
Abstract
Alzheimer’s disease (AD) is a complex and progressive neurological disorder, and amyloid-β (Aβ) has been recognized as the major cause of AD. Inhibiting Aβ production and/or enhancing the clearance of Aβ to reduce its levels are still the effective therapeutic strategies pursued in anti-AD research. In previous studies, we have reported that selenomethionine (Se-Met), a major form of selenium in animals and humans with significant antioxidant capacity, can reduce both amyloid-β (Aβ) deposition and tau hyperphosphorylation in a triple transgenic mouse model of AD. In this study, a Se-Met treatment significantly decreased the Aβ levels in Neuron-2a/AβPPswe (N2asw) cells, and the anti-amyloid effect of Se-Met was attributed to its ability to inhibit Aβ generation by suppressing the activity of BACE1. Furthermore, both the LC3-II/LC3-I ratio and the number of LC3-positive puncta were significantly decreased in Se-Met-treated cells, suggesting that Se-Met also promoted Aβ clearance by modulating the autophagy pathway. Subsequently, Se-Met inhibited the initiation of autophagy through the AKT-mTOR-p70S6K signaling pathway and enhanced autophagic turnover by promoting autophagosome-lysosome fusion and autophagic clearance. Our results further highlight the potential therapeutic effects of Se-Met on AD.
INTRODUCTION
As the most common form of dementia, Alzheimer’s disease (AD) is a global public issue that may contribute to 60–80% of dementia cases [1]. AD is characterized by cognitive impairment in clinical presentation with two classical neuropathological hallmarks: the accumulation of extracellular senile plaques [2] and intracellular neurofibrillary tangles in the brain [3]. Amyloid-β (Aβ) peptide, the main component of senile plaques, is predominantly generated as a 40–42-amino acid peptide by the sequential cleavage of the amyloid-β protein precursor (AβPP) by β- and γ-secretase [4]. Therefore, AβPP, β-site AβPP cleaving enzyme 1 (BACE1), and γ-secretase are involved in the process of Aβ production and mainly exert neurotoxic properties by generating Aβ. In addition, it has also been suggested that a failure to clear Aβ acts as an important mechanism by which Aβ accumulates in neuronal cells and exacerbates the pathogenesis of AD [5].
Autophagy is a highly conserved and lysosome-dependent degradation process that is primarily responsible for the degradation of long-lived or aggregated proteins and damaged organelles to maintain cellular homeostasis, and neurons are more dependent on high basal autophagy levels for survival than other cells [4 , 7]. Disturbed autophagy is an essential factor contributing to the pathogenesis of AD [8]. Based on accumulating evidence, an impairment in the autophagosomal-lysosomal degradation pathway disturbs AβPP processing and is associated with the increased Aβ levels observed in AD. Autophagy enhancers or the overexpression of autophagy-related genes, such as beclin-1, ameliorated Aβ deposition in AD mice [9, 10]. Reversal of the autophagy dysfunction (either by promoting autophagosome formation or enhancing lysosomal digestion) decreased amyloid pathologies and memory deficits in TgCRND8 mice [11]. Therefore, autophagy might play an important role in protecting against Aβ-induced neurotoxicity in AD.
Selenium (Se), an essential trace element nutrient with well-known antioxidant potential, protects the brain from oxidative damage in various neurodegeneration models [12, 13]. Moreover, experimental and clinical studies have reported a positive role for Se in cognitive performance in aging and AD [14 –16]. Sodium selenite and sodium selenate, two main inorganic Se compounds, have been shown to protect neurons against Aβ42-induced toxicity and mitigate cognitive deficits in AD models [17, 18]. Selenomethionine (Se-Met), the major organic form of selenium in yeasts, plants, and mammals, protected against Aβ- and iron/hydrogen peroxide-mediated death of primary cortical neurons [19]. Our previous studies also revealed a significant improvement in the spatial learning and memory deficits of 3×Tg-AD mice that had been treated with Se-Met for 3 months [20]. Se-Met also decreased the deposition of Aβ by modulating β-secretase levels in 3×Tg-AD mice [21]. In this study, we sought to determine whether Se-Met modulates the production and clearance of Aβ in an AD cell model, Neuron-2a/AβPPswe cells.
