
Abstract
Amyloid-β (Aβ), a major component of senile plaques, is generated via the proteolysis of amyloid-β protein precursor (AβPP). This cleavage also produces AβPP fragment-derived oligomers which can be highly neurotoxic. AβPP metabolism/processing is affected by many factors, one of which is oxidative stress (OS). Associated with aging, OS is an important risk factor for Alzheimer’s disease. In addition, the protein degradation systems, especially those involving cathepsins, are impaired in aging brains. Moreover, cathepsin B (CTSB) is a cysteine protease with potentially specific roles in AβPP proteolysis (β-secretase activity) and Aβ clearance (Aβ degradative activity). The present work examines the effect of OS and the involvement of CTSB in amyloid oligomer formation. The xanthine/xanthine oxidase (X-XOD) free radical generating system induced the partial inhibition of CTSB activity, which was accompanied by an increase in large amyloid oligomers. These were located throughout the cytosol and in endo-lysosomal vesicles. Cells treated with the CTSB inhibitor CA-074Me also showed increased amyloid oligomer levels, whereas those subjected to OS in the presence of the inhibitor showed no such increase. However, CTSB inhibition clearly modulated the AβPP metabolism/processing induced by X-XOD, as revealed by the increase in intracellular AβPP and secreted α-secretase-cleaved soluble AβPP. The present results suggest that CTSB participates in the changes of amyloid oligomer induced by mild OS.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder clinically characterized by cognitive dysfunction and failing memory. The major histochemical hallmarks of the disease are neurofibrillary tangles (NFTs) and plaques composed of aggregated amyloid-β (Aβ) [1]. Aβ peptides are produced during the sequential proteolytic processing of the amyloid-β protein precursor (AβPP) [2], a type 1 transmembrane protein with its amino terminus in the lumen/extracellular space and its carboxy terminus in the cytosol. AβPP processing toward either the amyloidogenic pathway (β- and γ-secretases), and thus the generation of Aβ, or toward the non-amyloidogenic pathway (α and γ-secretases), in which no Aβ is produced [3], is directed by cellular levels of secretases, plus the trafficking of AβPP to organelles that express these enzymes.
Aβ exists in two major peptide isoforms depending on its amino acids length, Aβ40 and Aβ42, with the latter markedly more prone to accumulation and aggregation [4]. Aβ42 firstly would form soluble oligomers, which later would come to produce insoluble aggregates that could accumulate as amyloid fibrils. In AD brains, these go on to form senile plaques [5]. Soluble Aβ oligomers formed during the first stages of AD are believed to be particularly toxic and responsible for early memory failure [6]. Recently, intermediate and large oligomeric Aβ assembly states have been associated with both aging and AD [7]. AβPP-C-terminal fragments (CTFs) generated by β-secretase can also form oligomers[8, 9].
Cathepsins reside in the endo-lysosome system [10] where β-secretase activity mainly occurs [11, 12]. Cathepsin B (CTSB) is a cysteine protease that cleaves AβPP at the same site as β–secretase [13] and also degrades the Aβ42 peptide, starting at its C-terminal [14]. Enzyme and proenzyme forms of CTSB have been identified in AD brains, and are particularly common in early endosomes [15].
Many studies have reported OS markers to exist in the brain and peripheral tissues of patients with AD, and indicate them to be among the earliest indications of the disease [16, 17]. Aging is an important risk factor for AD [18], and protein degradation systems, especially those involving the cathepsins, become impaired in aging brains [19, 20]. To study the involvement of OS in AD, our laboratory developed a human neuron model of mild OS induced by the xanthine/xanthine oxidase (X-XOD) free radical-generating system. This model allowed us to investigate the events preceding cell death [21], and the relationships between OS and AβPP metabolism/processing [22]. Moreover, it was used to show that OS influences AβPP processing, trafficking, and catabolism via the ubiquitin-proteasome system and autophagy-lysosomal pathway [23]. Defects in protein degradation might link aging to neurodegeneration [24, 25]. In addition, CTSB, with its potential function in AβPP proteolysis (β-secretase activity) and in the clearance of Aβ (Aβ degradative activity), plays likely an important role in the formation of amyloid oligomers.
The hypothesis of the present work was that mild OS regulates the formation of amyloid oligomers via CTSB, and that this might be a major step in the neurodegenerative process.
MATERIALS AND METHODS
Materials
SK-N-MC human neuroblastoma cells (HTB-10) were obtained from the American Type Culture Collection. Xanthine (X) was purchased from Sigma, xanthine oxidase (XOD) from Roche, Ca-074Me (a CTSB inhibitor) from Enzo and DAPT (a γ-secretase inhibitor) from Sigma. Culture medium components were purchased from Gibco Laboratories. Other chemicals were purchased from Merck or Sigma.
