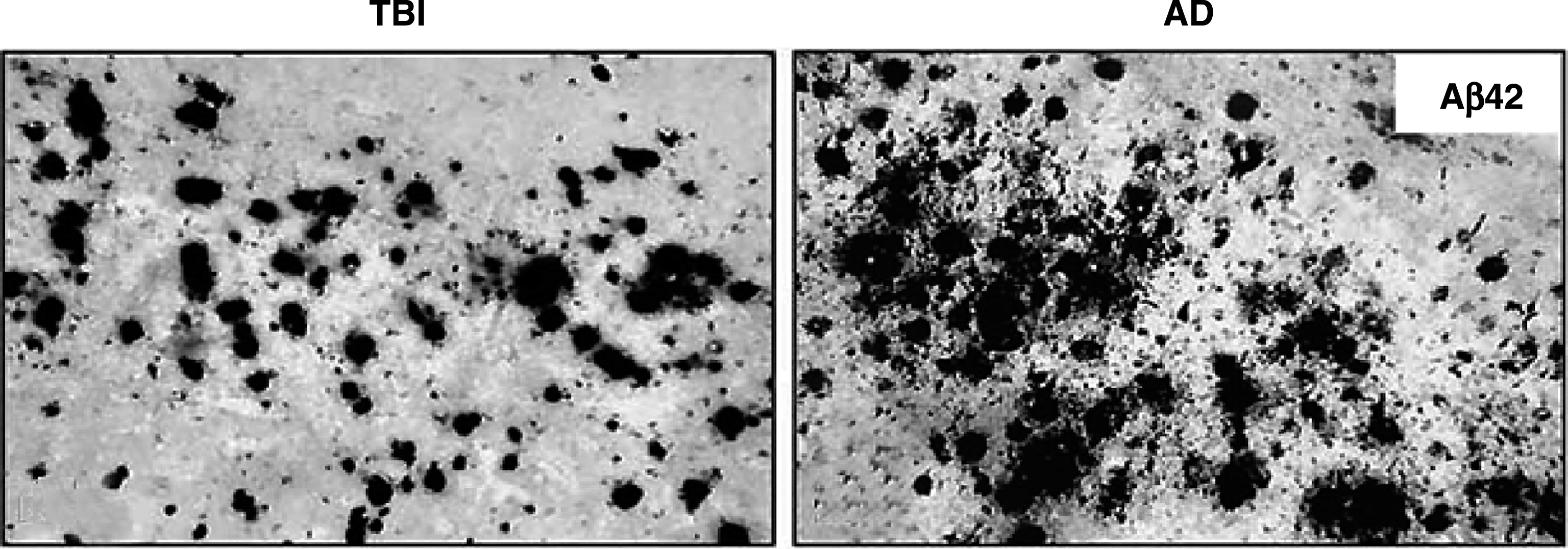
Amyloid (Aβ) deposition in traumatic brain injury (TBI) and Alzheimer’s disease (AD). Left: Tissue resected from temporal cortex of acute severe TBI patients showing Aβ staining. Right: Tissue form patient with AD showing similar Aβ staining. Reprinted from Experimental Neurology, Ikonomovic et al., 2004 [114], with permission from Elsevier.
In contrast to AD, most of the post-traumatic axonal Aβ may not be in fibrillar, thioflavin-S positive neuritic plaques characteristic of AD [120]. On autopsy, amyloid plaques do not occur in most patients with TBIs (70%) [81, 114]; only 30% of patients, who die of TBI, have actual Aβ plaques similar to those of AD [121].
In addition to the accumulation of Aβ, there is production of abnormal tau protein after a TBI [107, 122]. Although most tau transgenic mice exposed to multiple TBIs per day have not shown an accumulation of phospho-tau [123], which is seen in AD, investigators frequently find tau pathology among players of contact sports even in the absence of frank CTE [5, 124]. After an acute TBI, there is tau hyperphosphorylation as well as increased tau levels in the CSF, but not the tau-derived neurofibrillary tangles of AD [114]. Among 39 patients surviving severe TBI for up to 47 years, 34% of those less than 60 years of age have had tau pathology compared to only 9% of 47 control brains, and the tau among the TBI patients appears more widespread in the sulcal depths and superficial layers of the cortex, whereas it is limited to the entorhinal cortex and hippocampus in the control brains [120].
