
Abstract
INTRODUCTION
Alzheimer’s disease (AD) is the most common neurodegenerative disease, and the main cause of dementia in older people worldwide. AD is characterized by progressive loss of memory and cognitive dysfunction. The disease inexorably leads to death, with patients usually dying of complications of chronic illness [1, 2]. AD is proteinopathy-driven, principally by two types of abnormal protein deposition in the human brain: the extracellular accumulation of cytotoxic amyloid-β peptide (Aβ) in senile plaques, and the intracellular accumulation of hyperphosphorylated forms of a microtubule associated protein tau, in neurofibrillary tangles [3, 4]. Little to date is known of how these plaques trigger the onset of neurodegeneration. However, it is thought that heparan sulfate proteoglycans (HSPGs) may, on the one hand, promote Aβ or tau fibrillization, and, on the other, provide resistance against proteolytic breakdown [5]. In addition to Aβ, the most common AD-related subtype of senile plaques, called neuritic plaques (NPs), also contain accessory molecules, such as heparan sulfate (HS), apolipoprotein E, serum amyloid-P component, and α1-antichymotrypsin. Previous data indicates that the HS-Aβ interaction contributes to every stage of pathogenesis in AD, including production, clearance, accumulation, aggregation, and toxicity of Aβ [6–8].
Heparanase (HPSE) is an endo-β-D-glucuronidase that cleaves specific linkages in the structure of HS, yielding biologically active fragments [9]. By altering HSPGs on the cell surface, HPSE is thought to regulate the cellular response to external stimuli [10]. HPSE develops multiple functions in cancer, inflammation, infections, diabetes, and atherosclerosis [11–17], and in terms of neurodegeneration, recent studies have shown that overexpression of HPSE lowers the amyloid burden in transgenic mouse models of AD [18]. Heparanase 2 (HPSE2) is a homologue of HPSE that lacks HS-degrading activity, although it is able to interact with HS with high affinity [19, 20]. HPSE2 could act as a natural competitive inhibitor of HPSE due to is ability to associate with HPSE, and it appears, to modulate its enzymatic activity and signaling properties [14, 19].
In this study, we investigate for the first time the expression of two genes encoding heparanases in different stages of AD, using human brain tissue from different brain areas, principally following the Braak & Braak classification [21].
MATERIALS AND METHODS
Materials
The following materials were purchased from the manufacturers indicated: RNeasy Kit and RNase-Free DNase from Qiagen (Hilden, Germany); High-Capacity cDNA Reverse Transcription Kit and PowerSYBR Green PCR Master Mix from Applied Biosystems (Foster City, CA); GenElute PCR clean-up kit and 3–3’ diaminobenzidine from Sigma-Aldrich (St. Louis, MO); EnVisiontrademark Flex/HRP and high pH Envision FLEX target retrieval solution from Dako (Glostrup, Denmark). All other chemicals were obtained from commercial sources and were of analytical grade.
The following antibodies were used in this study: Goat anti-heparanase 1 polyclonal antibody (L-19), which recognizes the proteolytically-processed highly active HPSE (50 kDa active form of HPSE), was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) and rabbit anti-heparanase-2 polyclonal antibody, from GeneTex (Atlanta, GA). Both primary antibodies were properly validated as specific for the HPSE and HPSE2 proteins [14]. Anti-rabbit (sc-2004) and anti-goat (sc-2005) secondary antibodies were also from Santa Cruz Biotechnology (Santa Cruz, CA). Molecular Weight of proteolytically processed highly active HPA1:50 kDa.