MATERIALS AND METHODS
Cell culture
A murine neuroblastoma cell line stably transfected with the human AβPP-695 Swedish mutation (K595N/M596L) was used and is referred to here as N2asw. These cells were maintained in a selective and undifferentiated state by the antibiotic G-418 at a final concentration of 0.2 mg/ml in media containing 45% Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, Carlsbad, CA) and 55% Opti-MEM (Gibco) supplemented with 5% fetal bovine serum (FBS) in a humidified 5% CO2/95% air atmosphere at 37°C.
Transfection, rapamycin, and bafilomycinA1 treatment
The EGFP-LC3 and mTagRFP-mWasabi-LC3 plasmids were a generous gift from Dr. Cuihong Zhou (Peking University). Approximately 1.4×105 cells were plated in each well of a 24-well plate 24 h before transfection. Plasmid (500 ng/well) transfections were performed with Lipofectamine 2000 transfection reagent (Invitrogen, 11668) according to the manufacturer’s protocol. Cells were pretreated with rapamycin (100 nM, 2 h) or bafilomycin A1 (50 nM, 4 h) before being treated with Se-Met for 24 h to explore the effects of Se-Met on mTOR pathway or autophagic flux in N2asw cells.
Western blot
Cultured cells were collected after treatments, homogenized in RIPA buffer containing 1 mM PMSF, 25 μM leupeptin, and 1 μg/mL aprotinin, and centrifuged at 4°C for 0.5 h at 13,000 g. Then, the supernatant was collected. The protein concentration was determined using the BCA protein assay kit (Pierce, Rockford, IL). Protein samples (25 μg) were separated on a 10–15% sodium dodecyl sulfate-polyacrylamide gel and then transferred to 0.45 nm polyvinylidene difluoride membranes (Millipore, USA), followed by overnight incubation with the following primary antibodies at 4°C: anti-Aβ (6E10) from Covance (USA); anti-β-CTF (C-terminal fragment of AβPP generated by β-secretase) from Millipore (USA); anti-mammalian target of rapamycin (mTOR), anti-p-mTOR, anti-p70S6K, anti-p-p70S6K, anti-AMP-activated protein kinase (AMPK), anti-p-AMPK, anti-Akt, anti-p-Akt, anti-glycogen synthase kinase 3β (GSK3β), and anti-p-GSK3β from Cell Signaling Technology (USA); anti-AβPP and anti-BACE1 from Abcam (UK); anti-LC3-II from Sigma (USA); and anti-p62 and anti-cathepsin D from Santa Cruz Biotechnology (USA). In addition, the membranes were incubated with HRP-conjugated secondary antibodies (anti-mouse and anti-rabbit, NeoBioscience, China) in Tris-buffered saline with Tween (TBST) and 5% nonfat milk for 1 h at 37°C. The membranes were developed using an ECL kit and scanned with the Image Station 4000 M. The intensity of the bands was quantified by densitometric scanning with ImageJ and normalized to the β-actin or GAPDH levels (Proteintech, USA).
Immunofluorescence
Cells were cultured with the appropriate treatments in a glass-bottom cell culture dish, fixed with 4% paraformaldehyde in PBS (pH = 7.4) for 15 min and permeabilized with PBS containing 0.2% Triton X-100 for 10 min at room temperature. Then, cells were blocked with 5% bovine serum albumin (BSA) for 30 min, incubated with a primary antibody overnight at 4°C, and detected using Alexa Fluor fluorescent dye-conjugated secondary antibodies (anti-mouse and anti-rabbit; Alexa Fluor 488 and 695, Multi-Sciences Biotech). The immunoreactive areas in these sections were imaged with a fluorescence microscope for analysis and quantification (Zeiss, German).
Transmission electron microscopy
Transmission electron microscopy was performed using a previously described protocol [22, 23]. Briefly, cells were collected and then fixed with 2.5% glutaraldehyde for 2 h and 1% osmium tetroxide for 2 h at 4°C. After washing 3 times with distilled water, samples were dehydrated in a graded ethanol series and embedded in epoxy resin. Then, the samples were cut into ultrathin sections with a Leica ultramicrotome using a DiATOME knife, stained with 2% uranyl acetate and 2% lead citrate and analyzed under an FEI Tecnai G2 Spirit transmission electron microscope.
Statistical analysis
The data were analyzed using GraphPad Prism software. All data are expressed as the mean±standard error of the mean (SEM) and considered statistically significant at a level of p < 0.05. A t-test was used to analyze the data from the immunofluorescence staining and western blots.