Cell culture and treatments
Cells were cultured in minimal essential medium (MEM) supplemented with 10% fetal bovine serum and 50μg/ml gentamicin in a humidified 5% CO2 atmosphere. Exponentially growing cells at 80–90% confluence were placed in culture dishes with 6, 24, or 96 multiwells (M-6, M-24, or M-96) one day before treatment with X-XOD and/orCa-074Me.
On the day of treatment, cells were placed in fresh medium 1 h before addition of the compounds X (10μM) and XOD (50 mU/ml). The effects of their addition were measured 24 or 48 h later. For Ca-074Me treatments (concentrations are indicated in each experiment), the inhibitor was added for 1 h and the cell cultures then placed in fresh medium for the remainder of the experiment (24 h). The concentration of DMSO (vehicle) in the cell culture was 0.01% or lower.
Analysis of cell injury
The cell injury caused by exposure to OS was determined using the 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay [26] with minor modifications [21].
CTSB activity assay
CTSB activity was assessed following the method of Porter et al. [27] with modifications. Cells grown in a 6-well plate and incubated under different treatments were washed in PBS and lysed for 1 h at 4°C with shaking in 200μL of 50 mM sodium acetate (pH 5.5), 0.1 M NaCl, 1 mM EDTA, and 0.2% Triton X-100 (lysis buffer). Lysates were centrifuged at 13,000 g for 10 min and the supernatant (clarified lysate) used to determine proteolytic activity. For the activity assays, 50μg of clarified lysate were incubated at 37°C for 30 min in lysis buffer (100μL) in the presence of 20μM z-RR-AMC (P-137; Enzo Life Sciences) (an appropriate fluorogenic substrate). The fluorescence emitted as a result of proteolysis was recorded using an Infinite® 200 microplate reader (Tecan Trading AG)(excitation/emission wavelengths 360/430 nm).
Antibodies
The antibodies used in the present study were: anti-AβPP (22C11) (MAB348; Chemicon International, Inc.) directed against amino acids 66–81 of AβPP; monoclonal antibody 6E10 (SIG-39300; Covance) directed against amino acids 1-16 of human Aβ; anti-AβPP C-terminal fragment antibody (802803; BioLegend); anti-oligomer A11 (TA326459; OriGene); rabbit anti-CTSB (sc-13985; Santa Cruz Biotech.), rabbit anti-Rab7 (9367; Cell Signaling), mouse anti-LAMP1 (H4A3-C; DSHB Hybridoma Bank), mouse anti-CD222 (315902; BioLegend), and mouse anti-EEA1 (610457; BD Transduction Laboratories). α-tubulin levels were examined as an internal control (via the reaction with anti-α-tubulin [Sigma] in the same blots). Secondary antibodies for immunostaining included horseradish peroxidase-coupled antibodies (Vector), and antibodies labelled with Alexa Fluor 488 or Alexa Fluor 555 dye (Invitrogen). The ability of each antibody to recognize different metabolites according to their epitope location in AβPP has been described [22].
Western blot analysis
For AβPP and amyloid oligomer analysis, treated cells were washed in PBS and lysed in the lysis buffer containing PBS, 1% Triton X-100, and the Complete Mini protease inhibitor cocktail (Roche) on ice for 30 min, vortexing every 10 min. Supernatants were collected after centrifugation at 13,000 g for 30 min at 4°C. Samples of culture medium were prepared as previously described [22]. Blotting was then performed with the corresponding antibodies.
CTSB was analyzed in the lysates used in the above enzyme activity experiments, to complement the activity data observed and deepen into the effect of our treatments on this protease. These lysates were mixed with Laemmli buffer and heated for 3 min at 100°C. After electrophoretic separation, the gels were blotted onto a nitrocellulose membrane and stained with the appropriate antibodies and peroxidase-coupled secondary antibodies. Detection by enhanced chemiluminescence was performed using ECL western blotting detection reagents (Amersham Biosciences) according to the manufacturer’s instructions.
Immunoprecipitation
Immunoprecipitation of cell lysates was performed in PBS containing 1% Triton X-100, employing the SureBeads™ Protein G Trial Kit (1614821; BioRad), following the manufacturer’s instructions.
Protein concentrations
Protein concentrations were determined using the bicinchoninic acid assay kit (Pierce).