Patients and samples
Samples from 18 patients with AD and 6 controls were collected at the Brain Biobank of the Hospital Universitario de Araba between February 2013 and October 2015 following approval from the same institute’s Review Board on Ethical Procedures, and after informed consent for the research had been received from each patient or their relatives. The diagnosis of AD was based on morphological and immunohistochemical criteria according to the most up-to-date grading classification (NIA-RI Consensus 1997). The characteristics of the patients studied (age, gender) and the timing of the postmortem of each sample were recorded (Table 1). Samples from the different brain areas were collected following the Braak & Braak classification [21]. Additionally, samples from basal ganglia (pallidum) and the cerebellum were used as an internal control, since they are assumed to not be altered in this disease.
Patient characteristics and descriptive statistics
AD, Alzheimer’s disease; Mild AD, I-II Braak & Braak classification; Moderate AD, III-IV Braak & Braak classification; Severe AD, V-VI Braak & Braak classification.
Total RNA isolation and cDNA synthesis
To obtain the RNA, tissue fragments of between 20 and 30 mg in weight were homogenized using a polytron PT 2100 (Kinematica Inc.; Bohemia, NY), and RNA was isolated using the RNeasy kit, following the manufacturer’s specifications. To ensure removal of residual contaminating DNA, samples were subjected to treatment with RNase-free DNase during the purification process itself. The concentration of RNA obtained was determined spectrophotometrically by measuring absorbance using a Picodrop Microliter UV/Vis spectrophotometer (Picodrop Limited, UK). The samples were divided into aliquots of 10μl and either used for reverse transcription reactions or stored at – 20°C until furtheruse.
cDNA synthesis was carried out using the High Capacity cDNA Transcription Kit following the manufacturer’s guidelines. The reactions were performed using an iCycler IQ thermocycler (BioRad; Hercules, CA), using 2μg of RNA as starting material. The reaction products were cleaned using the PCR Clean-Up GenElute kit following the manufacturer’s instructions. Finally, the aliquots containing the cDNA were diluted 1:20 with water and used for qRT-PCR assays or stored at – 20°C until use.
qRT-PCR reactions
In all cases, specific oligonucleotides were designed on different exons or exon junctions, using the program Primer 3 (http://biotools.umassmed.edu/bioapps/primer3_www.cgi). The size of the amplicon in all cases was between 70 and 150 base pairs and wherever possible Tm was above 77°C. The theoretical Tm for each amplicon was determined using the program Biomath (http://www.promega.com/biomath/calc11.).
The primer sequences were: HPSE (Gene ID 10855) forward 5’ ATGCTCAGTTGCTCCTGGAC 3’, reverse 5’ CTCCTAACTGCGACCCATTG 3’; HPSE2 (Gene ID 60495) forward 5’ caccctgatgttatgctggag 3’, reverse 5’ tccagagcaatcagcaaagtta 3’.
A minimum four repetitions of all the qRT-PCR reactions were carried out in a final volume of 10μl, in accordance with the manufacturer’s specifications, using 1μl of the cDNA dilution as template, with 2μl of primer pair mix (200 nM final concentration) and 5μl of SYBR Green mix, assembled in 96-well microtiter plates. The plates were sealed with optical film and centrifuged at 2500 rpm for 5 min before being placed in a Real-Time ABI Prism Detection System device (Applied Biosystems; Foster City, CA) with the following cycling conditions: 95°C for 10 min, 40 cycles of 95°C for 15 s followed by 60°C for 60 s. Following the thermal cycling and data collection steps, amplifier products were analyzed using a melt curve program (95°C for 1 min, 55°C for 1 min, then increments of 0.5°C per cycle for 80 cycles of 10 s each). For each amplification, the presence of a single peak with a Tm corresponding to the theoretical value previously calculated was verified. Actin was included on each plate as a control gene in order to compare run variation and to normalize individual gene expression.
Data analysis
To calculate the efficiencies of amplification for each gene we used the program LinRegPCR (http://www.gene-quantification.de/download.html), taking the best correlation coefficient (after considering a minimum of 3 points within the window of linearity) and establishing the average of all positive amplifications. The expression values of the genes of interest were calculated as (1 + efficiency) –ΔCt (relative to actin as the housekeeping gene).
