
Abstract
While hypertension has been shown to be a risk factor for vascular dementia, several studies have also demonstrated that hypertension also increases the risk of Alzheimer’s disease (AD). Although the relationship between visit-to-visit blood pressure variability (VVV) and cognitive impairment, including AD, have been provided, the mechanisms remain poorly understood. This review paper focuses on the relationship of VVV with AD and summarizes the pathophysiology underlying that relationship, which appears to be mediated by arterial stiffness.
Keywords
INTRODUCTION
While hypertension is an important predictor of cardiovascular events [1], blood pressure (BP) fluctuation is caused by complex interactions of external environmental stimuli and internal physical status. BP variations can emerge when subjects are observed over the course of repeated clinical visits. Independent of the average BP level, visit-to-visit BP variability (VVV) was shown to be a predictor of stroke [2].
One major cause of death and disability is vascular disease of the brain [1]. In a magnetic resonance imaging (MRI) study, white matter lesions that are frequently seen in the elderly were shown to be associated with vascular dementia (VaD) [3]. While hypertension is known to be associated with VaD, several studies have also demonstrated the relationship of hypertension with Alzheimer’s disease (AD) [4, 5]. In fact, since Alois Alzheimer first described AD in 1906, white matter lesions have been well known to pathologists. However, a determination about whether white matter alterations are the cause of the neurodegenerative disease was not made at that time [6].
While several studies have shown that VVV had a relationship with cognitive impairment, we have observed independent associations of VVV with dementia [7 –9]. Cerebral small vessel disease was suggested to have a pivotal role in the pathophysiology of dementia [7 –9]. However, the impact of VVV on AD has received less attention. We hypothesized that higher VVV is an upstream risk factor of arterial stiffness and AD, and that arterial stiffness mediates the relationship between higher VVV and AD. From these standpoints, this review paper aims to assemble and evaluate the evidence regarding the relationship between VVV and AD, and summarize the impact of VVV on AD in relation to arterialstiffness.
In most studies, while the average BP was calculated as the average level of BP readings over multiple visits, the standard deviation (SD) and coefficients of variation (CV) [SD/average BP value over multiple visits×100 (%)] were representative measures of VVV [9].
References for this review were identified from a search of PubMed to March 2017 using “visit-to-visit blood pressure variability”, “cognitive impairment”, “dementia”, and/or “Alzheimer’s disease” as search terms.
VISIT-TO-VISIT BP VARIABILITY, COGNITIVE IMPAIRMENT, AND BLOOD PRESSURE LOWERING MEDICATION
VVV was shown to have an association with cognitive impairment in several studies [10 –15]. Recently, it has been investigated whether the relationship between VVV and cognitive impairment is moderated by BP-lowering medication (BPLM)[16, 17].
Visit-to-visit BP variability and cognitive impairment (Table 1)
In the HiroShima-Shobara-Soryo COhort (3SCO) study [10], we investigated the relationship between VVV and cognitive function among 201 elderly patients at high risk of cardiovascular disease. The coefficient of variation of systolic BP (CV SBP) and Δ SBP (maximum–minimum) values were significantly associated with lower Mini-Mental State Examination scores, independently of averageBP [10].
Visit-to-visit blood pressure variability and cognitive impairment
BP, blood pressure; CI, confidential interval; CV, coefficient of variation; MMSE, Mini-Mental State Examination; GDS, Global Deterioration Scale; SD, standard deviation; SBP, systolic blood pressure; ARV, average real variability; DBP, diastolic blood pressure; VVV, visit-to-visit variability.
In the PROspective Study of Pravastatin in the Elderly at Risk (PROSPER) study of 5,461 elderly participants at risk of cardiovascular disease, who were free from cognitive impairment at baseline, a higher VVV in SBP readings was associated with worse cognitive performance during an average of 3.2 years [11].
In the Three-City Study of 6,506 elderly individuals followed-up for 8 y, 474 participants developed dementia. The risk of dementia for those in the highest decile of CV SBP was higher than that for the lowest decile [12].