Immunofluorescence imaging
Immunofluorescence assays were performed as previously described [22]. Cells were examined using an LSM 710 laser scanning confocal microscope (Zeiss) coupled to a vertical M2 AxioImager (Zeiss) equipped with a 63X/1.4 Plan-Apochromat oil objective lens, or using a Zeiss Axiovert 200 fluorescence microscope equipped with a 63 X oil-immersion objective. Pictures were taken with a Spot RT digital camera (Diagnostic) using Zeiss ZEN 2010 imaging system software. Images were processed using Adobe Photoshop CS3.
Cell fractionation for endo-lysosome and cytosol enrichment
The method of Avrahami et al. [28] was used (with minor modifications) to isolate the different subcellular fractions. Cells were washed with PBS, placed in washing buffer (125 mM KCl, 30 mM Tris, pH 7.5, 5 mM MgOAc, 1 mM mercaptoethanol), and centrifuged at 800 g for 5 min. They were then resuspended in hypotonic buffer (10 mM KCl, 30 mM Tris, pH 7.5, 5 mM MgOAc, 1 mM mercaptoethanol, plus a Complete Mini protease inhibitor cocktail (Roche)) and broken open by 15 passes through a syringe with a 23 G needle. These homogenates were then centrifuged at 1000 g to precipitate the nuclei, debris, and any cells not destroyed. Supernatants were collected and centrifuged at 100,000 g for 1 h at 4°C to pellet the endosomes, lysosomes, and mitochondria (fraction L/LE). The final supernatant containing cytosol and microsomes (fraction Cyt) was also collected. Western blotting was then performed to detect organelle markers in the different fractions. AβPP, amyloid oligomers, and CTSB were similarly detected.
Statistical analysis
Results were expressed as means ± standard errors, and analyzed using the Student t test. Significance was set at p < 0.05.
RESULTS
X-XOD reduces CTSB activity
Cathepsin B (CTSB) activity was assessed in SK-N-MC cells treated with the X-XOD free radical generating system for 24 h [22]. To compare the effect of mild OS with the pharmacological inhibition of CTSB, cells were treated with CA-074Me (a specific inhibitor of this protease). CTSB activity was quantified using a fluorometric assay. X-XOD-treated cells showed CTSB activity to be reduced by up to 58% (Fig. 1A) (p < 0.001 compared to control culture). CA-074Me reduced CTSB activity in a dose-dependent manner (Supplementary Fig. 1A)— by 97% (p < 0.001) at its highest concentration (5μM), 71% (p = 0.0012) at an intermediate concentration (1μM), and 48% (p = 0.0115) at the lowest concentration (0.2μM). To confirm that the reduced activity was not due to any treatment-caused lessening of cell viability, MTT reduction assays were performed. Supplementary Fig. 2 (A and B) shows that none of the treatments reduced cell viability at 24 h of culture. Previous studies reported minimal cell injury at this time [21].

X-XOD reduces CTSB activity. SK-N-MC cells were treated with 10μM X/50 mU/ml XOD (X-XOD). After incubation for 24 h, CTSB activity (A) and CTSB protein levels (B) were measured. A) Proteolysis in the presence of the fluorogenic substrate z-RR-AMC was used to monitor CTSB activity. The graphs show the mean (±SEM) fluorescence values for each level of enzyme activity expressed (as a percentage of the control or vehicle value). *p < 0.05, **p < 0.01, and ***p < 0.01 (t-test, n = 3). B) Western blotting was performed using the anti-CTSB antibody. The bands correspond to the pro-enzyme (top) and enzyme (down) forms of CTSB. In the lower panel the band corresponds to α-tubulin. The data show the mean (± SEM) densitometry values (normalized against α-tubulin). Control or vehicle values were set at 1. *p < 0.05, **p < 0.01, and ***p < 0.01 (t-test, n = 3).
To deepen into the effect of these treatments on CTSB, the active enzyme levels were examined by western blotting in the same cell lysates, using a CTSB-specific antibody that recognizes the enzyme and its precursor (pro-enzyme). CTSB is synthesized as a pre-proenzyme, following removal of the signal peptide the inactive pro-enzyme undergoes further modifications (37–42 kDa) including removal of the pro region to result in the active enzyme (25 kDa) [29]. X-XOD (Fig. 1B) induced a significant reduction in CTSB mature/active-form levels (0.61±0.08-fold versus control; p = 0.098). The CTSB inhibitor CA-074Me (Supplementary Fig. 1B) also induced significant reductions [0.45±0.04-, 0.61±0.16- and 0.59±0.01-fold versus control for 5 (p < 0.001), 1 (p = 0.049) and 0.2 (p = 0.006) μM CA-074Me, respectively].