Immunohistochemistry
Paraffin embedded tissue sections were treated with xylene to render them diaphanous (the paraffin being removed later by passing it through decreasing alcohol concentrations until 100% water was reached). Rehydrated sections were rinsed in phosphate buffered saline (PBS) containing 1% tween-20. For the detection of HPSE2, sections were heated in high pH Envision FLEX target retrieval solution at 65°C for 20 min and then incubated for 20 min at room temperature in the same solution. For HPSE detection, the final step was omitted.
Endogenous peroxidase activity (3% H2O2) and non-specific binding (33% fetal calf serum) were blocked and the sections were incubated overnight at 4C with primary antibodies using a 1:100 dilution. Ready to use labelled polymer-HRP (DAKO, Spain) was used as secondary antibodies, and 3–3’ diaminobenzidine was employed as a chromogen. Finally, samples were counterstained with hematoxylin, dehydrated, and mounted in Entellan® (Merck, Germany). The sections were studied and photographed under a light microscope (Nikon - Eclipse 80i).
Immunohistochemistry assessment
The protein expression levels were evaluated by two independent experts (and in the case of disagreement, a third).
Statistical analysis
A non-parametric Wilcoxon test was used for the statistical analysis of the experiments with the level of significance set at p < 0.05. All analyses were performed using the Statistics for Windows program (Statsoft Inc.; Tulsa, OK).
RESULTS
Analysis of differential gene expression
Initially we used qRT-PCR to perform a quantitative analysis of the differential transcription of the coding genes using samples of 7 different tissue types from various encephalic areas. Mild AD cases (I-II Braak & Braak classification), Moderate AD cases (III-IV Braak & Braak classification), and Severe AD cases (V-VI Braak & Braak classification) were analyzed and compared to healthy controls. The samples corresponded to patients of both sexes aged between 52 and 103 years. Determination of the expression of HPSE and HPSE 2 from the different areas of AD brains was performed by immunohistochemistry.
Differential expression of heparanase
Analysis of the transcription levels of the HPSE gene by means of qRT-PCR detected expression levels in all samples studied. There were statistically significant differences in alterations between AD stages as defined by Braak & Braak staging. Additionally, significant overexpression of HPSE was noted in neurodegenerated areas of AD brains. Furthermore, significant HPSE underexpression was observed in pallidum in mild and moderate AD brains. The cerebellum of brains at different AD stages did not show any significant alterations in HPSE transcription levels (Fig. 1).
Compared with healthy tissue, immunohistochemistry showed the presence of intracellular HPSE deposits in atrophic neurons with neurofibrillary tangles. At the extracellular level, HPSE was mainly observed in NPs with fragmented core rather than those with a large dense core, pointing to the existence of a gradient of HPSE expression. Basal ganglia and cerebellum were mostly negative(Fig. 2).