In the Coronary Artery Risk Development in Young Adults (CARDIA) study of 2,326 healthy young adults for eight visits during 25 y, higher VVV was significantly associated with lower scores on cognitive function [13].
In the Ongoing Telmisartan Alone and in Combination with Ramipril Global Endpoint Trial (ONTARGET) and the Telmisartan Randomized Assessment Study in ACE Intolerant Subjects with Cardiovascular Disease (TRANSCEND), which enlisted 24,593 patients without preexisting cognitive dysfunction, the CV SBP was a significant predictor of cognitive dysfunction [14].
In the China Health and Nutrition Survey (CHNS) of 976 community-dwelling older adults, higher VVV in SBP was significantly associated with a faster decline of cognitive function[15].
In the elderly populations, VVV in BP has been shown to be significantly associated with cognitive impairment. In addition, VVV in BP in the young might serve as a predictor of cognitive impairment at middle age.
Visit-to-visit BP variability and cognitive impairment according to blood pressure-lowering medication
In the PROSPER study, patients taking calcium channel blockers (CCBs) had a high average score in the baseline Letter-Digit Coding test, while those with beta-blockers and renin-angiotensin system inhibitors had a steep decline in Stroop test [16]. Although VVV in SBP was higher in those with beta-blockers and renin-angiotensin system inhibitors, beta estimates in the association between VVV and cognitive function remained the same, even with adjustment for BPLM [16]. In the Three-City study, although non-dihydropyridine CCBs and loop diuretics were associated with a lower dementia risk, neither of CV SBP by BPLM interaction terms were statistically significant [17]. Thus, the association between VVV and cognitive impairment was not mediated by BPLM.
PATHOPHYSIOLOGICAL MECHANISMS INVOLVED IN THE RELATIONSHIPS OF INCREASED VISIT-TO-VISIT BP VARIABILITY WITH COGNITIVE IMPAIRMENT
Arterial stiffness
Higher VVV has been shown to have a significant relationship with cognitive impairment in the high-risk population with cardiovascular disease. The underlying mechanisms are not clearly understood.
The stiffness parameter β has frequently been used to examine the elastic properties of the carotid artery [18], and increased stiffness β-values are associated with coronary artery disease [19].