X-XOD increases amyloid oligomer levels
The effect of mild OS on soluble amyloid oligomer levels was examined in cell lysates by western blotting (Fig. 2) using the polyclonal antibody A11. This antibody specifically detects soluble amyloid assemblies distinct from fibrillar Aβ [30–32]. The effect of the CTSB inhibitor CA-074Me was also examined.

X-XOD increases amyloid oligomers levels. SK-N-MC cells were treated (A) with 10μM X/50 mU/ml XOD (X-XOD) and (B) with the indicated concentrations of CA-074Me. After incubation for 24 h, western blotting was performed using anti-N-terminal AβPP antibody (22C11) and anti-oligomer antibody (A11). Representative gels for X-XOD (A) and CA-074Me (B) treatments are shown. In the upper panel the band corresponds to high molecular weight amyloid oligomer, in the intermediate panel to hAβPP, and in the lower to α-tubulin. The data show the mean densitometry values (normalized against α-tubulin). Control or vehicle values were set at 1. *p < 0.05, **p < 0.01, and ***p < 0.01 (t-test, n = 4).
Figure 2A shows that the cells subjected to X-XOD treatment experienced a significant increase in a single band at 56 kDa (1.36±0.06-fold; p < 0.001), which also increased in the cells treated with CA-074Me (1.32±0.07; p = 0.005 and 1.53±0.40-fold; ns) at 5μM and 1μM, respectively (Fig. 2B).
Immunoprecipitation of this high molecular weight complex by the 6E10 antibody (an anti-Aβ, -βCTF and -holo AβPP [hAβPP] monoclonal antibody) but not by the C-terminal AβPP specific antibody (Supplementary Fig. 5) indicated it to be an Aβ oligomeric form derived from AβPP proteolysis. The possibility that this 56 kDa band, is derived from Aβ was reinforced by the observation that the inhibition of γ-secretase using DAPT led to a significant decrease in the levels of oligomers in the X-XOD treated cells (Supplementary Fig. 4).
Based on its electrophoretic mobility, this large amyloid oligomer most probably corresponds either to the 56 kDa soluble Aβ assembly known as Aβ*56 (detected in an AD transgenic rat brain model by Lesne et al. [33]) or to the more recently described oligomeric Aβ assemblies found in both ageing and AD brains [7].
Since the amyloid oligomer derives from AβPP proteolytic processing, AβPP protein was analyzed in the same cell lysates by western blotting using the 22C11 antibody which recognizes full-length AβPP (hAβPP) and AβPP fragments containing the N-terminal part (sAβPP). Figure 2A shows that treatment with X-XOD for 24 h led to a 3.85±1.07-fold increase in cellular AβPP levels (p = 0.037). The above results indicate that, during that period just before the transmission of cell death signals, the X-XOD system increases intracellular soluble amyloid oligomer and AβPP levels. In contrast, the levels of AβPP protein (Fig. 2B) were not significantly affected by CA-074Me treatment.
Together, these results show that a decrease in CTSB activity— via mild OS or the pharmacological inhibition of CTSB— leads to increased levels of amyloid oligomers, suggesting that the CTSB pathway is a participant in the changes of amyloid oligomer levels.
The intracellular location of amyloid oligomers was then investigated by double immunofluorescence analysis using the A11 and 6E10 antibodies. 6E10-immunoreactive protein species are probably composed of hAβPP, βCTF, and Aβ/βCTF assemblies (oligomers) plus a small amount of Aβ peptide. A11-positive staining reveals oligomeric forms but not monomers or fibrils [30].
In control cells, 6E10-positive structures (Fig. 3, green) appeared as small vesicles compatible with endosomes/lysosomes as previously described [23]. In contrast, A11-stained structures appeared as small points distributed throughout the cytoplasm (cytosol/microsomes); colocalization with 6E10-positive structures was minimal. In the X-XOD treated cells, the A11-stained structures distributed throughout the cytoplasm (cytosol/microsomes) were also present, but the number and size of structures positive for both antibodies increased. According to previous data of the laboratory [23], the large yellow signals probably correspond to vesicles of the endo-lysosomal system, and contain amyloid oligomers and AβPP/βCTF/Aβ.