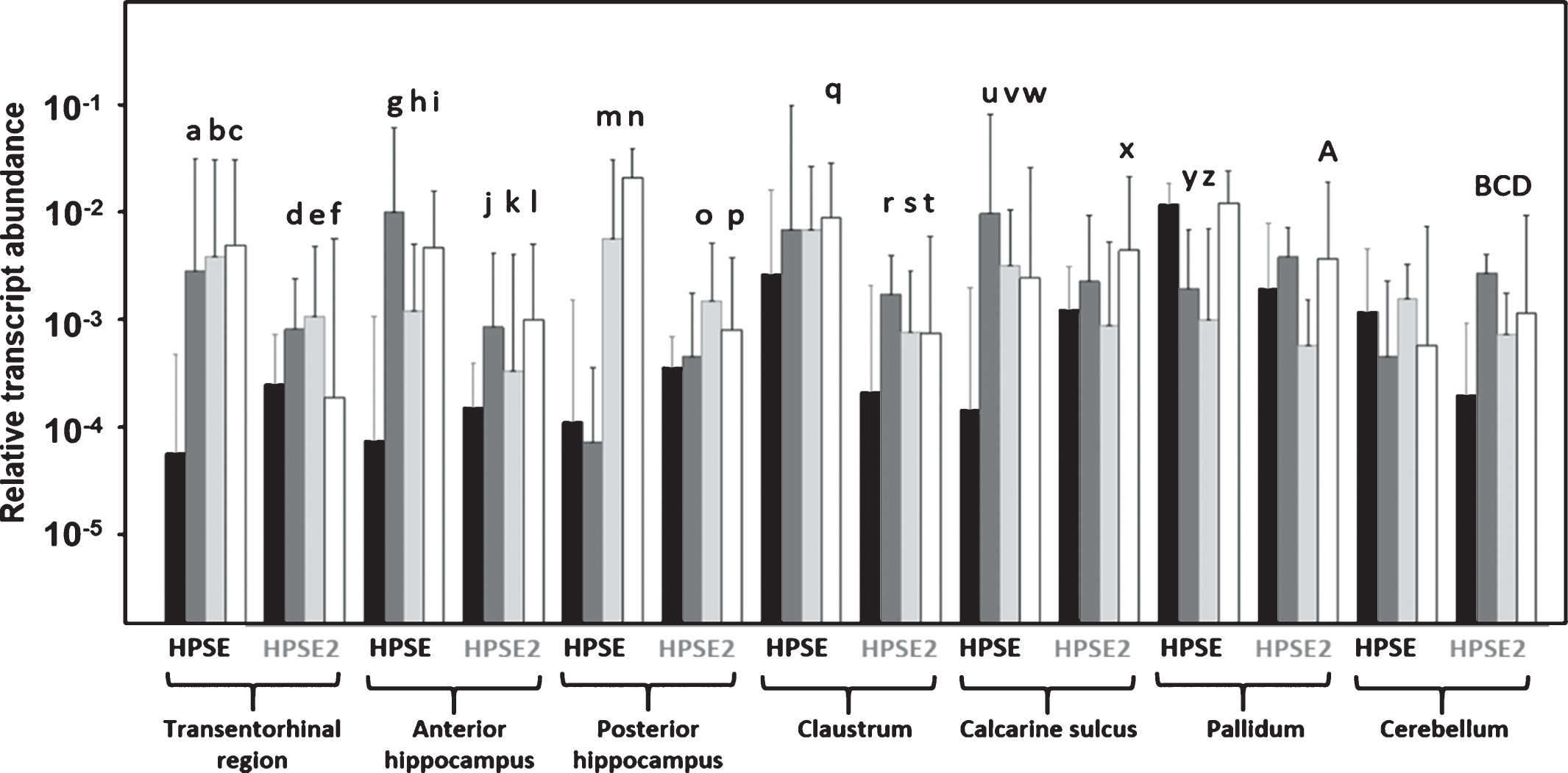
Differential transcription of heparanase and heparanase 2 in Alzheimer’s disease (AD). Relative abundance for controls (black bars), mild AD (dark gray bars), moderate AD (light gray bars) and severe AD (white bars) are plotted on a log scale for each gene assayed and the spreads represent standard deviations. Genes and zones that display significant differences in their transcription levels are highlighted. a: p = 0.000011; b: p = 0.000001; c: p = 0.000000; d: p = 0.013545; e: p = 0.01817; f: p = 0.026365; g: p = 0.000005; h: p = 0.002675; i: p = 0.000021; j: p = 0.043012; k: p = 0.013804; l: p = 0.000149; m: p = 0.000079; n: p = 0.00000; o: p = 0.025297; p: p = 0.048391; q: p = 0.024351; r: p = 0.001165; s: p = 0.041894; t: p = 0.037990; u: p = 0.000007; v: p = 0.000063; w: p = 0.005458; x: p = 0.005616; y: p = 0.000002; z: p = 0.000009; A: p = 0.012136; B: p = 0.001540; C: p = 0.004565; D: p = 0.007672. Values on the Y axis are represented on a logarithmic scale.
