
Abstract
Alzheimer’s disease prevalence has reached epidemic proportion with very few treatment options, which are associated with a multitude of side effects. A potential avenue of research for new therapies are protons, and their associated receptor: acid-sensing ion channels (ASIC). Protons are often overlooked neurotransmitters, and proton-gated currents have been identified in the brain. Furthermore, ASICs have been determined to be crucial for proper brain function. While there is more work to be done, this review is intended to highlight protons as neurotransmitters and their role along with the role of ASICs within physiological functioning of the brain. We will also cover the pathophysiological associations between ASICs and modulators of ASICs. Finally, this review will sum up how the studies of protons, ASICs and their modulators may generate new therapeutic molecules for Alzheimer’s disease and other neurodegenerative diseases.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive and irreversible disease that slowly impairs cognitive function, ranging from mild memory loss to a complete lack of independence to achieve the simplest of daily tasks [1, 2]. AD has become a very costly issue on multiple levels: (i) human cost: AD is now ranked as the 6th leading cause of death in the United States, and has been associated with an increased mortality rate in those aged 70 and older; caregivers are under high level of stress, suffer from depression, and put their health in second place; (ii) economical cost: AD care cost have been estimated at $236 billion in 2016 and are projected to grow to $1 trillion by 2050 impacting not only the nation but also the family of the patients [3, 4]. This disease is of global concern and has become a worldwide epidemic with increased prevalence also noted in Europe, Latin America, China and India [5–8].
This devastating disease is incurable and has been associated with a multitude of risk factors, aging being the highest [9]. While aducanumab, a new human monoclonal antibody that only targets the aggregated form of amyloid beta, has recently shown potential as an intervention for AD [10], only two types of treatments to alleviate cognitive symptoms associated with AD have been approved by the Food and Drug Administration and have been used alone or in combination: cholinesterase inhibitors (Donezepil, Galantamine, and Rivastigmine) for mild to moderate AD and N-methyl-D-aspartate antagonist (Memantine) for moderate to severe AD [11]. These treatments involve classical neurotransmitters (acetylcholine and glutamate), and have been associated with numerous adverse effects. Non-classical neurotransmitters may also have a role in early detection, symptom management, quality of life improvement, and slowing down the progression of the disease.
One overlooked non-classical neurotransmitter is the proton (H+). In order for protons to be classified as a neurotransmitter, they must meet specific criteria [12]. A neurotransmitter is defined by the following criteria: (1) be found in presynaptic neuron, (2) be released in response to presynaptic depolarization, and (3) elicit a response in a specific postsynaptic receptor. Protons have, at best, been considered “putative” or generally accepted neurotransmitters due to their effect on receptors and ion channels in vitro. In 2014, Du et al. provided a convincing and compelling argument using in vivo evidence to support the case for protons as neurotransmitters, as the agonist for a specific acid-sensitive ion channel [13]. In brief, Du et al. stimulated presynaptic neurons in lateral amygdala brain slices and observed the electrical response in postsynaptic neurons using whole-cell patch-clamp electrophysiology. Following stimulation, the postsynaptic acid-sensing ion channels were activated and a current was observed. This current was absent in ASIC1a –/– knockout mice brain slices. In this experiment, Du and colleagues provide evidence to support the consideration of protons as neurotransmitters since this cation was released following presynaptic stimulation and activated a postsynaptic receptor.
This review will provide a discussion on protons as neurotransmitters and their potential role in brain function and AD. We will first provide a detailed description of protons and the protein interaction with a receptor sensitive to these small ligands. Then, we will describe the role of protons, pH changes and how it affects receptor function and ultimately brain function in normal individual and in AD patients.
PROTONS
The proton (H+) is a hydrogen atom that has lost an electron and the remaining charge associated with this ion is found in the atomic nucleus. In chemistry textbooks, we see that a water molecule is in equilibrium with a hydrogen ion (H+) and a hydroxyl ion (OH–). In actuality, the presence of the simple proton is rare and the most represented species is the positively charged hydronium ion (H3O+). Changes in pH, i.e., H+ concentration, influence multiple facets of cell survival and organism subsistence. Therefore, pH regulation is vital to homeostasis and important for brain function as neuronal activity may be influenced. pH regulation is achieved intracellularly via buffering systems such as Na+/H+ exchange, Na+-driven HCO3– transport, Cl–/HCO3– exchange, and ATP-driven H+ pump [14].