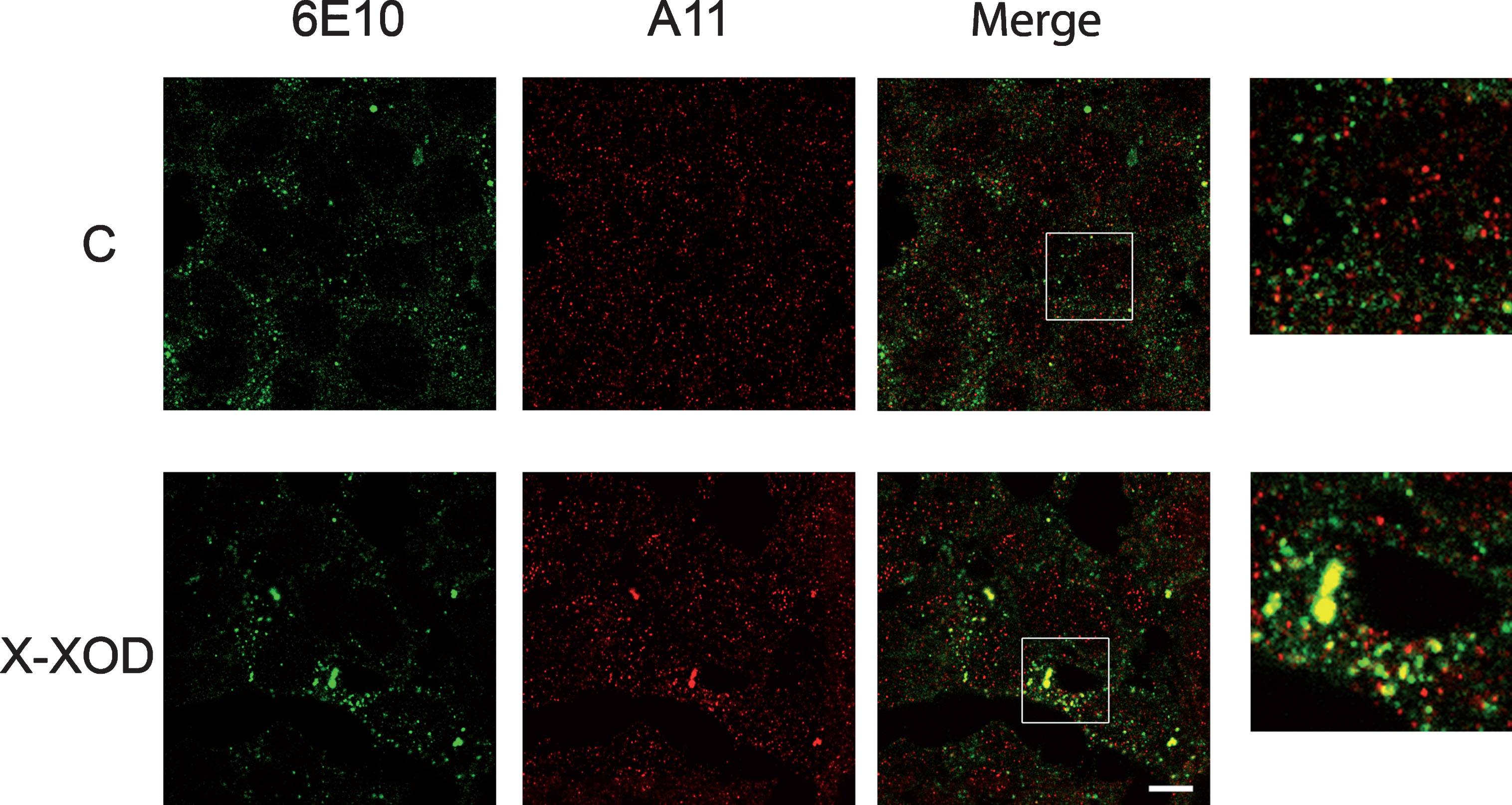
X-XOD produces the intracellular accumulation of amyloid oligomers. SK-N-MC cells subjected to 10μM X/50 mU/ml XOD (X-XOD) for 24 h were examined by double-label confocal microscopy. The representative panel shows immunofluorescence images for the anti-Aβ 6E10 (green) and anti-oligomer A11 (red) antibodies. The merged images show their overlay. For each cell culture, four microscopic fields from three independent cultures were analyzed. Original magnification: 63×. Scale bar: 10μm. No staining was observed when the primary antibodies were omitted.
Double immunofluorescence assays of the A11 antibody with markers of the endolysosomal system were performed and revealed the presence of A11 positive oligomers into CD222 positive organelles (late endosomes) (Fig. 4). Furthermore, a significant increase in the size of the late endosomes, some of which also contained A11 positive structures, was observed in the X-XOD treated cells.