RESULTS
Se-Met decreased the levels of Aβ oligomers in N2asw cells
We measured the Aβ levels in N2asw cells that had been incubated with or without Se-Met (1 μM) for 24 h using western blots to determine whether Se-Met decreased the Aβ content. Compared with the control cells (N2a cells), N2asw cells showed a significant increase in the levels of Aβ oligomers; Se-Met treatment decreased the levels of Aβ oligomers (at 8 kD, 24 kD, and 100 kD) in N2asw cells (p < 0.01, p < 0.05, p < 0.05, respectively; Fig. 1A, B). The results of the immunofluorescence analysis were consistent with the western blot; Se-Met reduced the deposition of Aβ in N2asw cells (Fig. 1C).
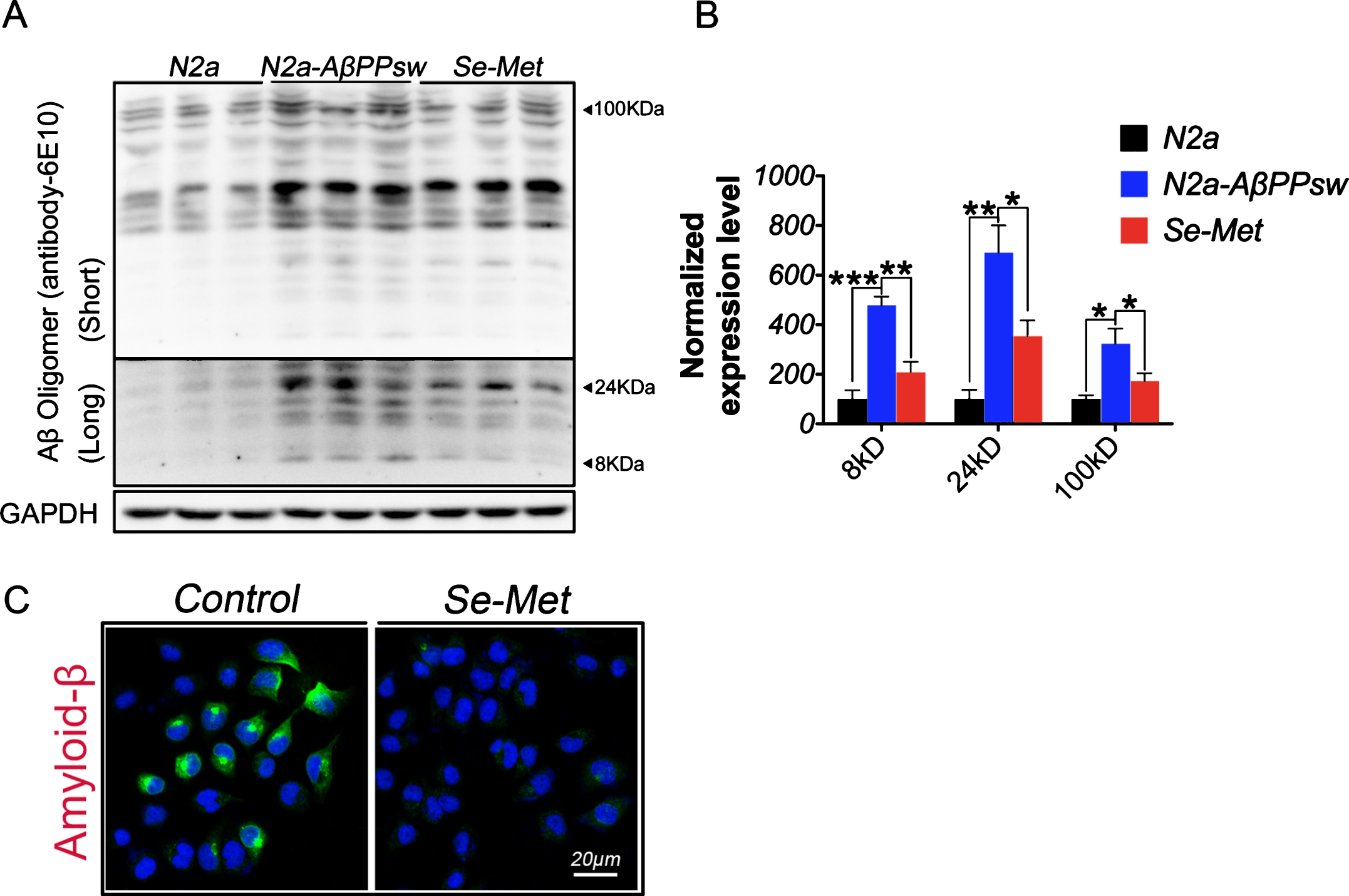
Se-Met reduced the Aβ levels in N2asw cells. N2asw cells were treated with vehicle (double-distilled water) or Se-Met (1 μM). A) The levels of Aβ oligomers in cell lysates were detected using western blot. B) The quantitative analysis showed that Se-Met significantly reduced the levels of Aβ oligomers (approximately 8 kD, 24 kD, and 100 kD) in N2asw cells. Quantitative data were normalized to the beta-actin levels. Values are expressed as percentages of the control (set to 100%) and are presented as the group mean±SEM (n = 6). * p < 0.05, ** p < 0.01, *** p < 0.001 versus the control group. C) Immunofluorescence staining using the 6E10 antibody revealed that Se-Met noticeably reduced Aβ deposition in cells. Scale bars, 20 μm.