In the 3SCO study, VVV was found to be a significant determinant of atherosclerosis and stiffness in the common carotid artery [20]. In addition, the relationship of VVV with stiffness in the common carotid artery was prospectively investigated in 164 elderly subjects with one or more cardiovascular risk factors [21]. In the mean 4.2-year follow-up, both SBP and its SD had significant associations with the stiffness parameter β at the follow-up, after adjusting for confounders. The patients with high SD SBP and high average SBP had an increased stiffness parameter β at follow-up, specifically in the group without CCBs (Fig. 1) [21, 22].
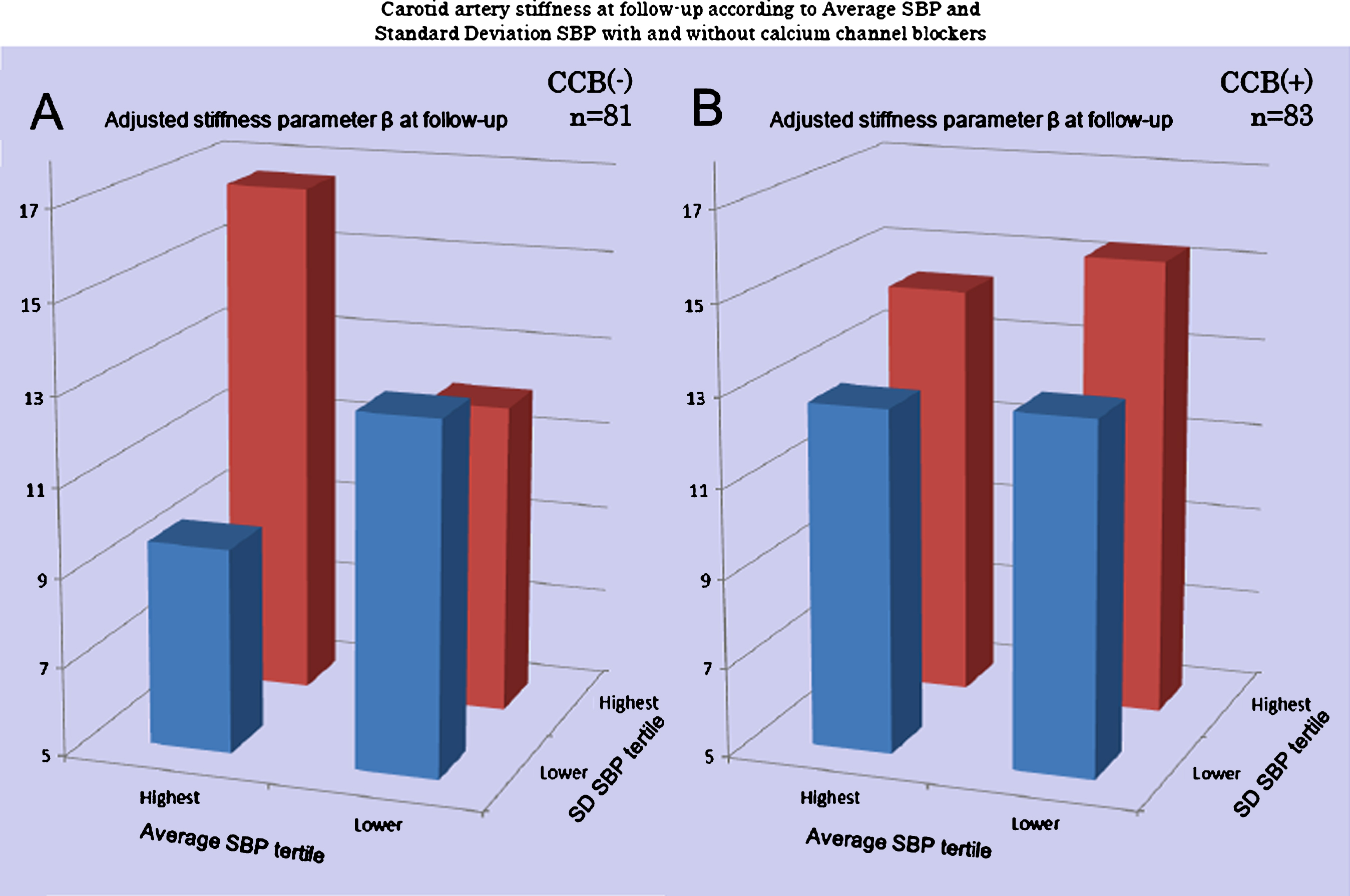
Carotid artery remodeling at follow-up according to average SBP and standard deviation of SBP. A significant difference in the stiffness parameter β between baseline and follow-up adjusted for age, smoking, HDL, follow-up period (p = 0.02) was observed in the non-CCB group (A) comparison of four groups according to the highest/lowest tertile of average SBP and SD SBP using analysis of covariance (ANCOVA), while no significant difference (p = 0.53) in the CCB group was observed. SBP, systolic blood pressure; CCB, calcium channel blocker; SD, standard deviation; CV, coefficient of variation; CI, confidence interval. From Nagai et al. [22].
In patients with type 2 diabetes, CV SBP has a positive correlation with brachial-ankle pulse wave velocity (PWV) [23]. In African Americans, a negative significant correlation between VVV in SBP and the flow-mediated/nitroglycerin-mediated dilation ratio was shown [24].
In the Multi-Ethnic Study of Atherosclerosis (MESA) of 2,640 participants, SD SBP was inversely associated with aortic distensibility independent of average SBP level and BPLM use in the cross-sectional analysis [25]. In addition, Tedla et al. [26] investigated the association between long-term VVV in SBP and 10-year percent change in arterial stiffness among 1122 middle-aged individuals who were not taking antihypertensive medications. In a multivariate linear regression model, individuals in the fifth quintile of SD, variability independent of the mean, and CV SBP had a higher decline in the distensibility coefficient and a higher progression in Young’s elastic modulus than those in the first quintile, respectively, after 10 years of follow-up, independent of average SBP [26].