A11 positive oligomers localized in late endosomes. SK-N-MC cells subjected to 10μM X/50 mU/ml XOD (X-XOD) for 24 h were examined by double-label confocal microscopy. The representative panel shows immunofluorescence images for the CD222 late endosome marker (green) and anti-oligomer A11 (red) antibodies. The merged images show their overlay. For each cell culture, four microscopic fields from two independent cultures were analyzed. Original magnification: 63×. Scale bar: 10μm.
X-XOD increases amyloid oligomer levels in partially purified cytosol and endo-lysosomal vesicles
Amyloidogenic processing of AβPP takes place mainly in the endo-lysosomes [12]. Based on the above immunofluorescence results, it was decided to further explore the subcellular location of the oligomers that accumulate under OS conditions. For this purpose, cells were fractionated following the method of Avrahami [28] to separate out a fraction enriched in lysosomes and late-endosomes (L/LE).
Figure 5 shows that the CTSB enzyme was enriched in the L/LE fraction, appearing alongside the endosome and lysosome markers (Rab7 and LAMP1). Large (56 kDa) and intermediate (a less reproducible band of approximately 40 kDa) molecular weight amyloid oligomers revealed by the A11 antibody were detected in the partially purified cytosol (Cyt) fraction, along with the specific marker for the early-endosome EEA1. However, the large oligomer form (56 kDa) and AβPP were preferentially located in the L/LE fraction.

X-XOD increases amyloid oligomer levels in partially purified cytosol and endo-lysosomal vesicles. SK-N-MC cells were treated with 10μM X/50 mU/ml XOD (X-XOD) for 24 h and then fractionated by differential centrifugation. Western blotting was performed using anti-oligomer (A11), anti-AβPP (22C11), anti-CTSB, anti-LAMP1, anti-Rab7, and anti-EEA1 antibodies. The protein quantity determined by a BCA assay was used as the loading control. A representative gel (of three independent experiments) is shown. Cyt, cytosol fraction; L/LE, lysosome-late endosome fraction.
In the X-XOD treated cells, both the 56 kDa oligomer and the AβPP were clearly increased in both fractions. The presence of A11-positive oligomers in the Cyt and L/LE is consistent with the staining pattern obtained in the immunofluorescence analysis (Figs. 3 and 4).
Inhibition of CTSB modulates the AβPP metabolism/processing induced by X-XOD
Although the two treatments studied in this work (X-XOD system and the CTSB inhibitor CA-074Me) induced an increase of amyloid oligomers, their effect on AβPP levels was clearly different, since X-XOD increased hAβPP levels but CTSB inhibitor did not (Fig. 2). This suggested that X-XOD induced OS and the CTSB inhibitor modulate AβPP metabolism by different mechanisms. To deepen into the complexity of AβPP metabolism modulation, we analyzed the levels of amyloid oligomer and hAβPP in cells treated with X-XOD in presence of CA-074Me. Firstly, the quantification of CTSB enzymatic activity with both treatments together (Fig. 6) revealed a complete inhibition (up to 98%; p < 0.001 versus control culture), whereas the results for X-XOD or CA-074Me alone were similar to those described above (Fig. 1 and Supplementary Fig.1). The MTT reduction assay was performed to confirm that this reduced activity was not due to a treatment-induced lessening of cell viability (Supplementary Fig. 3).

X-XOD and CA-074Me reduce CTSB activity. SK-N-MC cells were treated with 10μM X/50 mU/ml XOD (X-XOD) in the presence or absence of 5μM CA-074Me. After incubation for 24 h, CTSB activity was analyzed. Proteolysis in the presence of the fluorogenic substrate z-RR-AMC was used to monitor CTSB activity. The graph shows the mean (±SEM) fluorescence values for each activity level expressed as a percentage of the control value. *p < 0.05, **p < 0.01, and ***p < 0.01 (t-test, n = 4).
The effect of mild OS in the presence of the CTSB inhibitor on the levels of amyloid oligomers and AβPP was then examined by western blotting, using the A11 and 22C11 antibodies respectively. Figure 7A shows that the increase in the large amyloid oligomer induced by X-XOD was attenuated, but not significantly (p = 0.053) in the presence of CA-074Me (0.65±0.15-fold over X-XOD alone). The increase in AβPP protein induced by X-XOD was, however, significantly enhanced in the presence of CA-074Me (3.03±0.57-fold over X-XOD alone; p = 0.0083).