Effects of the Se-Met treatment on cellular Aβ production
Because Aβ levels are regulated by a dynamic equilibrium between Aβ generation and clearance, we first measured the AβPP levels in N2asw cells. Se-Met significantly reduced the expression of full-length AβPP in N2asw cells (p < 0.05, Fig. 2A, B).AβPP undergoes two well-known sequential steps of proteolytic processing. The first is mediated by BACE1/β-secretase to generate soluble AβPP peptides-β (sAβPPβ) and a β-C-terminal fragment (β-CTF, C99) in the amyloidogenic pathway, or α-secretase to generate soluble AβPP peptides-α (sAβPPα) and a α-C-terminal fragment (α-CTF, C83) favoring the non-amyloidogenic pathway; then the second is mediated by γ-secretase cleavage to finish AβPP process [24, 25]. Western blot analyses revealed a significant reduction in the sAβPPβ, β-CTF, and BACE1 levels in the Se-Met-treated cells (p < 0.01, p < 0.05, p < 0.05, respectively; Fig. 2A, B), However, Se-Met did not induce any changes in the levels of sAβPPα (Fig. 2A, B). Thus, Se-Met inhibited Aβ production by modulating AβPPprocessing.
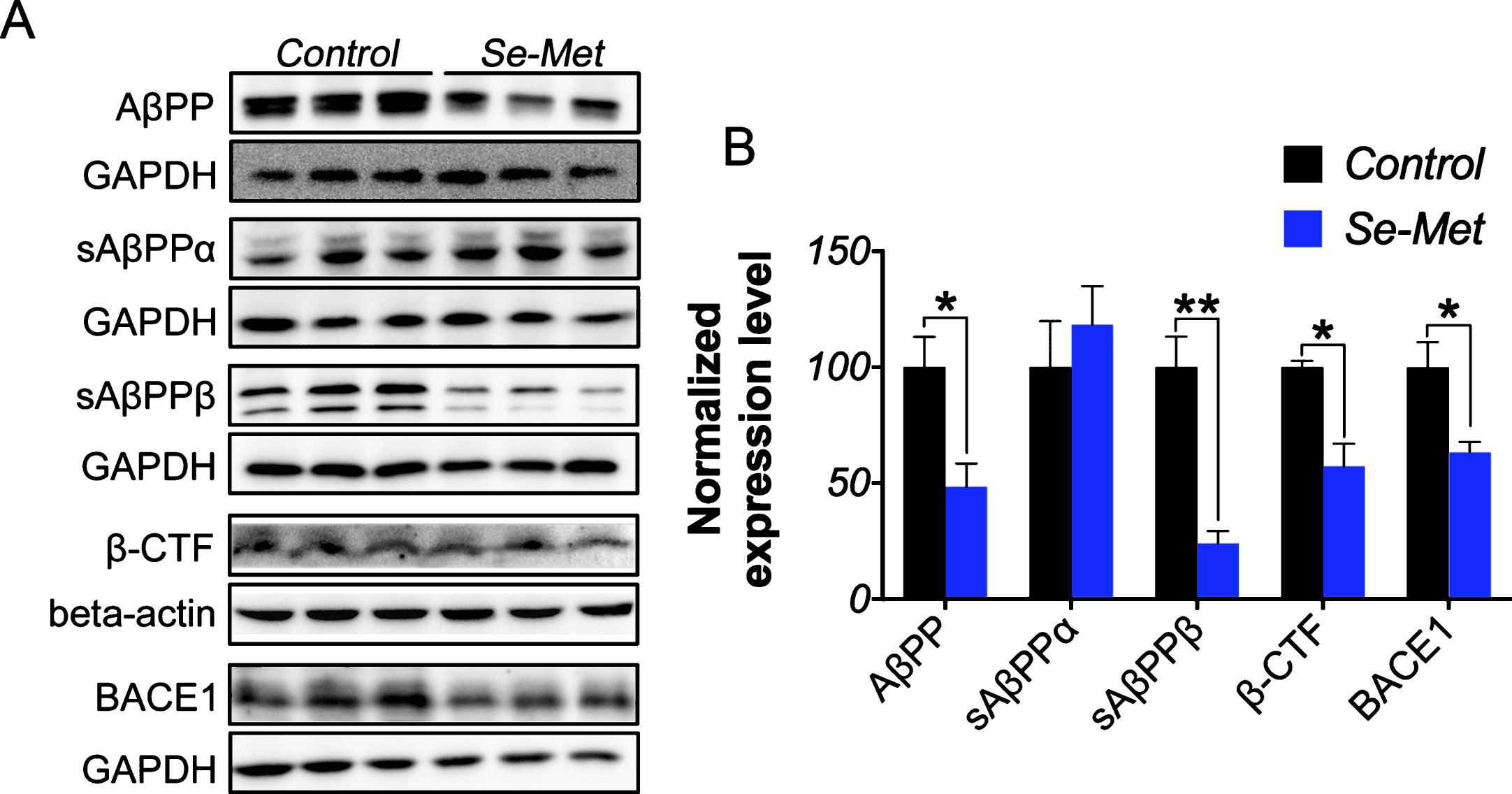
Se-Met downregulated the amyloidogenic pathway to decrease cellular Aβ generation. A) The expression levels of AβPP, sAβPPα, sAβPPβ, β-CTF, and BACE1 in cell lysates were detected using western blot. B) The quantitative analysis showed that Se-Met significantly reduced the AβPP, sAβPPβ, β-CTF, and BACE1 levels in N2asw cells. The quantitative data were normalized to the GAPDH levels. The values are expressed as percentages of the control (set to 100%) and are presented as the group mean±SEM (n = 4&6). * p < 0.05, ** p < 0.01 versus the control group.
Se-Met reduced the Aβ levels by promoting autophagy in N2asw cells
