
Abstract
Aggregation of amyloid-β (Aβ) and tau plays a crucial role in the onset and progression of Alzheimer’s disease (AD). Therefore, the inhibition of Aβ and tau aggregation may represent a potential therapeutic target for AD. Herein, we designed and synthesized both Aβ and tau dual aggregation inhibitors based on the structure of curcumin and developed the novel curcumin derivative PE859. In this study, we investigated the inhibitory activity of PE859 on Aβ aggregationin vitro and the therapeutic effects of PE859 on cognitive dysfunction via dual inhibition of Aβ and tau aggregation in vivo. PE859 inhibited Aβ aggregation in vitro and protected cultured cells from Aβ-induced cytotoxicity. Furthermore, PE859 ameliorated cognitive dysfunction and reduced the amount of aggregated Aβ and tau in brains of senescence-accelerated mouse prone 8 (SAMP8). These results warrant consideration of PE859 as a candidate drug for AD.
INTRODUCTION
The neurodegenerative disorder Alzheimer’s disease (AD) is the most common form of dementia. The number of patients with AD continues to increase globally [1]. Pathologically, senile plaques (SP) comprise the peptide amyloid-β (Aβ) [2] and neurofibrillary tangles (NFT) of tau protein [3], and these are the two major hallmarks of AD.
It is widely believed that Aβ aggregation is the primary cause of AD and that NFT, neural cell loss, and dementia occur after Aβ deposition [4]. Accordingly, aberrant Aβ deposition precedes other biomarker abnormalities [5]. In addition, many cases of familial AD are linked to mutations in the genes encoding the amyloid-β protein precursor (AβPP) or presenilin, which is a subunit of γ-secretase that is responsible for AβPP cleavage [6]. In these cases, Aβ overproduction in the brain results in early-onset of AD. The apolipoprotein E type 4 allele is also implicated in disorders of Aβ clearance and is a well-known risk factor of AD [7]. Aβ forms SP via intermediate aggregates including soluble oligomers, protofibrils, and insoluble fibrils, and oligomeric aggregates of Aβ are more cytotoxic than other intermediate aggregates [8]. Therefore, the inhibition of Aβ aggregation is considered a potential therapeutic approach for AD. However, Aβ aggregation inhibitors for AD treatment have not yet been successfully developed.
In addition, tau protein is also thought to be a therapeutic target for AD. Tau is a microtubule-associated protein that plays fundamental roles in the stabilization of microtubules [9]. In AD patients, tau is abnormally phosphorylated and dissociates from microtubules, and then aggregates into paired helical filaments, facilitating the formation of NFT [10, 11]. The severity of AD symptoms is positively correlated with the numbers of NFT [12, 13], and similar to Aβ, oligomeric aggregates of tau are more toxic than the other tau aggregate types [14]. Therefore, inhibition of tau aggregation may have therapeutic advantages in the treatment of AD, although no tau aggregation inhibitors have been developed to date.
We hypothesized that a dual inhibitor for both Aβ and tau cascades would be a more effective treatment for AD than existing inhibitors of Aβ or tau alone, and developed potent inhibitors of both Aβ and tau aggregation. Previously, we introduced the novel tau aggregation inhibitor PE859 [15]. This compound is a curcumin derivative that we designed and synthesized to be a dual inhibitor of Aβ and tau aggregation [16]. Curcumin is a natural polyphenol, and is a yellow spice that is derived from turmeric (Curcuma longa). Curcumin has antioxidant and anti-inflammatory activities, and reportedly inhibits aggregation of Aβ [17] and tau [18]. Accordingly, the incidence of AD is significantly greater in the US than in India, where curcumin is consumed on a daily basis [19]. However, curcumin is difficult to use as an AD drug because of its poor bioavailability [20]. In contrast with curcumin, PE859 exhibited higher inhibitory activity against the aggregation of both Aβ and tau in vitro, and higher absorption into the blood and penetration of the blood-brain barrier in vivo [16].
In these previous studies, cell-free thioflavin T (ThT) assays and in vivo motor function assessments showed beneficial effects of PE859, and these were related to inhibited tau aggregation. In the present study, we performed further investigations of the inhibitory activities of PE859 on Aβ aggregation. Specifically, we demonstrated protective effects against Aβ-induced cytotoxicity in neuroblastoma SH-SY5Y cells, and confirmed the dual aggregation inhibitory activity and beneficial effects of PE859 on cognitive dysfunction in senescence-accelerated mouse prone 8 (SAMP8). Because these mice show age-related cognitive dysfunction with Aβ and tau abnormalities, the present data indicate the potential of PE859 as a treatment for AD.
MATERIALS AND METHODS
Test compound
The test compound PE859 is synthesized by ChemGenesis (Tokyo, Japan) or NARD institute (Amagasaki, Japan).
Thioflavin T fluorescent assay
Aβ aggregation was monitored using the fluorescent dye Thioflavin T (ThT; Sigma-Aldrich Corp.,St. Louis, MO, USA), which specifically binds to the β-sheet structure of the protein [21]. Human Aβ1-40 or Aβ1-40 (Peptide Institute, Inc., Osaka, Japan) were dissolved in 0.1% NH3 (Nacalai tesque, Inc., Kyoto, Japan) at 0.5 mM and diluted to 20 μM with PBS (Nacalai tesque Inc.). The test compound solutions were prepared at 100 times of the final concentrations with dimethyl sulfoxide (DMSO; Nacalai tesque Inc.), and diluted 50-fold with PBS. To evaluate the inhibitory activity of PE859 on Aβ aggregation, the Aβ1-40 or Aβ1-40 solutions and each of the test compound dilutions were mixed in equal amounts. The final concentration of Aβ1-40 or Aβ1-40 were 10 μM, and those of PE859 were 0 (Aβ alone), 0.3, 1, 3, or 10 μM. The mixtures containing Aβ1-40 were incubated at 37°C for 0.5, 1, 4, 24, and 48 h. The mixtures containing Aβ1-40 were incubated at 37°C for 24, 48, 72, and 96 h. After the incubation, 6 μM ThT solution was added to the reaction mixtures in equal amounts and the fluorescence intensity was measured using a multilabel counter (ARVO; Perkin Elmer, Waltham, MA, USA), with excitation at 440 nm and emission at 486 nm. The sample without test compound was used as a negative control, and its fluorescence intensity was taken as 0% inhibition.
Quartz crystal microbalance assay
A quartz crystal microbalance (QCM) [22] was used for the detection of Aβ aggregation [23]. We used a 27 MHz QCM instrument (AFIINIX Q4; Initium Inc., Tokyo, Japan) featuring a 500-μL sensor cell equipped with a QCM plate (diameter of the quartz plate. 8.7 mm; area of the gold electrode, 4.9 mm2) at the bottom of the cell. It was calibrated to the change frequency by 1 Hz in response to a mass change of 0.62 ng/cm2 on the electrode (corresponding to a 1 Hz frequency decrease per 30-pg mass increase on the electrode). The binding of Aβ to the electrode and its aggregation were monitored though the frequency change measured (ΔF, Hz) according to the manufacturer’s protocol using the Sauerbrey equation:
where F0 represents the fundamental frequency of the QCM (27×106 Hz), Δm the mass change (g), A the electrode area (4.9 mm2), ρ q the density of quartz (2.65 g cm-3), and μ q the shear modulus of quartz (2.95×1011 dyn cm-2).
Before the assay, 490 μL of 2.5 μM PE859 solution in 50 mM Tris-HCl buffer (pH 7.6) was added into the sensor cell and incubated at 25°C with stirring until the frequency was stabilized. Aβ1-40 was dissolved in DMSO at 0.5 mM, and 10 μL of the solution was added into the sensor cell immediately. The final concentrations of Aβ and PE859 were 10 and 2.5 μM, respectively. The frequency change was monitored for 60 min after adding the Aβ to each cell.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blotting for Aβ
Aggregated Aβ was detected by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting. The solutions of Aβ1-40 with test compound were prepared in the same way as for the ThT assay, and incubated at 37°C (the final concentrations of PE859 were 1 or 10 μM). After 2- and 3-h incubation, the solutions were partly removed and mixed with an equal amount of Tris-SDS sample buffer (Cosmo Bio Co., Ltd., Tokyo, Japan) and stored at –30°C until just before electrophoresis. The 10-μL of each sample was electrophoresed at 100 V for 2 h on a 15–20% polyacrylamide tricine gel (Wako Pure Chemical Industries, Ltd., Osaka, Japan) and transferred to a 0.45 μm polyvinylidene difluoride (PVDF) membrane (Merck Millipore, Billerica, MA, USA), which was run at 15 V for 30 min. After blocking with 2.5% skim milk (Nacalai Tesque, Inc.) in Tris-buffered saline with 0.05% Tween 20 (TBS-T; Sigma-Aldrich Corp.) for 1 h, the blots were incubated with anti-Aβ antibody 6E10 (1:1,000 dilution; Covance Inc., Princeton, NJ, USA) overnight at 4°C. The blots were washed with TBS-T for 10 min three times, and incubated with horseradish peroxidase (HRP)-conjugated anti-mouse Immunoglobulin G (1:2,000 dilution; GE Healthcare, Little Chalfont, Buckinghamshire, UK) for 1 h at room temperature. After washing with TBS-T for 10 min three times, the aggregated Aβ was detected using Immobilon Western Chemiluminescent HRP substrate (Merck Millipore), and analyzed using an image analyzer (ImageQuant LAS4000; GE Healthcare).