CTE shares features with neurodegenerative dementias
As previously noted, repetitive mTBI, including blast-force mTBIs, can result in CTE, which is characterized by phospho-tau containing neurofibrillary tangles irregularly located in perivascular neurons and glia in the depth of sulci [5, 126]. In CTE, there is progressive deposition of phospho-tau and other neurodegenerative proteins in the brain, similar to neurodegenerative dementias [5, 128]. The neuronal hyperphosphorylated tau deposits in CTE have a3 R/4 R tau isoform ratio similar to AD [129]. Furthermore, among neuropathologically-diagnosed cases of CTE [126], over half also have Aβ deposition, which is present in AD, but which may not have a primary pathophysiological role in CTE [5, 130]. Patients with CTE may have TAR DNA-binding protein 43 and some have α-synuclein, which are other abnormal proteins seen in neurodegeneration [5, 130]. Additional pathology in CTE can include neuroinflammation with monocytes and activation of brain microglia, and traumatic blood-brain barrierdisruption.
CTE is a TBI-related neurodegeneration that is distinct despite sharing features with other neurodegenerative disorders. There is a predominant involvement and deposition of abnormal tau in cortical layers II and III rather than layers V and VI as in AD [131]. There is axonopathy from small blood vessel injury and extensive tau-immunoreactivity perivascularly in superficial cortical layers, especially in the depth of sulci in the frontal lobes [5, 39]. Many patients with a history of TBI have orbitofrontal and anterior temporal injury, and CTE starts as a focal neuropathology in frontal neocortex rather than mesolimbic or parietal changes as in typical AD [5, 132]. Positron emission tomography (PET) with radioligands that bind to fibrillary insoluble protein aggregates, although not established in terms of tau specificity or CTE neuropathology, are beginning to document increased PET binding in TBI patients with clinically probable CTE [133–135]. Investigators have also associated the neuropathological findings of CTE with the clinical and neuropathological changes of FTD, PD, and ALS [5].
TBI results in white matter tract and neural network disruptions
Acute brain damage is associated with diffuse axonal injury (DAI) and white matter pathway disruption [29, 137], evidenced by diffusely distributed and discontinuous areas of white matter abnormalities [138, 139]. During a TBI, the cortex moves at different speed relative to subcortical white matter, with consequent shear forces and deformation stress, axonal and vessel stretching, and microbleeds with resultant DAI. Post-TBI, DTI shows stretches of axons with damaged axonal membrane permeability and ionic shifts, with calcium damage to mitochondria, and resultant energy failure and breakdown of microtubules. Hence, where there is DAI, there is failure of axonal transport, increased axonal Aβ and other abnormal protein accumulation, and released Aβ and hyperphosphorylated tau [37, 140]. Recent work suggests, but does not establish, that these abnormal proteins could then spread in a prion-like fashion along white matter connections and functional neural networks [141]. Clearly, much more research is needed in order to establish this putative mechanism.