Synergistic impact of visit-to-visit BP variability and artery remodeling on cognitive impairment
Although a relationship between increased PWV and cognitive impairment has been reported [27 –29], the combined impact of VVV and arterial stiffness on cognitive impairment has not been analyzed. In the 3SCO study, patients with both high ΔSBP and high stiffness parameter β had a higher prevalence of cognitive impairment than patients with both a low ΔSBP and low stiffness parameter β [30]. A significant interaction was found in the relationship between ΔSBP and stiffness parameter β for cognitive impairment [30].
VISIT-TO-VISIT BP VARIABILITY AS A CAUSAL FACTOR OF ALZHEIMER’S DISEASE
Although VVV was suggested to have a close association with VaD, several studies have shown VVV as a causal factor of AD.
Qiu et al. [31] reported that BP markedly decreased over the 3 years before dementia set in. Among subjects with baseline SBP <160 mm Hg, SBP decline ≥15 mmHg occurring 3 to 6 years before diagnosis was associated with a relative risk of 3.1 forAD [31].
In a magnetic resonance imaging substudy of 553 participants in the PROSPER study, higher VVV in both SBP as well as DBP had significant association with lower hippocampal volume [11]. In the Three-City Study of 6,506 elderly subjects, a higher CV of SBP was significantly associated with an increased risk of AD [12].
In the Alzheimer’s Disease Neuroimaging Initiative (ADNI) study of 626 individuals with a screening diagnosis of mild cognitive impairment (MCI) or normal cognitive function, high VVV in SBP during the 3 years had a significant association with worse global and executive function and episodicmemory [32].
In a cohort of 70 patients with mild or moderate AD and 140 controls with normal cognitive function followed for 6 months [33], higher VVV in both SBP and DBP were observed in AD patients than in the controls. All of the VVV indices were significantly associated with AD in the regression models. While the VVV in SBP was associated with the rate of cognitive impairment in AD [34], it was not significantly associated with cognitive impairment in patients with frontotemporaldementia [35].
PATHOPHYSIOLOGY LINKING VISIT-TO-VISIT BP VARIABILITY AND ALZHEIMER’S DISEASE
Amyloid β and arterial stiffness
The pathogenetic cascade of neurotoxicity in AD starts from the extra and intraneuronal accumulation of amyloid β-peptide (Aβ) [36]. Aβ toxicity spreads from the entorhinal cortex, and then involves neurons of other areas. Neurofibrillary tangles (NFT) that consist of hyperphosphorylated microtubule-associated protein Tau are known to be a second histopathological hallmark inAD [36].
Neuroimaging [37] as well as postmortem histopathological [38] studies have reported that some degree of vascular pathology is observed in up to one-third of AD, while AD pathology was observed in a similar proportion of VaD cases. Vasoconstriction in the human cerebral arteries was enhanced by Aβ [39]. Because Aβ was shown to produce reactive oxygen radicals, a reduction in endothelium-mediated vasodilatation was shown to be related to Aβ in the cerebral arteries [40]. Angiotensin II was shown to induce SBP elevation, which resulted in impaired CBF in a transgenic model of AD [41, 42]. Wang et al. screened 55 clinically prescribed antihypertensive medications for AD-modifying activity using primary cortico-hippocampal neuron cultures generated from the Tg2576 AD mouse model. Through in vitro studies, only valsartan was found to be capable of attenuating oligomerization of Aβ peptides into high-molecular-weight oligomeric peptides, known to be involved in cognitive deterioration. Preventive treatment of Tg2576 mice with valsartan significantly reduced AD-type neuropathology and the content of soluble high-molecular-weight extracellular oligomeric Aβ peptides in the brain [43]. In addition, valsartan prevents AlCl3- and d-gal-induced cognitive decline, partly by restoring cholinergic function and attenuating oxidative damage [44]. In transgenic mice in which mutated forms of amyloid-β protein precursor (AβPP) were overexpressed, misfolded Aβ originating from AβPP was shown to reduce CBF [40, 45] and to impair autoregulation in the cerebral circulation [46]. In mice with bilateral common carotid artery stenosis, reduced cerebral perfusion resulted in accelerated Aβ deposition [46]. In addition, the APOE ɛ4 allele was shown to have a pivotal role in plaque formation [47]. The blood Aβ40 level had a significant positive association with cardiovascular death, the progression of arterial stiffness, and the incidence of subclinical atherosclerosis [48]. While hypoperfusion as well as hypoxia enhanced Aβ production, arterial stiffness and reduced microvascular function were shown to impair Aβ clearance and to elevate cerebral Aβlevels [49].