Transmission electron microscopy analysis
Fibrillar Aβ aggregates were observed by transmission electron microscopy with an electron microscope (JEM-1220; JEOL Ltd., Tokyo, Japan). The aggregation reaction mixtures containing 20 μM Aβ1-40 and 1 μM PE859 were incubated at 37°C for 24 h, and then 5-μL of each mixture was applied to carbon grid (Okenshoji Co., Ltd., Tokyo. Japan) followed by staining with 2% phosphotungstic acid (TAAB Laboratories, Equipment Ltd., Aldermaston, Berkshire, UK) for eight minutes. After washing with distilled water, each grid was observed at at 80 kV, 30,000×magnification. The number and length of Aβ aggregates in each image were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Amyloid-β-induced cytotoxicity
Human neuroblastoma SH-SY5Y cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Nacalai Tesque, Inc.) containing 100 U/mL penicillin, 100 μg/mL streptomycin (Nacalai Tesque, Inc.), and 10% fetal bovine serum (Life Technologies Carlsbad, CA, USA). The cells were seeded on 96-well tissue culture plates (Becton, Dickinson and Company, Franklin Lakes, NJ, USA), and incubated at 37°C under a saturating humidity atmosphere of 5% CO2/95% air. After 3-days incubation, the culture medium was removed, and serum-free DMEM with 10 μM Aβ1-40 and PE859 (0, 0.1, 0.3, 1, or 3 μM) was added. The treatment media were pre-incubated for 1 h before treatment. After a 72-h treatment, cytotoxicity was assessed by the leakage of lactate dehydrogenase (LDH) into the medium. The amount of LDH was determined using a Cytotoxicity Detection LDH Kit (Kyokuto Pharmaceutical Industrial Co., Ltd., Tokyo, Japan). The 50 μL of each culture medium was transferred into a new 96-well plate and mixed with 50 μL of the color regent in the kit. After a 30-min incubation, absorbance at 570 nm was measured with a microplate reader (Model 680; Bio-Rad Laboratories, Inc., Hercules, CA, USA). In addition, cell viability was determined using the methyl thiazolyltetrazolium (MTT) reduction assay. The remaining culture medium was replaced with serum-free medium containing 0.5 mg/mL MTT (Nacalai Tesque, Inc.) and incubated for 60 min. The culture medium was then removed, and 2-propanol (Nacalai Tesque, Inc.) was added. After shaking for 5 min, the absorbance at 570 nm was measured with a microplate reader (Model 680; Bio-Rad Laboratories, Inc., Hercules, CA, USA). The percentage viability was defined as the relative absorbance versus the control.
PE859 treatment in SAMP8 mice
Male SAMP8/TaSlc mice (2 months of age, n = 9 in the vehicle group and n = 8 in the other groups) were obtained from Japan SLC Inc. (Hamamatsu, Japan). PE859 was dissolved in 80% PEG400 (Nacalai Tesque, Inc.) and 20% water solution at 0- (vehicle group), 0.2- or 0.6-mg/mL, and orally administered in a volume of 5-mL/kg of body weight (at a dose of 0-, 1-, or 3-mg/kg/day) for 9 weeks (from 2-month of age to 4-month of age) using a flexible gastric tube (Fuchigami Kikai Co., Ltd., Mukou, Japan). The experimental procedures involving mice and their care were conducted in accordance with the ethical guidelines of the Kyoto University Animal Experimentation Committee and the guidelines of the Japanese Pharmacological Society.
Y-maze test
Working memory of the mice was evaluated using the Y-maze test at the start of the treatment, and at eighth week of the treatment. Each mouse was placed at one arm of Y maze, which has three arms 30 cm in length with equal angles between all arms (Bio Research Center, Nagoya, Japan). The mice were allowed to move freely in the maze for 8 min, and the sequence and number of arm entries were recorded. Spontaneous alternation behavior, which is used as a measure of spatial memory, was defined as entry into all three arms on consecutive choices. The percentage of spontaneous alternations was calculated as follows:
Morris water maze test
The spatial learning and memory of the mice was evaluated at the ninth week of treatment using a Morris water maze test. The Morris water maze, consisting of a circular pool 120 cm in diameter, was filled with water containing 1% skim milk to a depth of 20 cm. A circular platform (12 cm diameter) was placed in the pool. The temperature of the water was kept at 25±2°C. All mice were trained with two trials per day for 6 days consecutively. In each trial, the mice were allowed to swim until they reached the platform or for up to 90 s. If they reached the platform within 90 s, they stayed on it for 10 s. If they could not reach the platform within 90 s, they were moved to the platform and stayed on it for 15 s. On the day 0, the platform was placed 2 cm above the surface of the water (visible platform trial). From the day 1 to 5, the platform was placed 2 cm below the surface of the water (hidden platform trial). The mice swimming in the pool were tracked using a video tracking and analysis system (ACTIMAZE; Actimetrics, Wilmette, IL, USA), and their escape latencies (the time taken to reach the platform) were automatically recorded. The daily latency of each mouse in the hidden platform trial was obtained from the average latencies of the two trials undertaken each day. Furthermore, after the hidden platform trials in the day 5, the platform was removed and each mouse was allowed to swim for 90 s again (probe trial). In this trial, we counted the number that mice crossed the place where the platform had been located. After each trial, the mice were dried with paper towels before they were returned to the cage.
Motor function test
The ability of coordinated movements of the mice was evaluated using a rotarod treadmill MK-670 (Muromachi kikai Co., Ltd., Tokyo, Japan). Mice were placed on a rotating rod, which was slowly accelerated to 40 rounds per min in 300 s. The latency to fall from the rod was recorded for a maximum of 300 s. Mice were given two trials, and the mean of them was calculated. Grip strength of the mice was also measured at the ninth week of the treatment using a grip strength meter MK-380M (Muromachi kikai Co., Ltd.). Each mouse was placed on the metal mesh of the meter and its tail was pulled back in a horizontal direction. The grip strength of each mouse when it could no longer hold on to the mesh was recorded. Mice were given three trials, and the mean of them was calculated.
Protein extraction from the brain tissue
At the end of treatment, all mice were sacrificed under deep anesthesia with 100 mg/kg sodium pentobarbital (Kyoritsu Seiyaku Corp., Tokyo, Japan) administered intraperitoneally, and the whole brains were removed for biochemical analysis. The brain tissues were homogenized in ten volumes (w/v) of 2×RIPA buffer (Nacalai Tesque, Inc.) with 0.5 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich Corp.), 1×PhosSTOP (Roche, Basel, Switzerland), and 1% protease inhibitor cocktail (Nacalai Tesque, Inc.). To measure Aβ, we extracted soluble and insoluble protein by the recommended method of the Aβ enzyme-linked immunosorbent assay (ELISA) kits (Wako Pure Chemical Industries, Inc.). The homogenate was centrifuged at 100,000×g at 4°C for 20 min, and the supernatant was collected as a soluble fraction. The pellet was sonicated in 70% formic acid (Wako Pure Chemical Industries, Ltd.), neutralized with 20 volumes of 0.9 M Tris buffer (pH 11.0), and used as an insoluble fraction. To measure total tau and phosphorylated tau, we modified the method of extracting insoluble tau in the brain of human tau transgenic mice [24]. The homogenate was centrifuged at 250,000×g at 4°C for 20 min, and the supernatant was collected as a Tris buffer-soluble fraction. The pellet was sonicated in 10 mM Tris-HCl buffer (pH7.5) containing 1% sarkosyl, 0.5 M NaCl, and 10% sucrose (Wako Pure Chemical). After 1 h incubation at 25°C, the solution was centrifuged at 250,000×g at 4°C for 20 min. The supernatants were collected as the sarkosyl-soluble fraction. The pellet was sonicated in 1×PBS (Nacalai tesque, Inc.), and used as a sarkosyl-insoluble fraction. To measure AβPP, the Tris buffer-soluble fraction was used.
Enzyme-linked immunosorbent assay for amyloid-β
Aβ1-40 and Aβ1-40 levels in the brains were measured using human/rat Aβ ELISA Kits (Wako Pure Chemical Industries, Inc.) and a microplate reader (Model 680; Bio-Rad Laboratories, Inc.) according to the manufacture’s protocol. In the Aβ1-40 ELISA kit, anti-Aβ antibodies clone no. BNT77, which detects Aβ11 - 28, and clone no. BC05, which detects C-terminus of Aβ1-40, were used. In the Aβ1-40 ELISA kit, anti-Aβ antibodies clone no. BNT77 and clone no. BA27, which detects C-terminus of Aβ1-40, were used. These ELISA kits were available for the measurement of mouse Aβ. The insoluble fractions were diluted 1,000-fold with the dilution buffer provided in the kit. The soluble fractions were diluted 20-fold with the buffer.