Inhibition of CTSB modulates the AβPP metabolism/processing regulated by X-XOD. SK-N-MC cells were treated with 10μM X/50 mU/ml XOD (X-XOD) in the presence or absence of 5μM CA-074Me. After incubation for 24 h, cell cultures were examined A) intracellularly and B) extracellularly. A) Intracellular: Western blotting analysis was performed using anti-N-terminal AβPP antibody (22C11) and the anti-oligomer antibody A11. A representative gel is shown. In the upper panel, the band corresponds to high-molecular weight amyloid oligomer, in the intermediate to hAβPP, and in the lower panel to α-tubulin. The data (1) show the mean densitometry values (normalized against α-tubulin). The oligomer data for the representative gel are also shown (2). Values for X-XOD were set at 1. ***p < 0.001 (t-test, n = 5). B) Extracellular: secreted AβPP in the culture medium, derived from non-amyloidogenic α-cleavage (sAβPPα), was analyzed by western blotting with the 6E10 antibody. The data show the mean (± SEM) densitometry values. Values for control or X-XOD were set at 1. *p < 0.05, ***p < 0.01 (t-test, n = 4).
The effect of both treatments together on soluble AβPP (sAβPP) secretion was also examined by western blotting using the 6E10 antibody, which recognizes secreted AβPP derived from non-amyloidogenic α-cleavage (sAβPPα). As shown in Fig. 7B, an increase in sAβPPα at the culture medium of cells treated with X-XOD compared to control cells (1.25±0.03-fold; p = 0.011) was observed, being in concordance with previously published results [23]. This increase was further enhanced in the presence of CA-074Me (1.69±0.04-fold over control, p < 0.001; 1.37±0.05-fold over X-XOD alone; p = 0.027).
These results indicate that the AβPP metabolism/ processing induced by mild OS is modulated by inhibiting CTSB, leading to an increase in intracellular hAβPP and secreted sAβPPα.
DISCUSSION
Aβ peptides are produced during the sequential proteolytic processing of AβPP via canonical and no canonical secretases [34]. Their capacity to aggregate can lead to the formation of neurotoxic oligomers and to the accumulation of insoluble deposits in AD brains [35], which constitutes one of the hallmarks of the disease. It is widely accepted that Aβ oligomers are more potent neurotoxins than amyloid fibrils [36, 37], and several have been isolated and studied in detail [38]. However, there is no general consensus regarding the total number of Aβ oligomer types produced in the brain nor on which oligomers are the most important in the pathophysiology of AD [37]. Recently, large oligomeric assembly states of Aβ have been associated with both ageing and AD [7].
Numerous risk factors have been associated with the onset of AD, being one of them the ageing-associated OS [16, 17]. In addition to altering AβPP metabolism, OS affects protein degradation pathways [39]. Delving into this topic, we had previously reported the involvement of the ubiquitin-proteasome and the lysosome protein degradation systems in the regulation of AβPP metabolism/processing by the X-XOD free radical-generating system [23].
Furthermore, changes in the concentration, activity, and localization of the endo-lysosomal cathepsins are habitually seen in aging neurons, and are thought to be of age-related neuropathological significance [40, 41]. Interestingly, CTSB, a cysteine protease that has been associated with AD since it can be found together with Aβ peptides in the extracellular amyloid plaques of AD brains [42], has a potential role in the formation and in the clearance of Aβ peptides (AβPP β-secretase activity and Aβ degradative activity) [13, 14]. In agreement with these data, results of the present work support the involvement of CTSB in the regulation of amyloidoligomer levels.
Cathepsins activity is regulated in different ways— transcription, post-transcription/processing, translation, glycosylation, and trafficking, and also via its binding to endogenous inhibitors [43–45]. In the present study, mild OS induced by the free radical generating system X-XOD produced a reduction of CTSB activity and of CTSB protein levels, similarly to the pharmacological inhibitionby CA-074Me.
CA-074Me inhibition mechanisms are not completely characterized, but there are indeed some reports [46] describing that CA-074Me binds to CTSB in a substrate-like mode, occupying the S’ subsites. The reduction of CTSB levels in the cells treated with CA-074Me may result from the inhibition of its autoproteolytic activity [43]. The regulation of CTSB activity by mild OS is likely to be more complex than its pharmacological inhibition, since X-XOD disrupts the autophagic flux in this cell model [23, 47], probably affecting the trafficking of CTSB towards the endo-lysosomal vesicles.