Arterial stiffness and Alzheimer’s disease
Pulse pressure and Alzheimer’s disease
In the Kungsholmen study [50], there was a U-shaped relationship between level of pulse pressure (PP) and incidence of AD. In the retrospective cohort study, 877 participants without dementia (55–91 years of age) from the ADNI underwent a baseline health assessment [51]. Individuals with a cerebrospinal fluid P-tau-positive biomarker profile exhibited mean PP elevation regardless of age [51].
Higher PP is associated with increased risk for AD and dementia in the elderly, and is suggested to be caused by arterial stiffness and severe atherosclerosis [50]. Poor cerebral perfusion might explain the association between lower PP and increased risk for dementia [50].
Pulse wave velocity and Alzheimer’s disease
In 308 elderly subjects (AD, 41% : VaD, 6% : MCI, 27% : normal cognitive function, 26%), subjects with VaD or AD had significantly higher PWV than those without cognitive impairment, adjusted for confounders [52]. Adjusted odds ratios were 1.73 for AD and 3.52 for VaD for each 2 m/s increment in carotid-femoral PWV [52].
In the 1,101 dementia-free Framingham Offspring study participants [53], after adjustment for age and gender, higher carotid-femoral PWV predicted an increased risk of MCI and AD [53].
A relationship between arterial stiffness and cerebral Aβ deposition was reported in a group of 91 dementia-free elderly subjects [54]. Positron emission tomography scans using the Pittsburgh compound B showed 44 subjects with Aβ-positivity. For 1 SD increment in brachial-ankle PWV, a twofold increase in the odds ratio of Aβ-positive status was observed. For each 1 SD increment in PWV, a two- to four-fold increase in the odds ratio of being Aβ-positive and having high WMH burden was observed compared to Aβ-negative individuals having a low WMH burden. In addition, a higher carotid-femoral PWV was shown to be associated with medial temporal lobe atrophy [55] and reduced integrity in white matter and gray matter, all considered components of AD pathophysiology [56].
Cerebral blood flow and Alzheimer’s disease
Cerebral autoregulation aims to stabilize blood flow to the brain during variations in perfusion pressure, thus protecting the brain against the risks of low or high systemic BP. Roher et al. [57] revealed that total cerebral blood flow (CBF) was 20% lower in AD patients than in the non-demented control group and that these values were directly correlated with PP. The AD group had a significantly lower PP [57]. During repeated sit-stands, BP variability up to 20% of the baseline led to larger fluctuations of CBF in AD than in controls [58].
On the other hand, long-term angiotensin-converting enzyme inhibitor (ACE-I) (perindopril)-based therapy was shown to have a beneficial effect on cerebral circulation by improving the cerebral perfusion reserve in patients with previous minor stroke, although SBP/DBP and CBF during normocapnia showed no significant changes between entry and completion of the trial in the perindopril and placebo groups [59].
Zazulia et al. [60] investigated 20 patients with mild AD. Using 15O-positron emission tomography, the changes in CBF following a step decrease in systemic BP induced by intravenous infusion of CCB nicardipine were measured. Cerebral perfusion was well maintained in patients with AD despite a mean reduction in arterial pressure [60].