Western blotting for tau measurement
Tau and AβPP levels in the brains were measured by western blotting. Each fraction for tau measurement was mixed with an equal amount of Tris-SDS β ME sample buffer (Cosmo Bio Co., Ltd., Tokyo, Japan) and boiled at 100°C for 10 min. The 10 μL of each sample was electrophoresed at 200 V for 1 h on a 5–20% polyacrylamide gel (Wako Pure Chemical Industries, Ltd) and transferred to a 0.45 μm PVDF membrane (Merck Millipore), which was run at 15 V for 30 min. After blocking with 2.5% skim milk (Nacalai Tesque, Inc.) in TBS-T (Sigma-Aldrich Corp.) for 1 h, the blots were incubated with anti-total tau antibody TAU-5 (1:2,000 dilution; Thermo Fisher Scientific, Waltham, MA, USA) anti-phosphorylated (Ser202, Thr205) tau antibody AT8 (1:1,000 dilution; Thermo Fisher Scientific), anti-AβPP antibody (1:100 dilution; Immuno-Biological Laboratories Co., Ltd., Fujioka, Japan), or anti-sAβPPβ antibody (1:100 dilution; Immuno-Biological Laboratories Co., Ltd.) overnight at 4°C. The blots were washed with TBS-T for 10 min three times. To detect total tau and phosphorylated tau, the blots were incubated with HRP-conjugated anti-mouse Immunoglobulin G (1:2,000 dilution; GE Healthcare) for 1 h at room temperature. To detect AβPP and s AβPPβ, the blots were incubated with HRP-conjugated anti-rabbit Immunoglobulin G (1:3,000 dilution; GE Healthcare) for 1 h at room temperature and washed with TBS-T for 10 min three times. Total tau and AβPP were detected using Immobilon Western Chemiluminescent HRP Substrate (Merck Millipore), and phosphorylated tau and sAβPPβ were detected using Chemi-Lumi One Super (Nacalai Tesque, Inc.). Protein bands of the blots were analyzed using an image analyzer (ImageQuant LAS4000; GEHealthcare).
Data analysis
Data derived from ThT fluorescent assay, Y-maze test, and the escape latency of Morris water maze test were analyzed using two-way analysis of variance (ANOVA) and Bonferroni posttest. Data derived from transmission electron microscopy observation was analyzed using Student’s t-test. Data derived from the cytotoxicity assay, the probe trial of Morris water maze test, ELISA, and the western blotting were analyzed using one-way ANOVA and Dunnett’s multiple comparison posttests. The software Graphpad Prism (GraphPad Software Inc., San Diego, CA, USA) was used for these analyses, and p < 0.05 was considered statistically significant.
RESULTS
The inhibitory effects of PE859 on Aβ aggregation were monitored according to the fluorescence intensity of ThT (Fig. 1). In these experiments, both Aβ1-40 and Aβ1-40 increased ThT fluorescence and PE859 inhibited these increases in concentration-dependent manner, indicating that PE859 inhibits Aβ1-40 and Aβ1-40 aggregation. Subsequently, half-maximal inhibitory concentrations (IC50) of PE859 for Aβ1-40 and Aβ1-40 aggregation were calculated at the end of incubation periods, when fluorescence plateaued (48 h for Aβ1-40 and 96 h for Aβ1-40). These values were 5.0 μM and 2.9 μM, respectively. These results suggest that PE859 has greater inhibitory effects on Aβ1-40 aggregation than on Aβ1-40 aggregation.
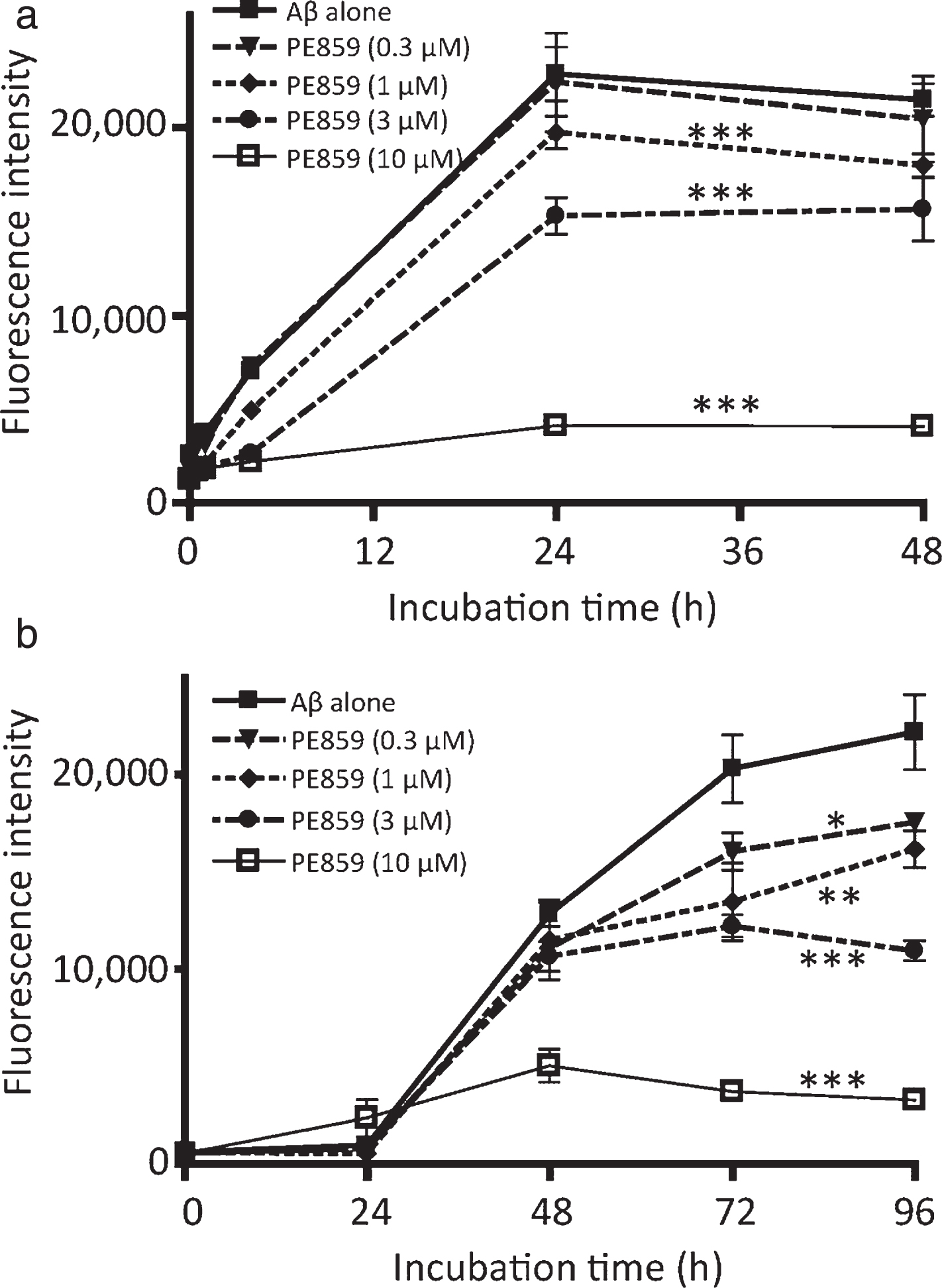
Inhibitory effect of PE859 on amyloid-β aggregation as monitored by the thioflavin T fluorescence assay. a) Aβ1-40 (10 μM) alone and Aβ1-40 with PE859 (0.3, 1, 3, or 10 μM) were incubated at 37°C for 48 h. b) Aβ1-40 (10 μM) alone and Aβ1-40 with PE859 (0.3, 1, 3, or 10 μM) were incubated at 37°C for 96 h. Mean±SD, n = 3. *p < 0.05, **p < 0.01, ***p < 0.001 versus Aβ alone.
Early stages of Aβ aggregation were monitored using QCM analysis (Fig. 2), which is a powerful tool for studies of Aβ aggregation, with high sensitivity (nanogram level) and a simple relationship between mass and QCM frequency [22]. In this technique, deposition of proteins or compounds onto the surface of piezoelectric quartz crystals, decrease fundamental oscillation frequencies in accordance with the Sauerbrey expression. In the present analyses, QCM frequencies decreased in association with Aβ aggregation on the electrode (Fig. 2). Moreover, the frequency change for Aβ with PE859 was smaller than that for Aβ alone, and no frequency change was detected in the presence of PE859 alone (data not shown). These results suggest that PE859 inhibits Aβ aggregation.