Protons come from metabolic reactions in cells and the solubilization of acids in water. What makes protons fascinating compared to ligands that are passed from cell to cell or used as signal messengers is that they are always present in the environment. The extracellular environment can be measured in pH units, the inverse log of proton concentration and can run along the pH continuum, from higher pH yielding an alkaline environment to lower pH producing an acidic environment. Even if the extracellular environment reaches as high as pH 8, there are still protons present (a low proton concentration of 0.01μM). In some aspects, the pH spectrum to a cell is similar to the range of membrane potential in excitable cells: some level of stimulus is always present. Membrane potential does not disappear, similar to pH values, but the quantity changes in response to external factors.
Classical neurotransmitters are packaged into vesicles through the use of vesicular transporters. In the case of neurotransmitter vesicle packing, these transporters, or antiporters, use a proton gradient to move neurotransmitters into the vesicle. Here, protons are pumped into the vesicle using vacuolar ATPases [15]. The influx of protons lowers intra-vesicular pH. Next, neurotransmitters are moved into the vesicle using antiporters that exploit the high concentration of protons in the vesicle as the driving force. The outward movement of protons provide the driving force for the shuttling of neurotransmitter in the vesicle. When these neurotransmitter vesicles release their contents into the synapse, the remaining protons in the vesicle are released [16]. These receptors are susceptible to the proton influence if they have acidic residues (e.g., glutamate and aspartate) or charged residues (e.g., histidine) in the protein’s extracellular domain.
Once released into the synapse, protons do not move like typical neurotransmitters. Neurotransmitters, like glutamate, diffuse across the synapse. Protons do not move this way. Due to their affinity for water molecules (H2O) to form the hydronium ion (H3O), C.J.T. de Grotthuss described protons moving along wires of water molecules [17, 18]. Instead of moving, de Grotthuss postulated that protons “hop” from one water molecule to the next. If we were to line up a series of six water molecules in a straight line and a proton were to bind to the first water molecule, a proton would come off the 6th water molecule in the line. Thus, protons once released from the presynaptic neuron would very quickly appear at the postsynaptic side and bind to receptors.
Protons can alter the activity of membrane receptors, but most of these interactions are indiscriminate. For protons to be considered a neurotransmitter, they require a specific postsynaptic receptor that is activated by proton binding. There is a postsynaptic receptor that serves this purpose for protons. These receptors are the acid-sensing ion channels.
Acid-sensing ion channel
Acid-sensing ion channels (ASICs) are sodium, and in some cases calcium, permeable channels that are formed from three protein subunits [19]. First identified as a proton mediated current in neurons [20] and subsequently cloned [21, 22], ASICs are members of the larger epithelial sodium channel/Degenerin (ENaC/DEG) family of ion channels, which are involved in sodium homeostasis [23]. Initially identified in the brain [22] and the periphery [24, 25], ASICs are ubiquitously expressed in the mammalian nervous system. There are five genes that encode 7 subunits (ACCN1 to ACCN5) [26–28]. The ASIC subunits are: ASIC1a and the splice variant 1b, ASIC2a and the splice variant 2b, ASIC3, ASIC4, and ASIC5 (the brain-liver sodium channel). The acid-sensing ion channel subunit compositionin vivo has been studied for several types of neurons [29–31]. However, the ASIC subunit composition in every neuron is not clear and could vary depending upon the neuron response to a dynamic extracellular environment. ASIC2b and ASIC4 do not form functional homomers and exert their influence through co-expression with other ASIC subunits [24, 32]. Acid-sensing ion channels sense changes in the extracellular pH environment, however individual ASIC pH sensitivity is dependent on the composition of the ASIC complex, or more specifically, which ASIC subunits are present in the functional channel [33]. ASIC1a, 1b, and 3 are activated by pH near physiological pH while ASIC2a requires much more acidic conditions (pH < 6.0) to be activated [33].
ASICs allow sodium influxes into a neuron upon activation, which then depolarize neurons and shift the membrane potential towards threshold. The resultant depolarization will increase activity of the depolarized neuron and generate one or more action potentials [34]. However, sojourns to low pH such as pH 6.0 that result in maximal ASIC activation inin vitro assays are not common in healthy individuals. Such low pH values are found in disease states, such as joint inflammation and pain [35]. Most relevant changes in pH are small and incremental (tenths of a pH unit) which may be associated with the release of protons into a synapse. These pH changes are sufficient to elicit a response in ASIC3, a response that does not desensitize and persists. For example, a shift in extracellular pH in ASIC3 from pH 7.4 to 7.2 is enough to activate the channel in a manner that does not desensitize [34]. Thus, there is a prolonged influx of sodium ions that maintain a depolarized membrane potential until the low pH environment returns to normal. The return to an inactivated ASIC will be delayed as well, limited by diffusion of protons from the region.