Cerebral autoregulation was shown to be impaired in AD patients, although some preliminary studies suggest that cerebral autoregulation is preserved in patients with AD [61]. In the report by Zazulia et al. [60], nicardipine was used to decrease BP, through systemic vasodilation including the cerebral vasculature. The stability in CBF in response to reduced BP was due to cerebral vasodilatation that was mediated directly by nicardipine. Nicardipine induced vasodilatation to a degree that matches exactly what would be needed to compensate for the reduction in perfusion pressure [61]. Use of some classes of BP-lowering agents such as ACE-Is or CCBs might have a protective impact on cerebral circulation that is independent of their BP-lowering effects [59, 61].
Antihypertensive medication, visit-to-visit BP variability, and Alzheimer’s disease
In a 291-subject postmortem study, Hoffman et al. [62] reported that there was substantially less AD neuropathology, as quantified by neuritic plaque and NFT densities, in the medicated hypertensives groups than in the non-hypertensives, suggesting the salutary effect of antihypertensive medication against AD-associated neuropathology [62].
In the nonrandomized Cache County Study, diuretics, specifically potassium-sparing diuretics, were associated with reduced incidence of AD [63]. Specifically, 3,417 participants had a mean of 7.1 years of follow-up. During this follow-up, 325 AD cases were ascertained with a total of 23,590 person-year. Use of any antihypertensive medication was associated with lower incidence of AD. Among different classes of antihypertensive medications, thiazide and potassium-sparing diuretics were associated with the greatest reduction of AD risk [64]. In the Systolic Hypertension in Europe (Syst-Eur) trial [65], median follow-up lasted only 2 years. Compared with the control group, antihypertensive therapy significantly reduced the risk of dementia by 55%. Interestingly, while the total incidence of dementia was 64 cases, 41 had AD. These findings suggest that BP-lowering therapy initiated with a CCB would protect against dementia, particularly AD, among the elderly with systolic hypertension [66].
POTENTIAL SCHEMA OF VISIT-TO-VISIT BLOOD PRESSURE VARIABILITY INVOLVEMENT IN ALZHEIMER’S DISEASE
The impact of visit-to-visit blood pressure variability on Alzheimer’s disease
An increased carotid arterial compliance was associated with attenuated pulsatile middle cerebral artery velocity, suggesting that Windkessel function was related with the reduction in the increased pulsatile component of cerebral perfusion induced by the enhanced left ventricular systolic function [67]. Cerebral matter is susceptible to fluctuations or inconsistent perfusion [68]. Thus, high VVV in BP might be associated with ischemic change in cerebral small vessel due to hypoperfusion during low-BP periods. Increased VVV had significant associations with white matter hyperintensity (WMH) [69] and cerebral atrophy [70], as well as microbleeds [71]. Increased carotid-femoral PWV also had a significant association with cerebral small vessel disease [72, 73]. Thus, the synergistic association of VVV and arterial stiffness with cognitive impairment might be mediated by cerebral microvascular remodeling in the elderly population.
The causes of abnormal VVV are still being debated. Increased sympathetic nervous system activity is suggested to be involved in the pathophysiology [74]. Increased hypothalamo-pituitary-adrenal axis activity might be associated with a sympathetic overdrive caused by factors such as emotional stress, environmental stress, and sleep dysregulation [75 –77]. Insomnia and long sleep duration, which were suggested to increase sympathetic nervous system activity, have been shown to have a relationship with increased VVV [78]. While the low frequency/high frequency ratio was significantly higher in AD patients compared to controls, the high frequency component was significantly decreased [79], suggesting that patients with AD manifested a predominantly higher sympathetic tone [80]. Thus, increased sympathetic nervous system activity might be a pivotal factor in the relationship between VVV and AD [81].
Baroreceptor sensitivity (BRS) is another major determinant of BP variability [82]. Increased large-arterial stiffness contributes to BRS depression in hypertensives [83] and has an important role for increased BP variability in response to changes in cardiac stroke volume. Thus, increased VVV is suggested to be associated with arterial remodeling, which reduces BRS and augments BP variability, leading to silent cerebral injury. BRS was shown to be reduced in patients with AD, suggesting that BRS is a factor in the relationship between vascular disorder and AD [84].