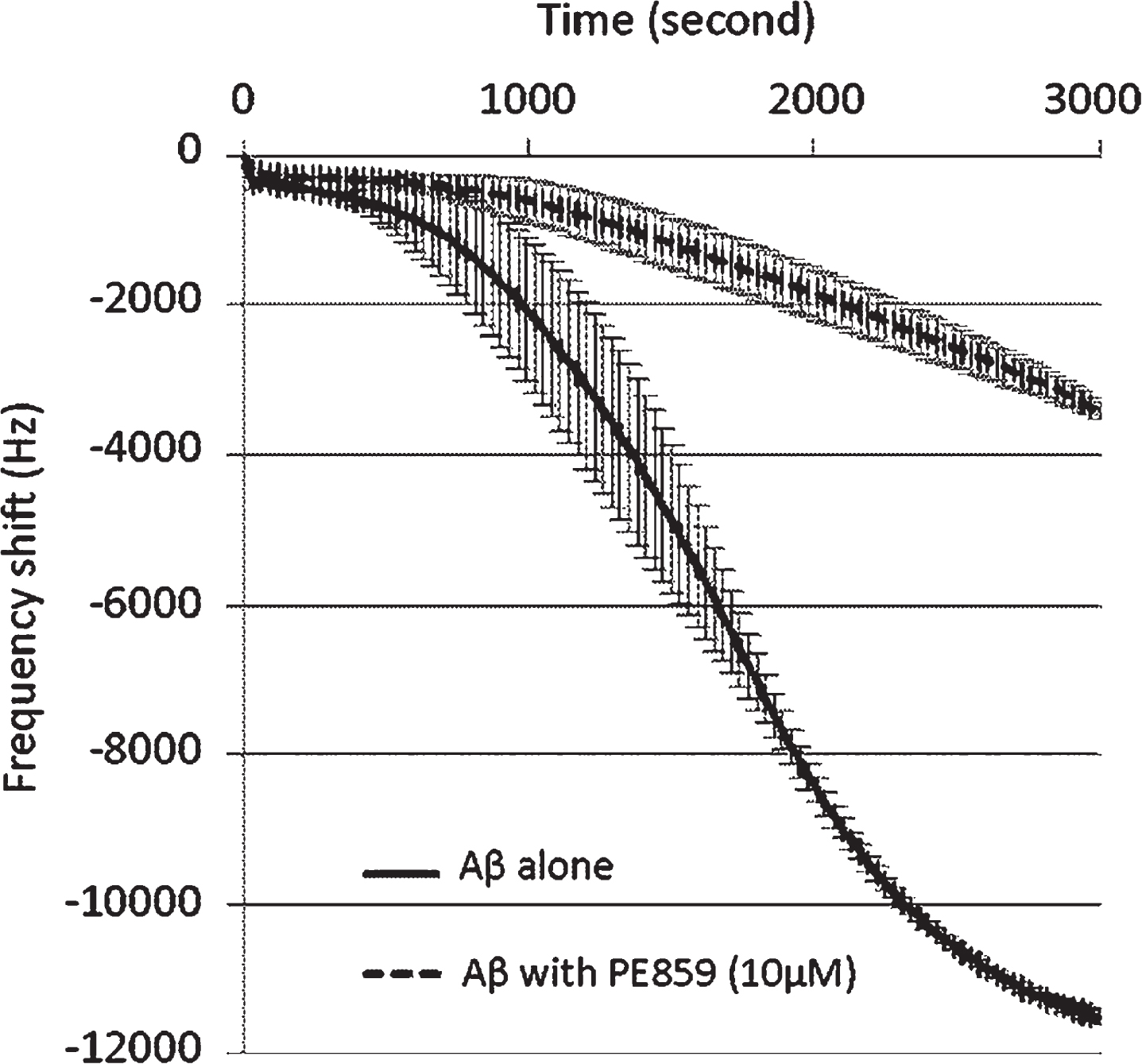
Inhibitory effect of PE859 on Aβ aggregation as monitored by QCM analysis. Aβ1-40 (10 μM) alone and Aβ1-40 with PE859 (2.5 μM) were incubated with stirring at 25°C for 3,000 sec. Mean±SD, n = 2.
In further experiments, inhibitory effects of PE859 on Aβ oligomerization were confirmed using western blotting (Fig. 3), and band densities of Aβ aggregation stages, including monomer, dimer, trimer, tetramer, intermediate aggregate, and large aggregate were quantified (Table 1). Before incubation, most Aβ peptide present in the monomeric form at 4 kDa (59.8% of total band density). In contrast, the amount of monomeric Aβ was decreased to nearly half (29.6%) after 3 h incubation, and more than two-thirds of monomeric Aβ remained (41.1%) in the presence of PE859. Highly aggregated Aβ contents increased by more than 20-fold during incubation (0.4% and 8.2% of total bands density at 0 h and at 3 h, respectively). In the presence of PE859, the amounts of highly aggregated Aβ were less than half those in its absence (Aβ with 1-μM PE859, 3.9% and Aβ with 10 μM PE859, 4.0%). Similarly, amounts of intermediate aggregates of Aβ in the presence of PE859 were also less than those in lanes with Aβ alone (Aβ alone, 4.7%, Aβ with 1-μM PE859:3.4% and Aβ with 10-μM PE859:2.6%). In the lanes containing Aβ with 1-μM PE859, amounts of trimer (19.7%) and tetramer (28.9%) were greater than those in lanes with Aβ alone (trimer, 12.3% and tetramer, 28.8%). These data suggest that PE859 inhibits Aβ aggregation, in particular at the stages of Aβ tetramers or smaller multimers.
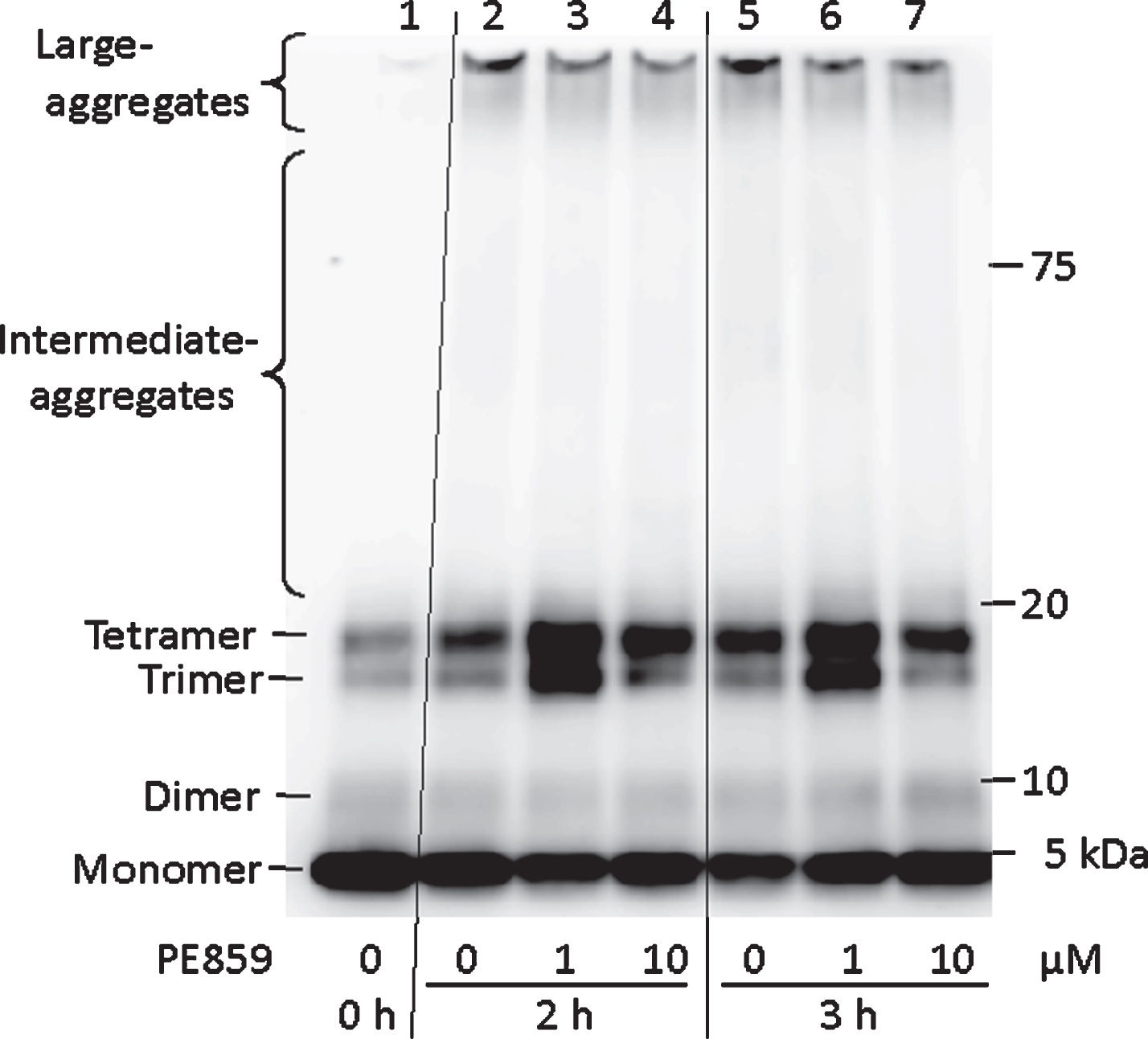
Inhibitory activity of PE859 against Aβ aggregation as detected by western blotting. Aβ1-40 (10 μM) alone or Aβ1-40 (10 μM) with PE859 (1 and 10 μM) were detected by western blotting. Lane 1, Aβ alone (before incubation); Lane 2–4, Aβ alone, Aβ with PE859 (1 μM), and Aβ with PE859 (10 μM) after a2 h incubation, respectively. Lane 5–7, Aβ alone, Aβ with PE859 (1 μM), and Aβ with PE859 (10 μM) after a 3 h incubation, respectively.