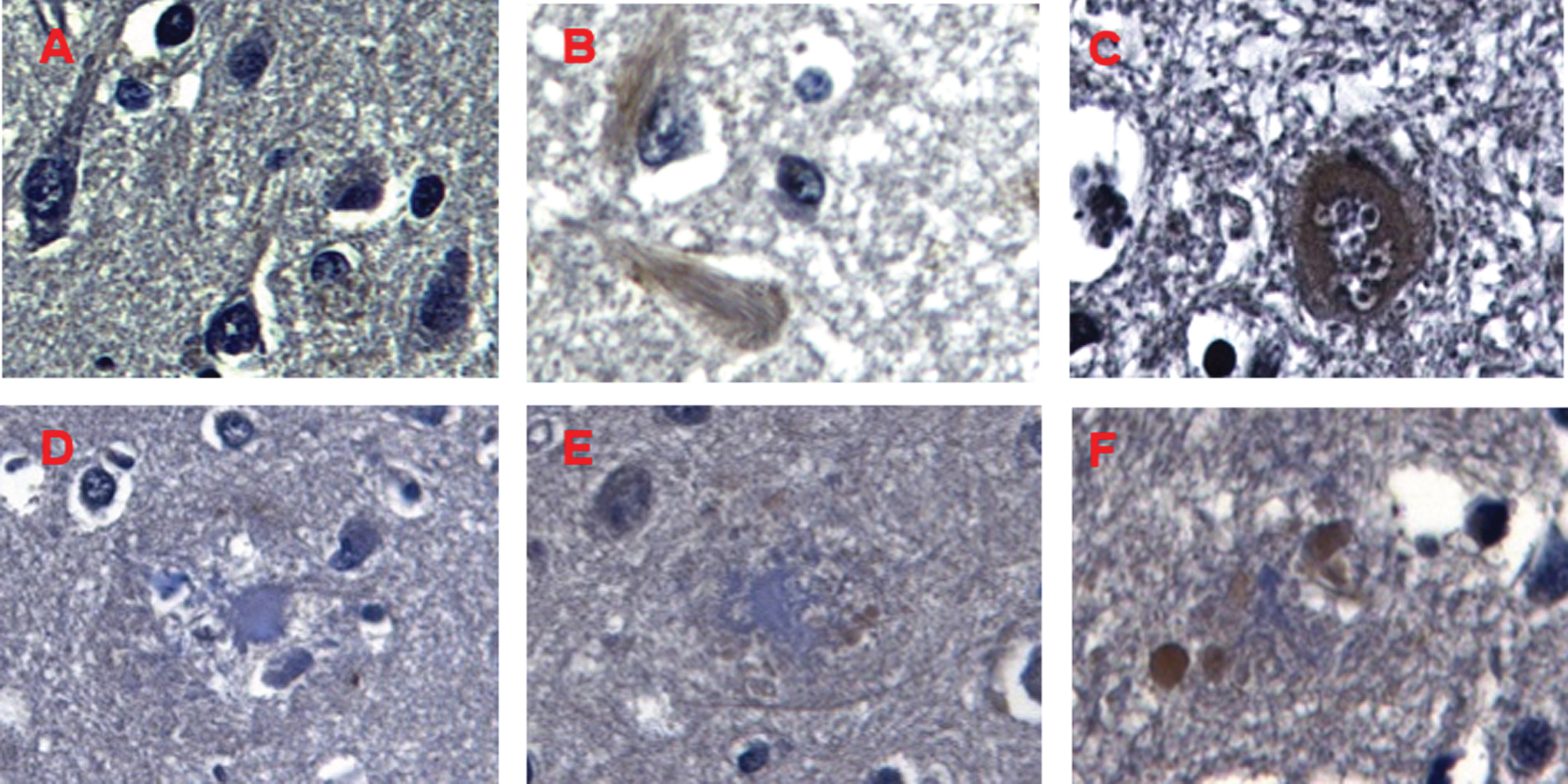
Immunohistochemical staining of HPSE in Alzheimer’s disease. A) Negative expression of HPSE in neurons and glial cells in control brain samples. B, C) HPSE overexpression in neurons with neurofibrillary tangles (B) and granulovacuolar degeneration (C). D-F) NPs showing different core size, from big to medium-sized and small (white arrows); in inverse correlation to HPSE deposits from mild (D) to moderate (E) and high levels (F) (black arrow). Magnification 600X.