Cerebral hypoperfusion due to arterial remodeling enhances the production of Aβ. Arterial stiffening as well as microvascular dysfunction impair Aβ clearance and elevate brain Aβ levels [49]. Increased large arterial stiffness might provide a direct effect on cerebral penetrating arteries associated with an altered structure and function. Subsequently, this process has a harmful role on perivascular Aβ from the brain along to the perivascular space via the cerebrospinal fluid drainage. Thus, a disruption of vascular dynamics and reduced perivascular flow of Aβ causes decreased Aβ clearance [85]. As a consequence, arterial remodeling has a relationship with cerebral Aβ deposition. In addition, regarding the central autonomic nervous system, NFT deposition in the insular cortex (Ic) might increase VVV. These factors might be associated with the genesis of AD [86, 87]. Figure 2 illustrates the possible pathophysiology of VVV for both VaD and AD in relation to Aβ.
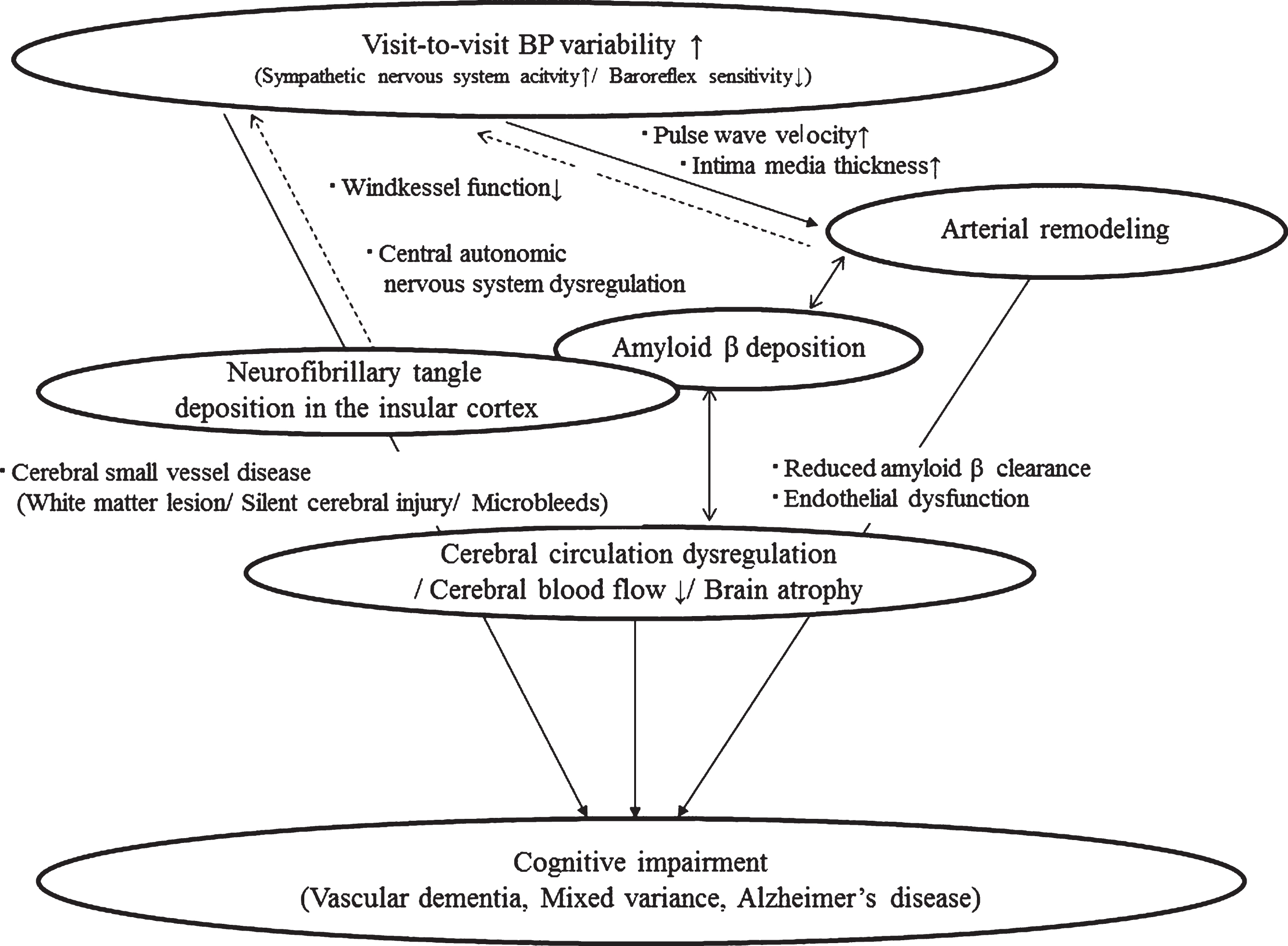
The pathophysiology of visit-to-visit BP variability for Alzheimer’s disease. The relationship between visit-to-visit BP variability and artery remodeling suggests that visit-to-visit BP variability is a risk factor of vascular dementia and Alzheimer’s disease. The continuous-line arrow shows known relationships: the solid-line arrows indicate possible relationships.
Alzheimer’s disease pathology in the insular cortex and visit-to-visit blood pressure variability: A possible insight into the brain-heart axis
Recent studies have supported the notion that the cardiovascular system is regulated by a cortical network consisting of the Ic, anterior cingulate gyrus, and amygdala, and that this network regulates the central autonomic nervous system in relation to emotional stress [88]. Because the Ic is located in the region of the middle cerebral arteries, its structure tends to carry a higher risk of cerebrovascular disease [88]. In clinical studies, Ic damage has been associated with arrhythmia, diurnal BP variation disruption (e.g., a non-dipper or riser pattern), and myocardial injury, as well as higher plasma levels of brain natriuretic peptide and catecholamine [88].