Quantitative value of aggregated Aβ in the western blotting image of Fig. 3. Each band density was presented as a relative proportion to the whole bands density in a lane
To confirm inhibitory activities of PE859 on Aβ fibril formation, we observed fibril formation using electron microscopy (Fig. 4). After 24-h incubation, Aβ alone formed many fibrous aggregates (Fig. 4a), whereas Aβ with 1-μM PE859 formed few aggregates (Fig. 4b). Specifically, 1,021 Aβ aggregates were observed in the image of Aβ alone (73.5 aggregates per μm2) and only 191 were observed in the presence of 10-μM PE859 was 191 (13.7 aggregates per μm2). The length of Aβ aggregates were then measured and stratified by size (Fig. 4c, d), and in the images of Aβ alone, 101–200-nm aggregates comprised 41% of all aggregates (419/1021), whereas more than 90% (181/191) of aggregates were smaller than 100 nm in images of Aβ with 10-μM PE859. Moreover, the average length of Aβ aggregates in the image of Aβ alone (142±95 nm) differed significantly from that of Aβ with 10-μM PE859 (40±41 nm; p < 0.001, t-test).
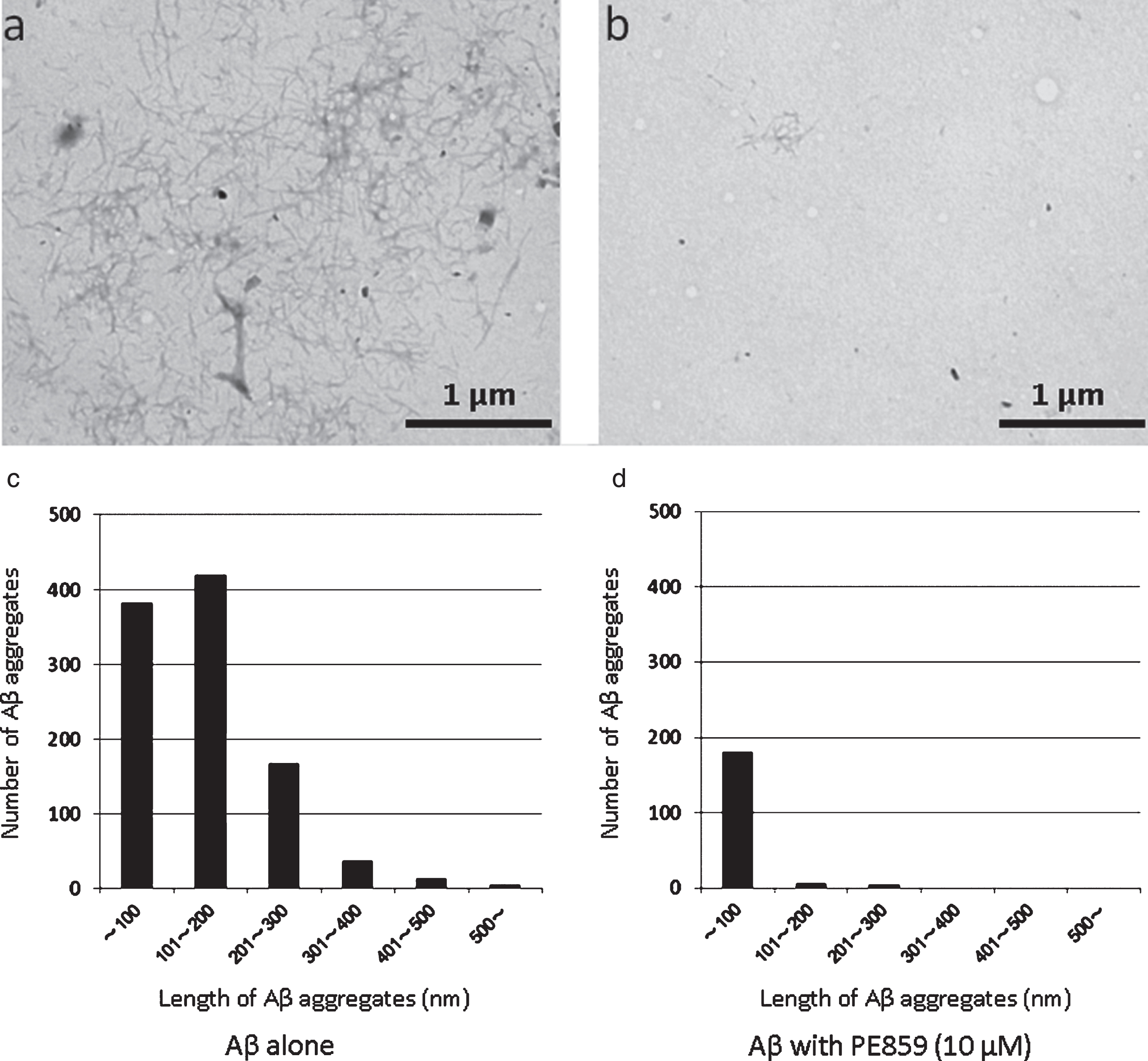
Inhibitory activity of PE859 against Aβ aggregation as observed by transmission electron microscopy. Aβ1-40 (10 μM) alone (a) and Aβ1-40 with 1 μM PE859 (b) were incubated at 37°C for 24 h, and stained with 2% phosphortungstic acid. Scale bar = 1 μm. The length and number of Aβ aggregates in the image of Aβ alone (c) and Aβ with 10 μM PE859 (d).
As shown in Fig. 2, the amounts of trimeric and tetrameric Aβ seemed to increase in the presence of PE859. Because these forms of Aβ may be cytotoxic, we evaluated the protective effects of PE859 on Aβ-induced cytotoxicity in cultured cells (Fig. 5). In these experiments, SH-SY5Y cells were exposed to 10-μM Aβ1-40 with PE859 (0.1–3 μM) for 72 h and cell viability was determined using MTT assay. Cells treated with Aβ alone had significantly decreased viability and PE859 restored viability in aconcentration-dependent manner (Fig. 5a). Moreover, in LDH assays of Aβ-induced cell death (Fig. 5b), increases in LDH levels in the medium of Aβ-treated cells were suppressed by PE859, indicating restrained Aβ-induced LDH leakage. Cells treated with PE859 alone showed no signs of cytotoxicity in either assay, suggesting that PE859 protects cultured cells from Aβ-induced cytotoxicity.
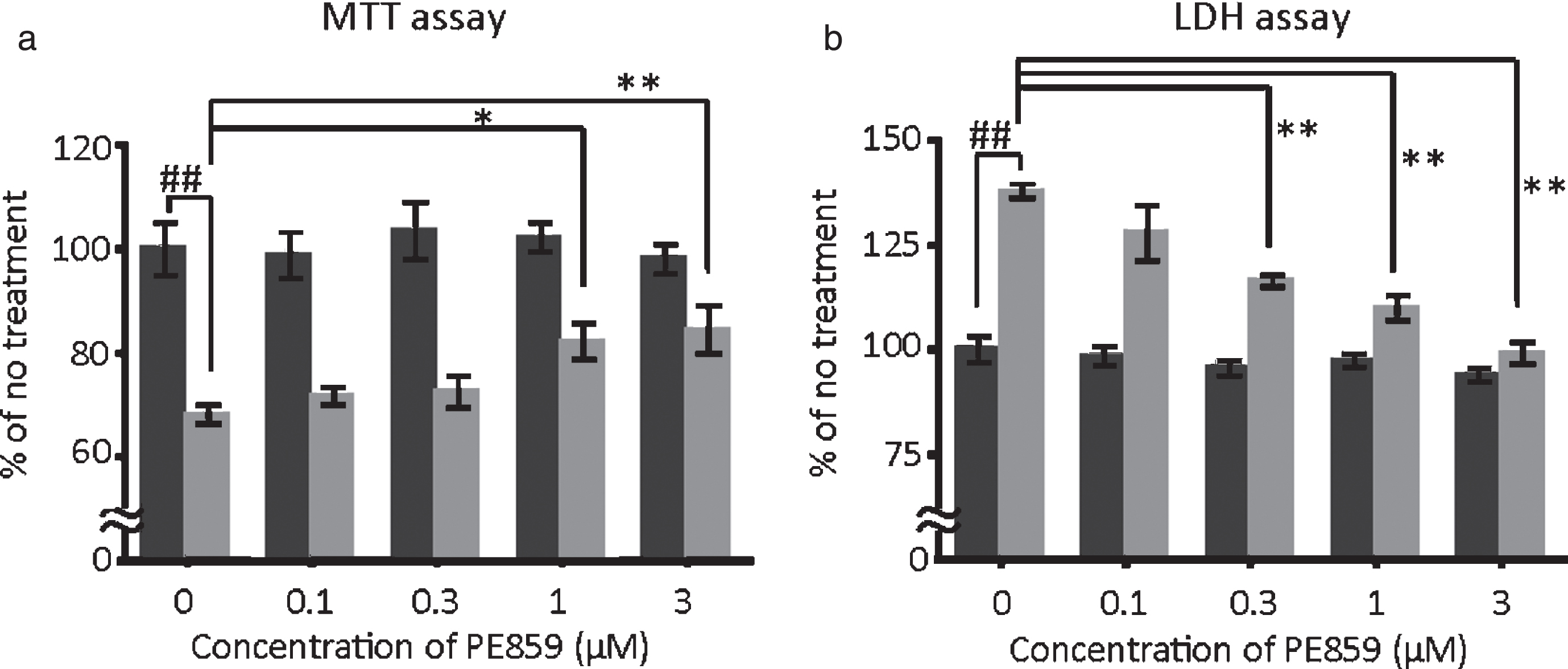
Protective effect of PE859 on Aβ-induced cytotoxicity. SH-SY5Y cells were treated with Aβ1-40 (10 μM) alone or with Aβ1-40 (10 μM) +PE859 (0, 0.1, 0.3, 1, or 3 μM) for 72 h. Mean±SEM, n = 5. Black bar, PE859 alone; gray bar, PE859 with Aβ. a) Cell viability was determined using the MTT assay (p < 0.001, one-way ANOVA, ##p<0.01 versus no treatment, *p < 0.05 and **p < 0.01 versus Aβ alone, Dunnett’s posttest). b) Cytotoxicity was determined by LDH leakage into medium. (p < 0.001, one-way ANOVA, ##p < 0.01 versus no treatment, **p < 0.01 versus Aβ alone, Dunnett’s posttest).