Action potential generation through ASIC was observed following modest incremental pH stimuli in cardiac dorsal root ganglia [34]. However, the extent of ASIC-mediated influence on generating action potential is limited and may be ASIC subtype-dependent. Low pH antagonism of voltage-gated sodium channel (Nav1.7) prevented action potential generation in naked mole rat nociceptors despite having ASIC1 present [36]. ASIC subunit dependence plays a role in the availability of protein to respond to low pH stimuli. These proton sensitive proteins are influenced by steady-state inactivation, which describes what fraction of ASIC protein is available to respond to an acidic stimulus [37–39]. All ASIC subtypes are governed by steady-state inactivation. As the extracellular environment becomes more acidic, the number of ASIC protein that will be unresponsive to the lower pH will increase. Furthermore, ASICs undergo tachyphylaxis, a reduced response to repeated low pH exposures. This ASIC proton tachyphylaxis is observed in homomeric ASIC1a, when the protein is exposed to repeated low pH stimuli (pH < 6.7) and is absent in the other ASIC subtypes [40]. Proton tachyphylaxis is absent in ASIC1a/2 heteromers and could influence neurons in vivo where both ASIC subtypes are expressed [41]. ASIC influence on neuron activity, ultimately, depends on the ASIC subtype expressed and different for each type of neuron.
Along with their permeability to sodium, ASICs, especially ASIC1a, have been shown to also flux calcium ions [42, 43]. Influx of calcium into the cytosol creating an overload lead to cytotoxicity and cell death, as seen in ischemic conditions [44]. This underlying mechanism supports a potential role of ASICs in the development of AD, as ASIC1a as particularly abundant in the brain and cell death is associated with AD. The following section will provide support for a possible role of ASICin AD.
SPECULATIVE INFLUENCE OF PROTONS IN ALZHEIMER’S DISEASE
While the notion of protons as neurotransmitters is relatively new, studies have already established a link between protons and their potential contributions to the functioning of the central nervous system and involvement in several pathological conditions. Acid-mediated currents have been measured in neurons [45], with ASIC1a being crucial as a channel subunit for these neuronal currents [46]. This particular subunit was detected in dendrites, cell body and at postsynaptic localization suggesting a potential role in synaptic transmission [47]. Using ASIC1a knock-out mice, Wemmie et al. established the importance of ASICs and protons in proper long-term potentiation [46]. Furthermore, ASIC1a–/– mice exhibited mild spatial learning and memory impairment which was reversed by intensive training, had impaired eyeblink conditioning [46] and had slower fear conditioning learning [48]. The absence of ASIC1a led to a mild impairment, which is counterintuitive to their speculated involvement in AD development. However, it does suggest that ASICs are involved in learning and memory, and that perhaps their role is more complex than a simple activation/inhibition scheme. In a normal setting, ASIC1a might be necessary to counterbalance proton-dependent inhibition of NMDA receptors, thereby allowing for proper synaptic transmission. However, under acidotic conditions, the activation of ASICs might contribute to development and furthering of pathologies. Overall, these studies suggest that ASICs and protons may be involved in various aspects of brain functioning governed by different brain regions such as hippocampus, cerebellum and amygdala which are affected in AD patients.
Protons and pH homeostasis are crucial to proper cellular function, and acidosis has been linked to a variety of conditions such as ischemia, cellular degeneration, chronic pain and seizures [49, 50]. In cell culture and rat models of ischemia, acidosis and ASICs have been implicated in the resulting injuries [43, 44]. Furthermore, ASICs and localized acidosis have been implicated in various pathologies such as Parkinson’s disease, Huntington’s disease, glioma, seizures, and others previously reviewed [51, 52]. While there has not been a direct link between AD and acidosis/ASIC, studies of neuronal death and brain function suggest that pH imbalance may play a role in AD and modulation of ASIC activities could serve as new therapeutic models. Furthermore, conditions present in AD patients have also been linked to ASIC activation. AD risk factors are associated with hypoperfusion leading to hypoxia. A study demonstrated that hypoxia treatment can potentiate AD pathogenesis, observed with increased amyloid-beta deposition and further memory deficits in a mouse model of AD [53]. Hypoxic condition leads to the production of protons and inflammatory markers such as nitric oxide, arachidonic acid, and IL-1, all of which can activate and upregulate ASICs [54]. This link between hypoxia/inflammation and ASIC activation supports the potential role of protons and ASICs in the pathogenesis of AD.