Differential expression of heparanase 2
We analyzed the levels of transcription of HPSE2 in cerebral tissue using primers designed to detect wild-type transcripts (HPSE2c). Analysis of the transcription levels of HPSE2 detected expression in all samples studied. Stage-dependent (based on Braak & Braak) statistically significant differences were found in AD brains. Additionally, significant HPSE2 overexpression was noted, mainly in neurodegenerated areas of AD brains and in the cerebellum in all AD cases. Pallidum, on the other hand, showed no significant alterations of HPSE2 transcription levels (Fig. 1).
HPSE2 protein expression was analyzed by immunohistochemistry, showing the presence of intracellular HPSE2 deposits in atrophic neurons with neurofibrillary degeneration. At the extracellular level, HPSE2 was observed in almost all NPs. HPSE2 was undetectable in pallidum and cerebellum, although the latter showed a very mild diffuse positivity (Fig. 3).
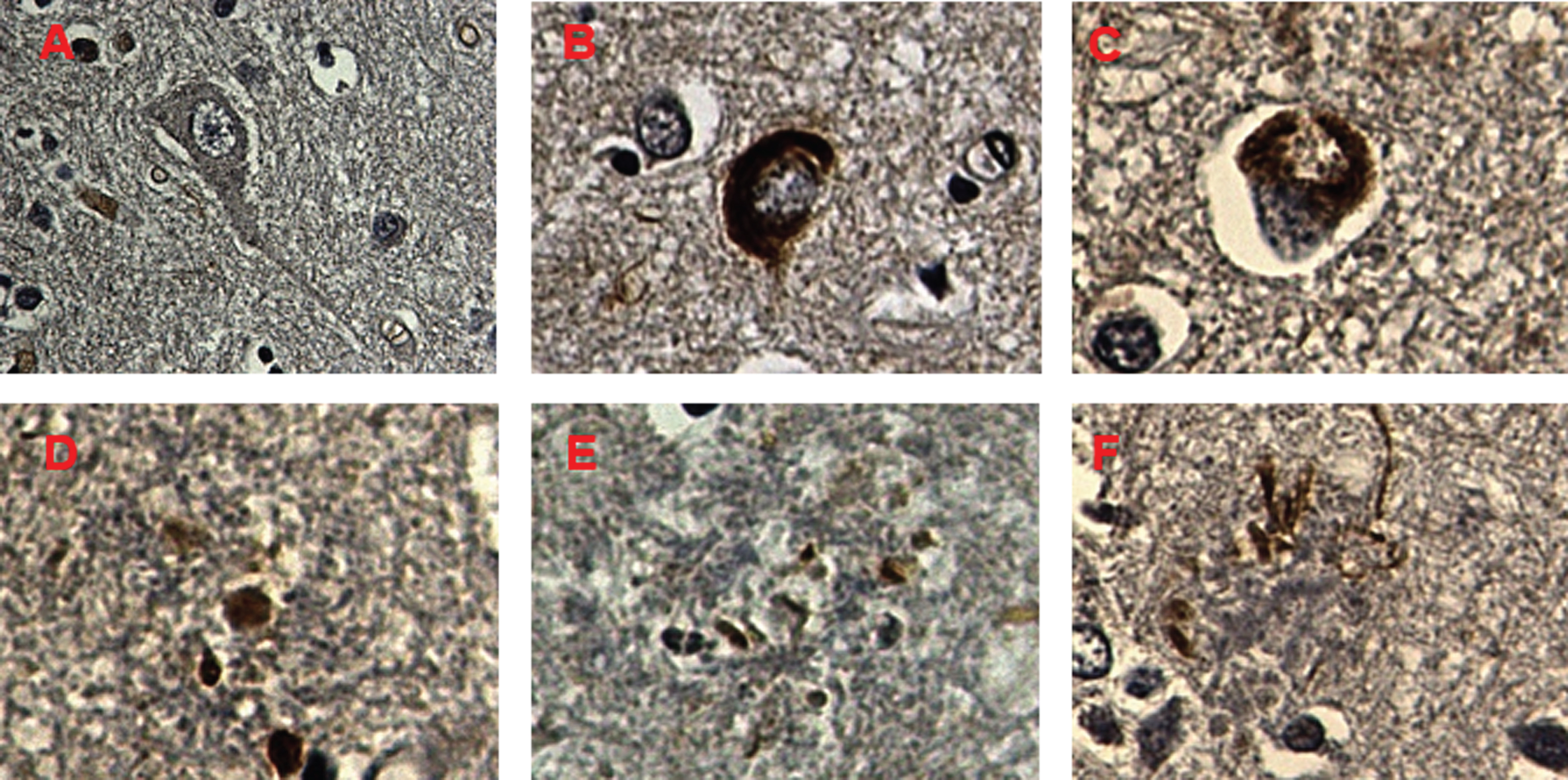
Immunohistochemical staining of HPSE 2 in Alzheimer’s disease. A) Negative expression of HPSE2 in neurons and glial cells in control brain samples. B, C) HPSE2 overexpression in neurons with neurofibrillary tangles (B) and granulovacuolar degeneration (C). D-F) NPs showing different forms of HPSE 2 deposits. Concentrated deposits in unfragmented core of NPs (D), dispersed deposits (E), and filament-like deposits are indicated (F) (blue arrows). Magnification 600X.
DISCUSSION
AD is a fatal progressive neurodegenerative disorder mainly caused by extracellular accumulation of Aβ protein in plaques, which is thought to account for the onset of neurodegeneration. Several in vitro studies have demonstrated the interaction of Aβ with HS [22, 23]. In addition, studies have demonstrated that HS molecules are preferentially colocalized with Aβ around the dense cores of senile plaques [18, 24] and it has been