We explored the effect of the Se-Met treatment on autophagy to determine whether the protein clearance mechanism also played a role in the Se-Met-induced reduction of the Aβ levels. Light chain 3 (LC3), a selective marker for autophagy, is used to identify specific subpopulations of autophagosomes. The processing of full-length LC3 (LC3-I) to its cleaved form (LC3-II) is an important step in autophagosome membrane formation and autophagic flux [26]. The western blot analysis showed a significant increase in the LC3-II (16 kD)/LC3-I (18 kD) ratio in N2asw cells compared with N2a cells (p < 0.05, Fig. 3A, B). Whereas the LC3-II/LC3-I ratio was significantly decreased in the Se-Met-treated N2asw cells compared with the untreated control cells (p < 0.01, Fig. 3A, B). Thus, Se-Met decreased the formation of autophagosomes. We transfected N2asw cells with an LC3-GFP plasmid to further validate this result and found that the density of LC3-positive puncta in the Se-Met-treated cells was obviously less than the density in the control cells (Fig. 3C). The subsequent transmission electron microscopy analysis also revealed fewer autophagosomes in the Se-Met-treated group (Fig. 3C, D). We detected the colocalization of LC3 and Aβ in N2asw cells by immunofluorescence using specific antibodies against LC3II and Aβ to clarify the relationship between the decreased number of autophagosomes and the reduced Aβ levels. As shown in Fig. 3E, the numbers of both the Aβ-positive and LC3-positive puncta were obviously reduced and the percentage of Aβ and LC3 colocalization was increased in the Se-Met-treated cells relative to the control cells. The subsequent super-resolution immunofluorescence imaging revealed an accurate positional relationship between LC3 and Aβ: Aβ puncta were directly surrounded by LC3 puncta. Thus, Aβ was phagocytized byautophagosomes and thereby cleared through the autophagic pathway (Fig. 3F).