Similar TBI and AD effects on white matter tracts on diffusion tensor imaging (DTI) suggest a reduction of functional long-distance connectivity between frontal and caudal brain regions and a possible mechanism for the synergy of TBI with AD and other dementias (see Fig. 4). DTI measures the integrity of white matter, and, although functional anisotropy measures of DTI may decrease, increase, or remain unchanged after TBI [142–144], there are frequent reductions of functional anisotropy in the corpus callosum, cingulum, and other major fiber bundles [145]. On DTI, some investigators report diffusely distributed and discontinuous areas of white matter abnormalities in patients with mild TBIs [125], as well as in veterans who have had blast-force TBIs [139, 147]. Among boxers, investigators report abnormalities in alternate DTI measures of white matter integrity (mean diffusivity, axial diffusivity, radial diffusivity) compared to controls [148, 149], and among soccer players and other athletes, there may be increased (abnormal) axial diffusivity in the corpus callosum and other major tracts [145, 151]. Investigators have found significantly more DTI abnormalities among veterans heavily exposed to bombardment compared to those with limited exposure, demonstrating a potential cumulative effect of injury [151]. In AD and mild cognitive impairment, DTI has also shown early disruption of whole-brain white matter connectivity and long-range tracts such as the cingulum, uncinate fasciculus, superior longitudinal fasciculus, inferior fronto-occipital fasciculus, and corpus callosum [152–157].
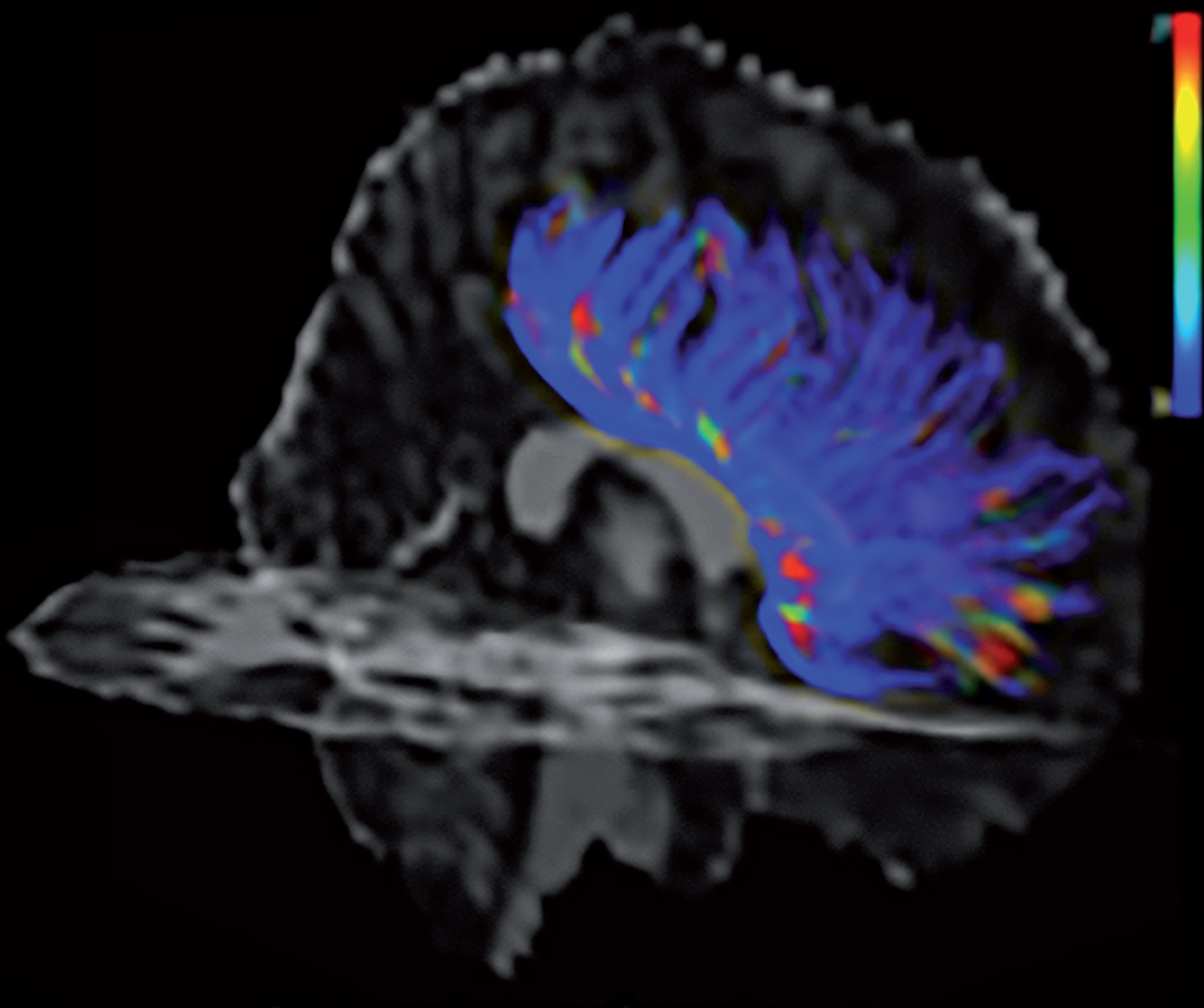
Diffusion tensor imaging showing axon injury and white matter tract disruption in frontal corpus callosum region due to traumatic brain injury. Blue indicates greater disruption. From Emily Dennis, Laboratory of Neuro Imaging Keck School of Medicine, and Paul M. Thompson, Ph.D., University of Southern California.