proposed that Aβ-HS interaction is mutually protective, in that Aβ is protected from protease degradation and HS is protected from HPSE degradation [22, 25]. HPSE is a multitasking protein characterized by involvement in enzymatic and non-enzymatic activities. The former activity gives rise to fragments of HSPG chains which are biologically involved in multiple physiological and pathological functions [9]. Thanks to the cleavage of HS, HPSE also promotes the release and diffusion of several HS-linked molecules, such as growth factors, cytokines, and enzymes. In addition to degrading HS chains, HPSE has non-enzymatic functions that trigger several signaling pathways [26]. In the present work, we have focused on the study of the expression levels of 2 genes which encode heparanases at different stages of AD using RT-PCR, and then localized the proteins in the tissue through immunohistochemistry.
Concerning HPSE, a significant overexpression was noted in neurodegenerated areas of AD brains, which mostly correlated well with the Braak & Braak stage for each case. We speculate that HPSE overexpression in AD could be a consequence of the disease itself, rather than being part of the cause. Under this scenario, different brain cells would release HPSE in order to reduce the existing AD pathology by degrading HS chains, exposing Aβ deposits to degradation. Many different treatment approaches have tried to focus on inhibiting tau-aggregation or reducing Aβ deposits by decreasing production, preventing aggregation, or increasing removal [27, 28]. There have been many clinical trials for AD treatments, and although the majority have proved inconclusive [29–34], these treatment options continue to be pursued. However, it is our view that the lack of progress in AD treatment is due to the majority of studies having underestimated either the functional or structural roles of HS molecules in AD [35].