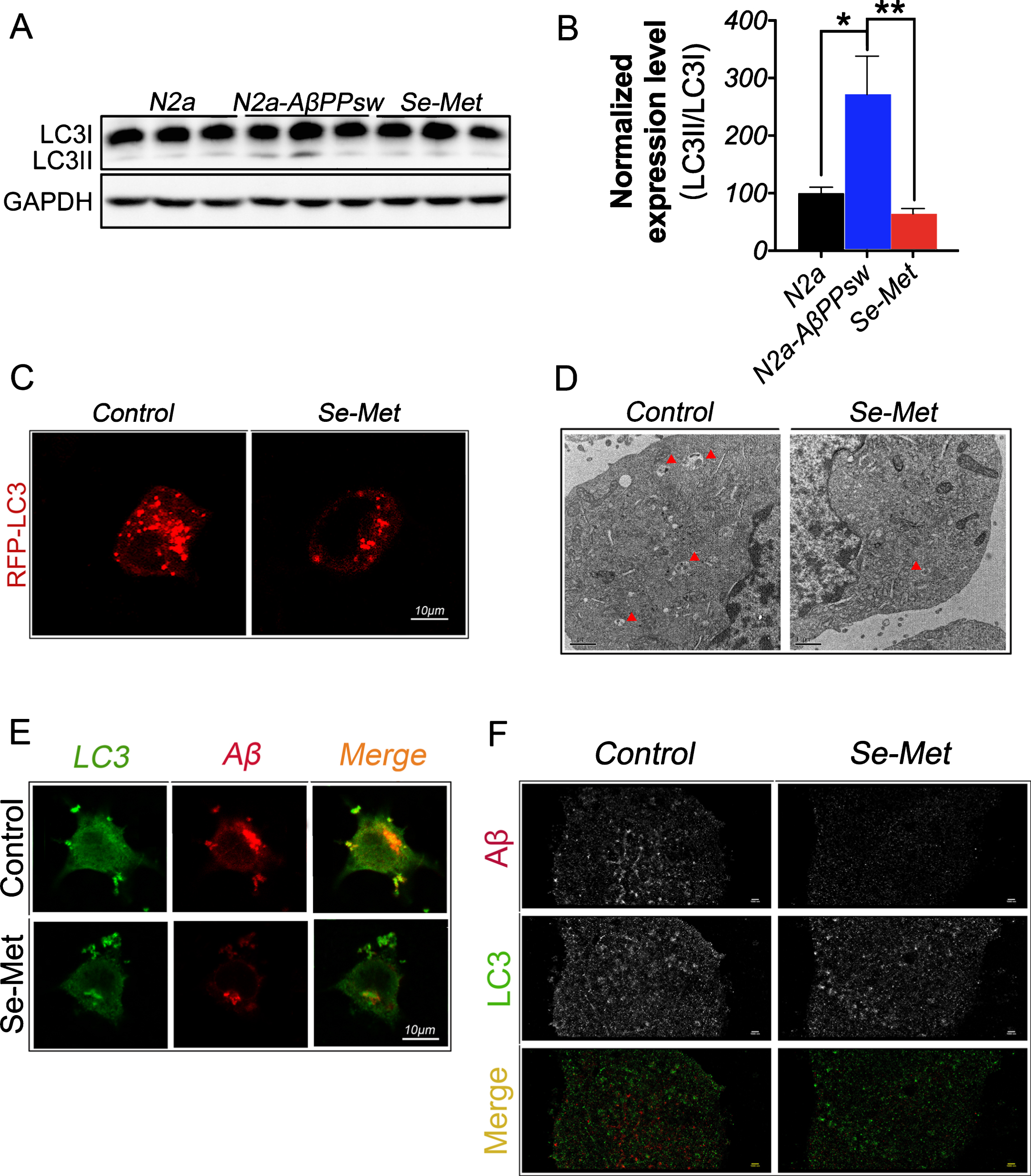
Se-Met decreased the level of autophagy and increased the colocalization of LC3 and Aβ in N2asw cells. A, B) The LC3II/LC3I ratio was significantly decreased in the Se-Met-treated group. The quantitative data were normalized to the GAPDH levels. The values are expressed as percentages of the control (set to 100%) and are presented as the group mean±SEM (n = 6). * p < 0.05, ** p < 0.01 versus the control group. C) Representative fluorescence images of LC3-GFP-transfected N2asw cells (red) show that Se-Met significantly decreased the number of LC3-positive puncta. Scale bars, 10 μm. D) The double membrane-bound autophagosomes in N2asw cells were observed using transmission electron microscopy. Scale bars, 1 μm. E) Immunofluorescence staining revealed that Se-Met significantly reduced the LC3 levels and the Aβ deposition in cells but increased the colocalization of LC3 and Aβ. Scale bars, 10 μm. F) Super-resolution imaging of the immunofluorescence staining for Aβ and LC3. Scale bars, 1 μm.