To examine the beneficial effects of PE859 on cognitive deficits, the compound was orally administered to 2-month-old male SAMP8 mice, which is a substrain of the senescence-accelerated mouse model of accelerated aging [25]. SAMP8 mice showed age-related deficits in memory and learning [26, 27] and Aβ and tau pathology in brains [28, 29]. Furthermore, increased levels of insoluble (aggregated) Aβ and tau were observed in the brains of cognitively impaired SAMP8 mice in this study and others (data not shown), validating this model for assessments of PE859. Thus, in further experiments, spatial working memories of the mice were evaluated using Y-maze tests (Fig. 6). At the start of treatment(2 months of age), spontaneous alternations occurred with a frequency of about 69% in all treatment groups (69.1±1.8% in the vehicle group, 69.3±1.5% in the 1-mg/kg PE859 group, and 69.1±1.5% in the 3-mg/kg PE859 group). Subsequently, percentages of spontaneous alternations were reduced with age to 51.5±2.4% after eight weeks of treatment. In contrast, in the percentages of spontaneous alternations remained unchanged at 65.9±4.0% in the 1-mg/kg PE859 group and 64.1±4.0% in the 3-mg/kg PE859 group. These differences between the PE859-treated groups and the vehicle group were significant (F (2, 44) = 5.23 and p = 0.014, two-way ANOVA, p < 0.001 vehicle versus 1-mg/kg PE859 and p < 0.01 vehicle versus 3-mg/kg PE859, Bonferroni posttest). For comparison, we evaluated the performance of non-treated control SAMR1 mice (male, n = 5) in the same Y-maze test. These animals showed spontaneous alternations of 65.9±5.1% at 2 months of age and 72.9±4.8% at 4 months of age, suggesting that spontaneous alternations of cognitively-normal mice are more than about 60% and that PE859 treatment prevent the development of cognitive impairments in SAMP8 mice. In agreement, numbers of total arm entries in Y-maze tests tended to be reduced in PE859-treated SAMP8 mice (Fig. 6b), but these alterations were not significant (F (2, 44) = 1.85 and p = 0.18). Subsequently, we assessed spatial learning using the Morris water maze test at the ninth week of treatment. In these experiments, escape latencies on day 5 were 44.5±12.1 s in the vehicle group, 44.7±15.3 s in the 1-mg/kg PE859 group, and 19.4±4.9 s in the 3-mg/kg PE859 group. Although escape latencies tended to be shorter in the 3-mg/kg PE859 group, no significant differences were identified between groups (F (2, 86) = 0.74 and p = 0.49). Numbers of times that mice crossed the platform location in probe trials tended to increase dose-dependently (1.4±0.3 times in the vehicle group, 1.7±0.5 times in the 1-mg/kg PE859 group and 2.2±0.4 times in the 3-mg/kg PE859 group), but again no significant differences were identified between groups (F (2, 22) = 0.74 and p = 0.28, one-way ANOVA).
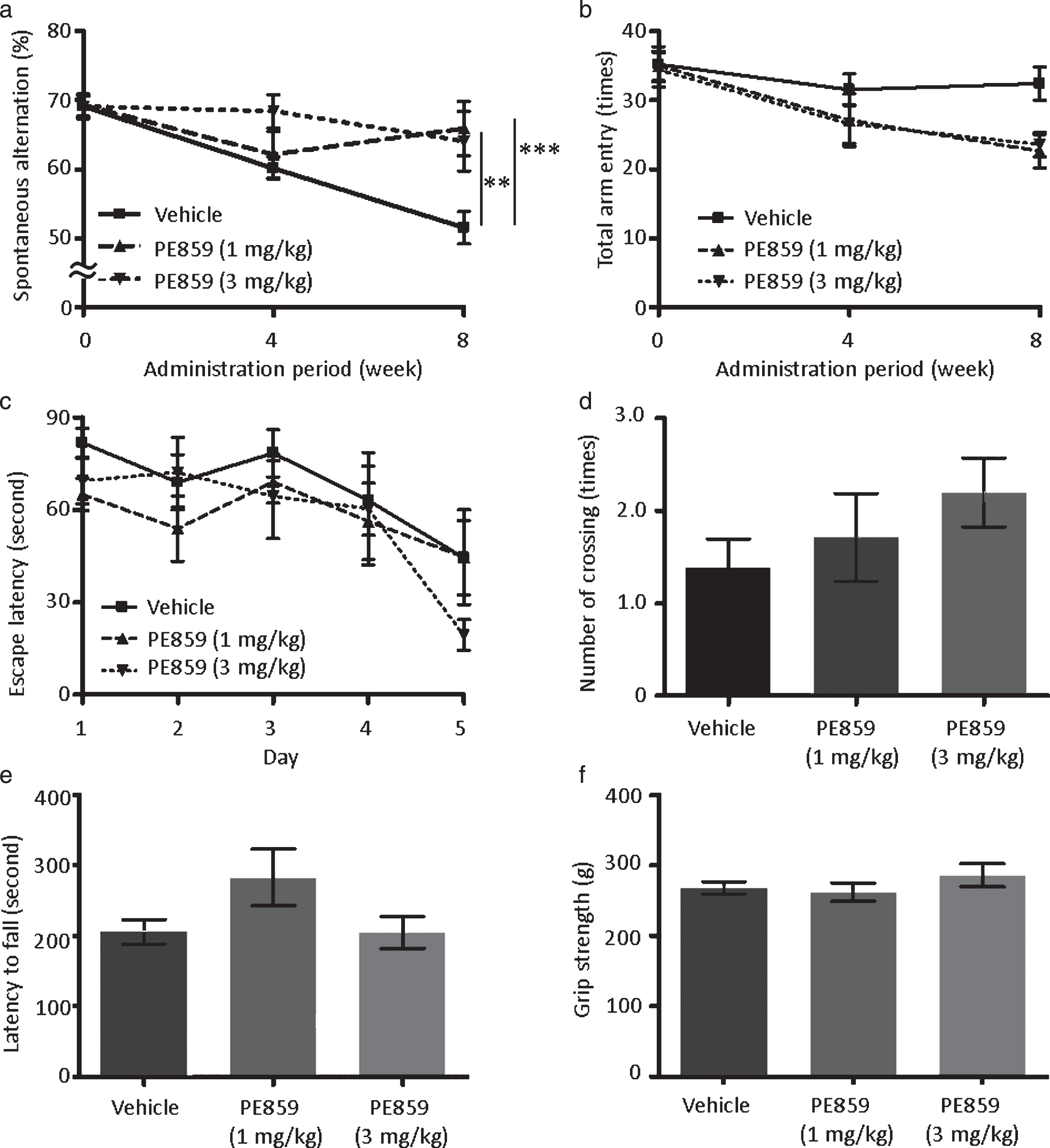
Effects of PE859 on cognitive dysfunction in SAMP8 mice. a) Percentage of spontaneous alternation behavior in the Y-maze test at the start, four weeks, and eight weeks of the PE859 treatment (p = 0.014, two-way ANOVA, ***p < 0.001 vehicle versus 1-mg/kg PE859 and **p < 0.01 vehicle versus 3-mg/kg PE859, Bonferroni posttest). b) Total arm entry in the Y-maze test (p = 0.18, two-way ANOVA). c) Escape latency in the Morris water maze test after nine weeks of the PE859 treatment (p = 0.49, two-way ANOVA). d) Number of platform crossing in the probe trial of the Morris water maze test (p = 0.28, one-way ANOVA). e) Latency to fall in the rotarod test after nine weeks of the PE859 treatment (p = 0.11, one-way ANOVA). (f) Grip strength after nine weeks of the PE859 treatment (p = 0.44, one-way ANOVA). Mean±SEM, n = 9 (vehicle group) or 8 (1-mg/kg and 3-mg/kg PE859 groups).