There is growing evidence that both TBI and AD target functional neural networks with disconnection occurring before other neuropathology [158–164]. In AD, resting state functional MRI classically shows early dysfunction of the default mode network (DMN) in the precuneus/posterior cingulate and the mesial temporal lobe, even before any clinical symptoms [165–173], with an eventual loss of cross-network relations as the severity of AD increases [174]. After mild TBI, resting state functional MRI also shows abnormalities in neural networks [125, 175]; there is decreased connectivity of the DMN immediately after TBI which then increases during later stages, possibly as a form of compensation for injury to other neural networks. Investigators report the development of hyperconnectivity of the DMN associated with repetitive subconcussive mTBI [176]. In contrast, there may be hyperconnectivity in frontally-based executive and salience networks in AD [177–179]. A possible explanation for these findings is that TBIs promote AD through enhancing the early impairment of the DMN, but later diverge with subsequent opposite effects as other neural networks become involved.
CONCLUSIONS
This literature clarifies several aspects of the TBI-dementia relationship. First, the risk of dementia increases with increased severity, frequency, and repetitiveness of TBIs, but does not dependent on the type of physical force to the head. This finding suggests a cumulative effect of violent head displacement with consequent brain deformation, shear forces on axons, and DAI. Second, the impact of these forces depends on individual vulnerabilities of the patient’s brain. Factors affecting vulnerability include cognitive and neuronal reserve, age at time of TBI, and the presence or absence of an APOE4 allele. Third, the effects of TBI are more general than effects on AD, extend to other neurodegenerative disorders, and may suggest a less specific pathophysiological mechanism. TBI is a risk factor for FTD, PD, and possibly ALS as well for AD. This is also suggested by a general effect of lowering of the age of onset of dementia and the addition of TBI cognitive-behavioral features. Fourth, the pathophysiology of TBI is reflected in the neuropathology of CTE, the occurrence of abnormal proteinopathies, and the disruption of white matter tracts and neural networks. One putative mechanism from these results is the release and accumulation of Aβ, hyperphosphorylated tau, and other abnormal proteins along white matter tracts and networks. Additional potential mechanisms include neural network disruption, neuroinflammation, abnormal calcium signaling, vascular abnormalities, and others.
This synthesis of research on the relationship of TBI and dementia is a preliminary assessment of a rapidly growing literature. The conclusions from this review are mitigated by significant reservations, including recall bias on reporting the presence, number, and severity of TBIs as well as major methodological hurdles in establishing causality and not just association between TBI and dementia. Future research is need in order to establish whether dose-dependent effects of head displacement in vulnerable brains damages axons and, thereby, initiates a neurodegenerative process. More work is specifically needed on post-traumatic biomarkers that may predict cognitive decline, the relationship of clinical dementia syndromes with the neuropathology of CTE, and the effects of neural network disruption and potential abnormal protein propagation. In sum, this review can stimulate future studies on the important relationship between TBI and AD, CTE, and other forms of neurodegeneration with potentialdementia.