In multiple sclerosis patients, increased ASIC expression was associated with damage and neurodegeneration [55], and suggested that the use of amiloride, an antagonist of ASICs, reduced atrophy, implying a neuroprotective role for ASIC modulators. Other factors released during injury have been shown to further the toxic effects of acidosis by activating ASICs [50]. Some of these compounds may be present in the brains of AD patients and could be contributing to the associated pathologies by activating ASICs [56]. For example, lactate levels were higher in the brains of early AD patients compared to controls [57] and lactate has the capacity to modulate ASIC1a and ASIC3 activities by chelating divalent ions [58]. Certain natural compounds have been determined to lower neurotoxicity perhaps via inhibition of ASIC1a [59, 60], while others such as nitric oxide [61], arachidonic acid [62], and spermine [63] have been shown to activate ASICs and potentiate injuries. Interestingly, drugs acting on other receptors affected during AD have been shown to also interact with ASICs, i.e. memantine, an NMDA receptor antagonist, also inhibited ASIC1 [64]. Thus far, all these studies only infer that ASIC might be involved in AD due to the physiological importance in synaptic plasticity and learning and memory, and evidence of their activation in pathophysiological conditions. However, with current treatments being associated with a multitude of adverse events [65, 66], it remains important to find alternative approaches and ASIC antagonists/modulators have the potential to improve synaptic transmission and cognitive status with possibly less side effects.
To understand pH changes in relation to normal condition and disease states, non-invasive methods are needed [67]. With advances in technology, it is now possible to obtain indirect information on intracellular pH in the brain non-invasively and a few studies have used this new methodologies. In 2012, Mandal et al. derived information about cellular environment, such as pH, using multi-voxel 31P magnetic resonance spectroscopy [68], however this method has major limitations including the need for special hardware. Nevertheless, they identified differences in pH between controls and AD subjects within the hippocampal area, a region particularly affected in AD patients. More recently, 1H magnetic resonance imaging have been determined to have a higher spatial and temporal resolution that 31P spectroscopy, and changes in relaxation times, especially T1 in the rotating frame (T1ρ) have been associated with ischemic events and changes in intracellular pH [69, 70]. Furthermore, Magnotta et al. have shown that T1ρ can be used as a marker indicative of pH changes in mouse and human brains [71]. Using subjects displaying a range of cognitive impairments from control to mild AD, Ponto et al. have used T1ρ measurements and determined that poorer cognitive performance was associated with higher T1ρ, reflecting a more acidic environment (estimating about 0.5 pH unit difference between controls and mild AD based on data by Magnotta et al.) [72]. Their research also associated increased pathology (impaired glucose metabolism and amyloid burden) with declining cognitive performance (MMSE and delayed logical memory score) and increased T1ρ [73]. While these data are very preliminary, they are in synch with other results suggesting an association between pH fluctuations, cognitive performance and pathologies associated with AD, and definitely warrant morestudies.
CONCLUDING REMARKS
Research indicate that protons/pH and ASICs in particular are good candidates for intervention in AD and other neurodegenerative diseases. However, more studies are needed to decipher their role in AD, and to determine whether manipulating cellular pH, i.e. H+ concentration, can be considered a promising new avenue of research.
While directly manipulating the number of protons released or reducing the acidity of the brain might be difficult to achieve, modulating acid-sensing ion channels and other ion channel targets of protons may be a better avenue for therapeutics for AD and neurodegenerative diseases. Currently, there is one clinically approved antagonist for acid-sensing ion channels: amiloride, a diuretic that blocks epithelial sodium channels in the tubules of kidneys in an effort to control hypertension [74, 75]. However, amiloride is non-selective, does not cross the blood-brain barrier, and would have little utility for modulating ASICs expressed in the brain. Natural sources for ASIC antagonists do exist, although large-scale synthesis and administration may prove to be cost-prohibitive. For example, natural venom toxins have been identified that target and alter ASIC activity. The Trinidad chevron tarantula, Psalmopoeus cambridgei, produces a venom peptide (Psalmotoxin 1) that selectively antagonizes ASIC1a [76]. ASIC3 is antagonized by APETx2, a sea anemone toxin from the Anthopleura elegantissima [77]. The black mamba venomous snake produces Mambalgin-1 and 2 [78] while the green mamba produces Mambalgin-3 [79]. Each of these mambalgin peptides inhibit ASIC1a and 2a activity. Conversely, the Texas coral snake produces MitTx venom peptide that does not antagonize ASICs, but activates or potentiates the current generated from ASIC1a, 2a, 3 and heteromeric channel combinations [80]. These natural peptides are selective, but they may be cost prohibitive for long term therapy. Furthermore, these peptides are subject to high first-pass metabolism, and may not cross the blood-brain barrier leaving intracranial injections the only potential route of administration, limiting their chronic dosing.