Motor function of mice often affects the results of Morris water maze tests. Thus, we assessed motor functions of the mice using rotarod (Fig. 6e) and grip strength tests (Fig. 6f), but found no significant differences between treatment groups (F (2, 22) = 2.49 and p = 0.11 in rotarod tests, F (2, 22) = 0.86 and p = 0.44 in grip strength tests, one-way ANOVA). These data suggest that PE859 does not affect motor function but ameliorates cognitive dysfunctions in SAMP8 mice.
To determine amounts of insoluble and soluble Aβ in mice brains after PE859 treatments, we performed enzyme linked immunosorbent assays (ELISA; Fig. 7) and showed significant reductions in insoluble Aβ1-40 contents in PE859-treated groups (Fig. 7a) compared with the vehicle group (9082±1551 pmol/g-tissue in the vehicle group, 4728±566 pmol/g-tissue in the 1-mg/kg PE859 group, and 5620±789 pmol/g-tissue in the 3-mg/kg PE859 group, F (2, 22) = 4.24 and p = 0.029, one-way ANOVA. p < 0.05 vehicle versus 1-mg/kg and 3-mg/kg PE859 groups, Dunnett’s posttest). In contrast, soluble Aβ1-40 (F (2, 22) = 0.09 and p = 0.91, Fig. 7b), insoluble Aβ1-40 (F (2, 22) = 0.46 and p = 0.63, Fig. 7c) and soluble Aβ1-40 (F (2, 22) = 0.45 and p = 0.64, Fig. 7d) contents did not differ significantly between treatment groups. In further experiments, we evaluated the effects of PE859 on AβPP processing by detecting AβPP (Fig. 7e) and sAβPPβ (Fig. 7f) proteins using western blotting analyses. In these experiments, AβPP and sAβPPβ contents did not differ significantly between treatment groups (AβPP, F (2, 22) = 0.64 and p = 0.54; s AβPPβ, F (2, 22) = 0.36 and p = 0.70), suggesting that PE859 does not affect Aβ production but does inhibit Aβ aggregation.

The amount of Aβ and AβPP in the brains of SAMP8 mice. a) Insoluble Aβ1-40, (p = 0.029, one-way ANOVA, *p < 0.05 vehicle versus 1-mg/kg PE859 and 3-mg/kg PE859, Dunnett’s posttest). b) Soluble Aβ1-40 (p = 0.91, one-way ANOVA). c) Insoluble Aβ1-40 (p = 0.63, one-way ANOVA). d) Soluble Aβ1-40 (p = 0.64, one-way ANOVA). e) AβPP (p = 0.54, one-way ANOVA). f) sAβPPβ (p = 0.70, one-way ANOVA). Mean±SEM, n = 9 (vehicle group) or 8 (1-mg/kg and 3-mg/kg PE859 groups).
In further studies, we quantified insoluble and soluble tau proteins in brains after PE859 treatment using western blotting (Fig. 8). Similar to observations in AD patients and tau transgenic mice, highly-aggregated sarkosyl-insoluble tau was detected in the brains of SAMP8 mice (Fig. 8a). In addition, PE859-treated SAMP8 mice showed significant reductions in sarkosyl-insoluble tau protein contents compared with the vehicle group (F (2, 22) = 6.95 and p = 0.005, one-way ANOVA, p < 0.05 vehicle versus 1-mg/kg PE859 and p < 0.01 vehicle versus 3-mg/kg PE859, Dunnett’s posttest). In contrast, amounts of moderately aggregated, sarkosyl-soluble tau (Fig. 8b) and non-aggregated, Tris buffer-soluble tau (Fig. 8c) did not differ significantly between treatment groups (sarkosyl-soluble tau, F (2, 22) = 1.16 and p = 0.33, Tris buffer-soluble tau, F (2, 22) = 0.59 and p = 0.56).
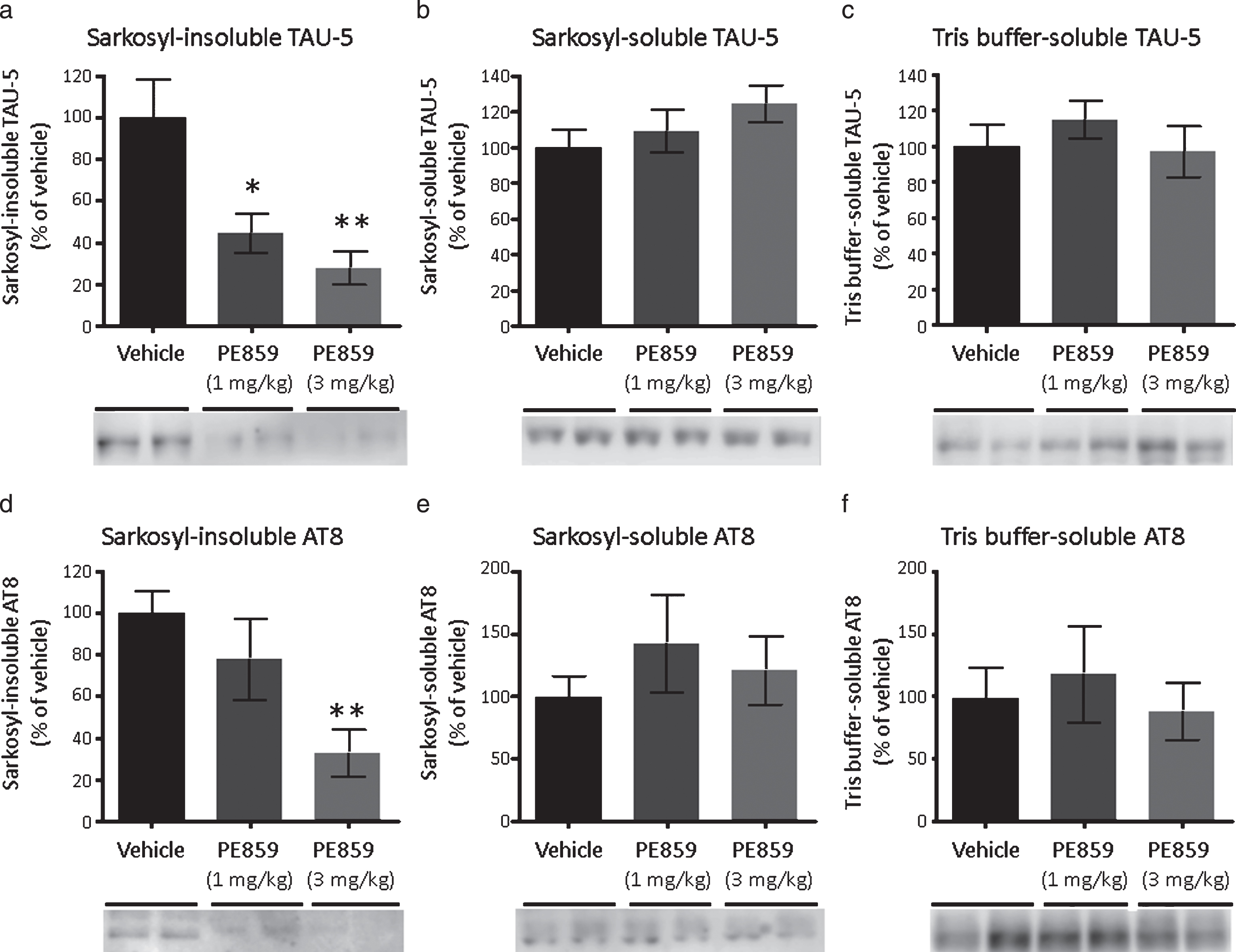
The amount of tau in the brains of SAMP8 mice. (a) Sarkosyl-insoluble tau detected by TAU-5 (p = 0.005, one-way ANOVA, *p < 0.05 vehicle versus 1-mg/kg PE859 and **p < 0.01 vehicle versus 3-mg/kg PE859, Dunnett’s posttest). (b) Sarkosyl-soluble tau detected by TAU-5 (p = 0.33, one-way ANOVA). (c) Tris buffer-soluble tau detected by TAU-5 (p = 0.56, one-way ANOVA). (d) Sarkosyl-insoluble phosphorylated tau detected by AT8 (p = 0.012, one-way ANOVA and **p < 0.01 vehicle versus 3-mg/kg PE859, Dunnett’s posttest). (e) Sarkosyl-soluble phosphorylated tau detected by AT8 (p = 0.55, one-way ANOVA). (f) Tris buffer-soluble phosphorylated tau detected by AT8 (p = 0.78, one-way ANOVA). Mean±SEM, n = 9 (vehicle group) or 8 (1-mg/kg and 3-mg/kg PE859 groups).