Western blotting with the A11 antibody (which recognizes oligomeric structures) showed both X-XOD and CA-074Me to affect the levels of soluble amyloid oligomers, revealing a significantly increased large molecular weight soluble amyloid oligomer (56 kDa). This oligomer was formed by Aβ but not by βCTFs [8, 33] since it was recognized by the 6E10 monoclonal antibody (which attaches to Aβ) but not by the AβPP C-terminal specific antibody. Nevertheless, the immunoreactivity of A11 in the 6E10 immunoprecipitated material decreased in X-XOD treated compared to the control conditions, in contrast with the increase observed in whole cell lysates (Fig. 2). This apparent contradiction could be explained by the presence of post-translational modifications in the Aβ oligomers induced by oxidative stress [48, 49] that would provoke a decrease of the affinity of 6E10 antibody. In this respect, molecular modifications in Aβ peptide induced by oxidative stress have been reported (for recent review, see [50, 51]). Besides, in X-XOD treated cells, the levels of the oligomer diminished in the presence of the γ-secretase inhibitor DAPT, further supporting the idea that it contains mainly Aβ peptides and not C-terminal fragments of AβPP. On the other hand, that decrease of A11 specific band levels observed in presence of γ-secretase inhibitor was not very high/significant what reinforces the notion that 56 KDa Aβ oligomer generation is not dependent only on gamma-secretase cleavage, but may involve additional unknown mechanism(s).
Regarding the intracellular location of the oligomer, a punctate pattern of A11-positive structures distributed throughout the cytoplasm was detected both in control and X-XOD treated cells whereas this mild OS led to the appearance of enlarged vesicles, compatible with endo-lysosomes, positive for both A11 and 6E10; they therefore would contain amyloid oligomers, and probably AβPP/βCTF/Aβ too. Actually, vesicles stained by a CD222 specific antibody, corresponding to late endosomes, were found to contain A11 positive structures and to be significantly enlarged in X-XOD treated cells. In summary, these experiments revealed that A11 positive oligomers were distributed throughout the cell, both outside and inside endolysosomal vesicles, being this last location compatible with that described for the enzymes and products of the amyloidogenic proteolysis of AβPP [12, 52].
The data obtained after subcellular fractionation of the SK-N-MC cells revealed the presence of the 56 kDa oligomer and AβPP in the cell fraction enriched in endo-lysosomal vesicles. Moreover, the present results also showed CTSB to be mainly localized in the endo-lysosome enriched fraction, which is in line with data reported by other authors [43] and reinforces the probability of its involvement in the formation and/or degradation of amyloid oligomers. Interestingly, previous studies in AD and Down’s syndrome have revealed activated or enlarged endosomes containing soluble Aβ, prior to the deposition of amyloid plaques [53].
Since the previous results suggested that the modulation of AβPP metabolism/processing by X-XOD and by the CTSB inhibitor CA-074Me occurs via different mechanisms, we aim to further characterize the role of CTSB in the modulation of AβPP metabolism induced by X-XOD, by analyzing the changes of amyloid oligomer and hAβPP levels in the presence of both treatments. If X-XOD modulates oligomer levels via the same mechanism as CA-074Me, a higher increase in oligomer levels after the combination of both treatments would be expected; nonetheless, we observed no such effect, which reinforced our above-mentioned conclusion that the modulation of AβPP metabolism/processing by both treatments takes place via different mechanism. In contrast to the absence of a combined effect on the levels of oligomers, we found a marked increase in intracellular AβPP and extracellular sAβPPα levels when cells were treated with X-XOD in the presence of CA-074Me, suggesting that CTSB participates in different steps of AβPP metabolism induced by mild OS.
In the light of these results, we propose that the modulation of AβPP metabolism by mild OS is partly mediated by CTSB, but that the effects of OS most probably involve additional mechanisms, like its involvement on the ubiquitin proteasome pathway [23], or its general effect on the lysosomal function [23, 47] that we reported previously.
Although the mechanisms underlying the relations among Aβ/CTF aggregation, free radical damage and cell death remain unclear, recent reports support the involvement of CTSB in the induction of inflammatory response by Aβ oligomers [54] or in the degradation of the key AD proteins [41].
In summary, the present results support the participation of CTSB in the regulation of AβPP metabolism by mild OS, although further research on its specific role and on the contribution of additional lysosomal activities in that regulation is guaranteed. This work also suggests that CTSB could serve as a therapeutic target for the treatment of AD, in line with previous studies in cellular and animal models of the disease [13, 55]. Besides, knowing the relationship between lysosomal function, OS, and AβPP metabolism/processing could help us to better understand the pathophysiology of the disease.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the Ministerio de Ciencia e Innovación (SAF2014-53954-R). Llorente P is supported via the Fundación Severo Ochoa. The institutional grants of Fundación Ramón Areces and Banco de Santander to the Centro de Biología Molecular Severo Ochoa is gratefully acknowledged.