Another option is the identification of novel molecules that may antagonize ASICs. An ideal candidate for attenuating the effects of ASIC would be a compound that is blood-brain barrier permeable, is selective for ASIC, and can be administered in a way that patient compliance is met. At this time, there are no such compounds available. Efforts are underway to identify compounds that influence ASIC activity and to synthesize and generate novel molecules to antagonize ASICs.
In summary, more research is needed to identify novel molecules modulating ASICs to help reducing pathologies and cognitive dysfunction associated with AD and other neurodegenerative diseases (Fig. 1).
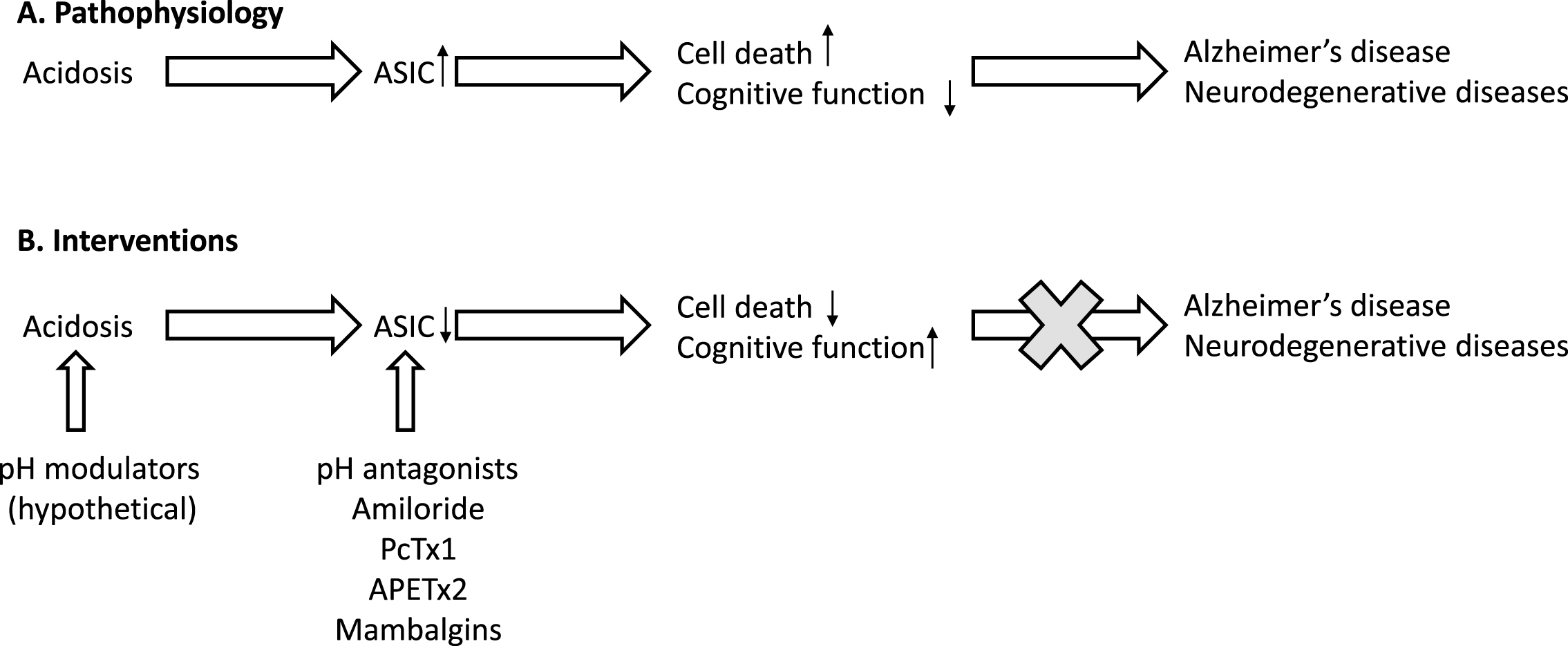
Relationship between acidosis, ASIC, and Alzheimer’s disease.
ACKNOWLEDGMENTS
This work was supported by institutional funds (EBG and NS) and P01 AG027956 (NS).
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-1131r2).