Published data indicates that intact HS chains are required at multiple sites in order to mediate neurodegeneration, further supporting the notion that HPSE is possibly a crucial element that may have potential treatment implications in AD [36]. Additionally, recent experimental evidence suggests that transcellular propagation of fibrillar protein aggregates drives the progression of neurodegenerative diseases in a prion-like manner [37]. Likewise, the mechanism by which aggregated extracellular proteins, such as tau, bind and enter cells to trigger intracellular fibril formation occurs via HSPG binding [38]. HPSE could, therefore, act to modulate or impede this process, as heparinase III does in vitro in order to slow down neurodegeneration [38]. However, there are potentially contradictory roles that can be ascribed to HPSE in AD: On the one hand, HPSE could impair inflammatory response and macrophage-mediated clearance of Aβ favoring AD progression. On the other, HPSE could block the internalization and propagation of pathologic tau throughout the brain [38, 39]. The significant overexpression of HPSE in calcarine sulcus (neocortex) at early stages of AD could be explained by the existence of extracellular deposits related to fragmented NPs. Likewise, the significant underexpression of HPSE in the pallidum could be the result of strong HPSE diffusion to neurodegenerative areas, even at early stages of disease. And finally, we have found the presence of HPSE principally in those NPs with a fragmented core, which may possibly contribute to breaking down plaques and exposing Aβ to degradation [40–42].
We observed that HPSE2 was significantly overexpressed in AD cases following neurodegeneration, the same as HPSE, albeit with lower levels of ARNm expression. Similar again to the findings for HPSE, significant changes in HPSE2 were mostly AD-stage-dependent. Histologically, HPSE2 was found to be associated to NPs and neurofibrillary tangles. Furthermore, a mild diffuse neurophilic immunostaining was noted in AD-related areas and cerebellum. In this sense, HPSE2 binds to HSPGs but, unlike HPSE, it fails to become internalized, and remains on the cell surface for a relatively long period of time [20]. Detailed characterization of HPSE2 at the biochemical, cellular, and clinical levels is not available to date, and its role in normal physiology and pathological disorders is unclear. However, recent studies have shown that HPSE2 is an endogenous inhibitor of HPSE activity, likely due to its high affinity to HS and its ability to associate physically with HPSE [20]. In this light, HPSE2 could inhibit HPSE activity, facilitating the progression of AD pathology, by impeding the clearance of Aβ deposits and giving rise to more internalization and propagation of pathologic tau throughout the brain.
In conclusion, our study reveals details of the behavior of both heparanases in AD, pointing to the combined overexpression of HPSE and HPSE2, mostly related to AD pathology. While HPSE activity would seem to either release Aβ deposits or block tau intracellular fibril formation and propagation, HPSE2 appears to act as an inhibitor of HPSE. Finally, the relevance, both biological and clinical, for studying both heparanases is based on the fact that no other molecules are capable of performing the same function, making both enzymes an attractive potential target for medical treatment. In this regard, considering that HS-Aβ interaction contributes to every stage of the Aβ pathogenesis in AD, including production, clearance, accumulation, aggregation, and toxicity, it is rational to hypothesize that interfering in the HS-Aβ interaction may have multiple beneficialeffects.
Footnotes
ACKNOWLEDGMENTS
This study was funded by the Health Department of the Basque Country [grant 2014111060 to IFV]; the Government of the Principado de Asturias (Spain) for consolidated research groups [grant FC-15-GRUPIN14-141 to BG]; and the Fundación de Investigación Oftalmológica through the Fundación Cristina Masaveu Peterson [Instituto Universitario Fernández-Vega; to BG, JM, LQ].