Finally, we compared the level of phosphorylated tau in each fraction. As shown in Fig. 8d, sarkosyl-insoluble phosphorylated tau was significantly decreased in the 3-mg/kg PE859 group compared with the vehicle group (F (2, 22) = 5.55 and p = 0.012, one-way ANOVA, p < 0.01 vehicle versus 3-mg/kg PE859, Dunnett’s posttest). In contrast, sarkosyl-soluble phosphorylated tau (Fig. 8e) and Tris buffer-soluble phosphorylated tau (Fig. 8f) contents did not differ significantly between treatment groups (sarkosyl-soluble tau, F (2, 22) = 0.60 and p = 0.55, Tris buffer-soluble tau, F (2, 22) = 0.25 and p = 0.78), suggesting that PE859 does not affect tau phosphorylation but inhibits tau aggregation.
DISCUSSION
SP formation is a major hallmark of AD and has occurs in the brain many years prior to the onset of AD [12], and the main component of SP is Aβ. Oligomeric Aβ aggregates, especially dimers and trimers, cause neurotoxicity [8], reduce hippocampal long-term potentiation [30], and disrupt cognitive function [31]. Larger dodecamers also induce neuronal loss and disrupt memory [32]. Thus, Aβ aggregation is considered key to the onset and progression of AD.
Previously, we designed and synthesized the compound PE859 from curcumin as a seed compound [16]. Curcumin reportedly reduced Aβ accumulation in AβPP transgenic mice model [17] and inhibited Aβ deposition in a rat model with intraventricular infusion of Aβ [33]. Therefore, curcumin is a candidate inhibitor of Aβ aggregation. However, curcumin has poor bioavailability when administered orally because of its low absorption [34] and rapid metabolism [35]. Thus, we developed PE859 to overcome these disadvantages of curcumin and showed that PE859 is a more potent inhibitor of Aβ aggregation than curcumin in vitro, and has better pharmacokinetic properties and blood-brain barrier permeability than curcumin [16]. Because curcumin is a well-known antioxidant and anti-inflammatory agent [36, 37], PE859 may synergistically improve cognitive dysfunction via similar mechanisms, although further research is necessary to confirm these assertions.
In the present study, PE859 inhibited Aβ oligomerization in some in vitro assays (Figs. 1–4). Specifically, PE859 inhibited the increase in ThT fluorescence due to Aβ aggregation (Fig. 1). Because ThT fluorescence is enhanced upon binding to β-sheet structures of proteins, these data indicate that PE859 binds Aβ and inhibits its β-sheet formation aggregation. In addition, PE859 prevented Aβ aggregation into trimeric and tetrameric forms (Fig. 3). Moreover, PE859 inhibited Aβ-induced cytotoxicity in SH-SY5Y cells, (Fig. 5), further suggesting that PE859 binds Aβ and decreases the cytotoxicity of oligomeric Aβ. However, the ensuring molecular basis of the interactions between PE859 and Aβ remain unclear, and further research is required.
Oral treatments with PE859 in SAMP8 mice resulted in significant improvements in cognitive dysfunction in Y-maze tests (Fig. 6). Moreover, in the Morris water maze tests, PE859 tended to improve cognitive dysfunction (Fig. 6c, d), although no significant differences were identified, potentially indicating that the present mouse numbers (8–9 per group) were insufficient for water maze tests and that cognitive function of SAMP8 mice should be evaluated by other methods.
In Y-maze tests, total numbers of arm entries tended to decrease in PE859-treated groups (Fig. 6b). SAMP8 mice are normally restless and mice of the vehicle group ran around frequently in the Y-maze whereas PE859-treated SAMP8 mice were relatively sedentary. However, decreased total numbers of arm entries of PE859-treated mice were unlikely due to differences in motor function because this did not differ between treatment groups in rotarod tests (Fig. 6e) and grip strength tests (Fig. 6f). Hence, PE859 might have calming effects that lead to decreased total numbers of arm entries.
Analyses of proteins in SAMP8 mice revealed that PE859 treatments led to decreased insoluble Aβ (Fig. 7a) and insoluble tau (Fig. 8a) levels. In contrast, no significant differences between soluble Aβ (Fig. 7b) and soluble tau protein levels (Fig. 8b, c) were observed. Moreover, PE859 had no effects on AβPP processing (Fig. 7e, f) or tau phosphorylation (Fig. 8f). These results suggest that PE859 may protect nerve cells from damage and prevent subsequent cognitive dysfunction by inhibiting the aggregation of Aβ and tau. In this study, we could not detect SP or NFT in brain sections using immunostaining with the anti-mouse Aβ antibody D12B2 or the anti-phosphorylated tau antibody AT8 (data not shown). Hence, aggregates of Aβ and tau are likely present at undetectable levels in the brains of these animals. Alternatively, SP and NFT might not be formed in SAMP8 mice brains. Thus, further evaluations of the efficacy of PE859 may be required in transgenic mice models that have detectable form SP and NFT in the brain. In brains of PE859-treated mice, amounts of insoluble Aβ1-40 were dramatically decreased compared with those of insoluble Aβ1-40 (Fig. 7). Moreover, in ThT assays, PE859 showed a higher inhibitory activity against Aβ1-40 aggregation than against Aβ1-40 aggregation (Fig. 1). Thus, PE859 may have higher binding affinity for Aβ1-40 than for Aβ1-40. In addition, soluble Aβ1-40 was present at three times the concentration of Aβ1-40 in SAMP8 mice brains (Fig. 7b, d), suggesting that most PE859 binds Aβ1-40 preferentially, with little inhibition of Aβ1-40 aggregation. Aβ1-40 is considered more neurotoxic than Aβ1-40, warranting the development of novel compounds with higher specificity for Aβ1-40 aggregation, inhibitory effects against Aβ1-40 and tau aggregation, and favorable pharmacokinetic and safety profiles. Such compounds may be more suitable candidates for AD therapy than PE859; therefore, we will continue to develop novel compounds.
In the last 15 years, more than 100 clinical trials have been conducted in AD patients [38] and about 100 new drugs are currently in clinical trials [39]. Among these blockers of Aβ cascades such as β/γ-secretase inhibitors and Aβ immunotherapies remain largely unsuccessful. Most clinical trials of AD have targeted mild-moderate AD patients. However, histochemical analyses of brains from AD have shown that numbers and sizes of SP do not continue to increase throughout the illness [40], indicating that Aβ plays a pathogenic role in the early stages of AD, but not in later stages. Therefore, AD drugs that target only Aβ may only have beneficial effects at very early stages, in which cognitive impairments are mild or undetectable.
The other important AD target tau is a main component of NFT, and similar to Aβ, tau oligomers are more toxic than tau monomers or fibrils [41]. Although it remains contentious whether NFT is the cause of neural cell death or an effect of protective neuronal responses, tau is considered a less important target than Aβ because Aβ pathology precedes tau pathology [5]. Moreover, Aβ reportedly promotes abnormal phosphorylation of tau [42, 43], although anti-Aβ therapies may not ameliorate tau pathologies because tau accumulation occurs independently of Aβ cascades. In addition, tau accumulates without Aβ in the brains of patient with tauopathies, and time-space distribution patterns of Aβ plaques and NFT differ in AD brains [12, 44]. Cognitive declines in MCI and AD correlate with early pretangle formation of tau [45] and previous studies suggest that tau is essential for Aβ-induced neurodegeneration in AD. Specifically, knockout of tau protein conferred resistance to Aβ toxicity in cultured neurons [46] and in AD model mice [47]. Hence, tau-targeting drugs may be more suitable for the prevention of cognitive impairments than Aβ-targeting agents.
Methylene blue and LMTX® are well-known tau aggregation inhibitors [48], but have not yet been developed as therapeutic agents. Moreover, drugs that target only tau may not fully address AD because Aβ induces neural disorders via mechanisms other than tau phosphorylation. Among these, Aβ induces synapse loss by downregulating cAMP regulatory element binding protein [49] or by activating N-methyl-D-aspartate-type glutamate receptors [50]. Aβ also activates inflammatory pathways by upregulating macrophage-colony stimulating factor [51] and inducing mitochondrial oxidative damage [52]. Therefore, inhibition of both Aβ and tau pathologies is required for effective treatment of AD. Accordingly, the best-known Aβ aggregation inhibitor tramiprosate failed to show sufficient efficacy in a clinical trial of AD patients [53], potentially reflecting inhibition of Aβ aggregation but promoting tau aggregation [54]. In contrast, PE859 inhibits the aggregation of both Aβ and tau and has shown a trend for improvement in cognitive dysfunction in vivo. Hence, we suggest that dual inhibitors of Aβ and tau aggregation, such as PE859, will be more effective for the treatment of AD than inhibitors of Aβ or tau alone.