AD is associated with both insular pathology and autonomic dysfunction [89, 90]. Braak and Braak [91] demonstrated that NFT progress through the brain in a highly-structured sequence that begins in the mesiotemporal cortex and progressively invades the association cortex in the frontal, temporal, and parietal lobes. AD is merely the final stage of a pathological process that spans decades. Recent studies have demonstrated a hierarchichal sequence of AD pathology that includes the Ic [89, 90]. This may explain why AD has effects on BP and central autonomic cardio-regulatory functions. Brainstem nuclei are affected too late in the Braak sequence to explain preclinical dysautonomic symptoms. Instead, AD pathology in the Ic is a more likely origin of autonomic dyscontrol in early AD [89, 90]. The Ic is affected at a preclinical stage in the Braak sequence (i.e., stage III of six). AD reaches the Ic at a “preclinical” stage in its development (i.e., before “dementia” can be diagnosed). Thus, AD pathology should also be considered a possible explanation for autonomic morbidity and mortality in the non-demented elderly [89, 90]. It is hypothesized that autonomic dyscontrol, commonly seen in non-demented well elderly persons without significant cardiovascular disease, reflects subclinical stages of AD pathology affecting the insular cortex [89, 90]. If true, then preclinical AD pathology should be considered a possible explanation for higher VVV in the elderly. These points even suggest the possibility of a reverse causality between higher VVV and AD. However, higher VVV has been shown to be a predictor of cognitive decline and arterial remodeling. Thus, higher VVV would be an upstream risk factor of AD incidence, although AD pathology mediates this relationship to some degree.
CONCLUSION
The recent literature has confirmed that silent cerebral injury is responsible for the close relationship between VVV and cognitive impairment. Arterial stiffness might serve as a common pathophysiology in the relationship between VVV and AD. Until now, there have been no studies regarding the direct relationship of VVV with Aβ deposition or Aβ clearance in the human brain. Future studies are warranted to determine the impact of VVV on the pathogenesis of AD.
DISCLOSURE STATEMENT
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-1172r1).