The Akt/GSK3β and Akt/mTOR/p70S6K signaling pathways were both involved in Se-Met-induced inhibition of autophagy
GSK3β has been reported to regulate Aβ production, a GSK3β-specific inhibitor reduces BACE1-mediated AβPP cleavage and favors AβPP degradation via autophagy [27]. Thus, the expression levels of GSK3β and GSK3β phosphorylation at Ser9 (p-GSK3β, the inactive form of GSK3β) were detected by western blot to determine whether GSK3β was involved in the Se-Met-induced inhibition of the amyloidogenic pathway. Se-Met significantly increased the level of p-GSK3β without changing the level of GSK3β (p < 0.05, Fig. 4A, B),indicating that Se-Met inhibited GSK3β activity in N2asw cells. As an important upstream protein kinase that phosphorylates GSK3β at Ser9 [28], the levels of Akt and Akt phosphorylation at Ser473 (p-Akt, the active form of Akt) were also detected. As shown in Fig. 4A and C, the levels of both Akt and p-Akt were significantly increased in the Se-Met-treated cells (p < 0.01 and p < 0.05, respectively;Fig. 4A, C).

Se-Met activated Akt, mTOR, and p70S6K while inhibiting GSK3β in N2asw cells. A) Representative western blots showing the GSK3β, pGSK3β (Ser9), Akt, p-Akt (Ser473), mTOR, p-mTOR (Ser2448), p70S6K, and p-p70S6K (Thr389) levels in control and Se-Met-treated N2asw cells. B-E) The quantitative analysis of the expression levels of these proteins showed that Se-Met significantly reduced the pGSK3β (Ser9) levels without changing the GSK3β levels and increased the levels of Akt, p-Akt (Ser473), mTOR, p-mTOR (Ser2448), p70S6K, and p-p70S6K (Thr389). The quantitative data were normalized to the α-tubulin or beta-actin levels. The values are expressed as percentages of the control (set to 100%) and are presented as the group mean±SEM (n = 6). * p < 0.05, ** p < 0.01.
The mTOR/p70S6K pathway, another downstream target of Akt, plays an important role in regulating the initiation of autophagy [29, 30]. Se-Met significantly increased the expression levels of mTOR, p-mTOR (Ser2448, the active form of mTOR), p70S6K, and p-p70S6K (Thr389, the active form of p70S6K) (p < 0.01, p < 0.05, p < 0.05, and p < 0.01, respectively; Fig. 4A, D, E). To further determine whether the mTOR pathway was involved in Se-Met-induced inhibition of autophagy, a specific inhibitor of mTOR (Rapamycin, 100 nM/2 h), was used before Se-Met treatment. The western blot analysis showed rapamycin significantly reduced the p-mTOR levels (p < 0.001, Fig. 5A, B) without changing the mTOR levels in N2asw cells, which indicated rapamycin could inhibit the mTOR activity. Although the mTOR level significantly increased (p < 0.001, Fig. 5A, B), Se-Met could not improve the p-mTOR levels or decrease LC3-II/LC3-I ratio in rapamycin pretreated N2asw cells, suggesting that the effect of Se-Met-induced inhibition of autophagy was impeded when the mTOR activity was blocked by rapamycin in N2asw cells. Therefore, Se-Met decreased the Aβ content in N2asw cells by modulating both the Akt/GSK3β and mTOR/p70S6Kpathways.
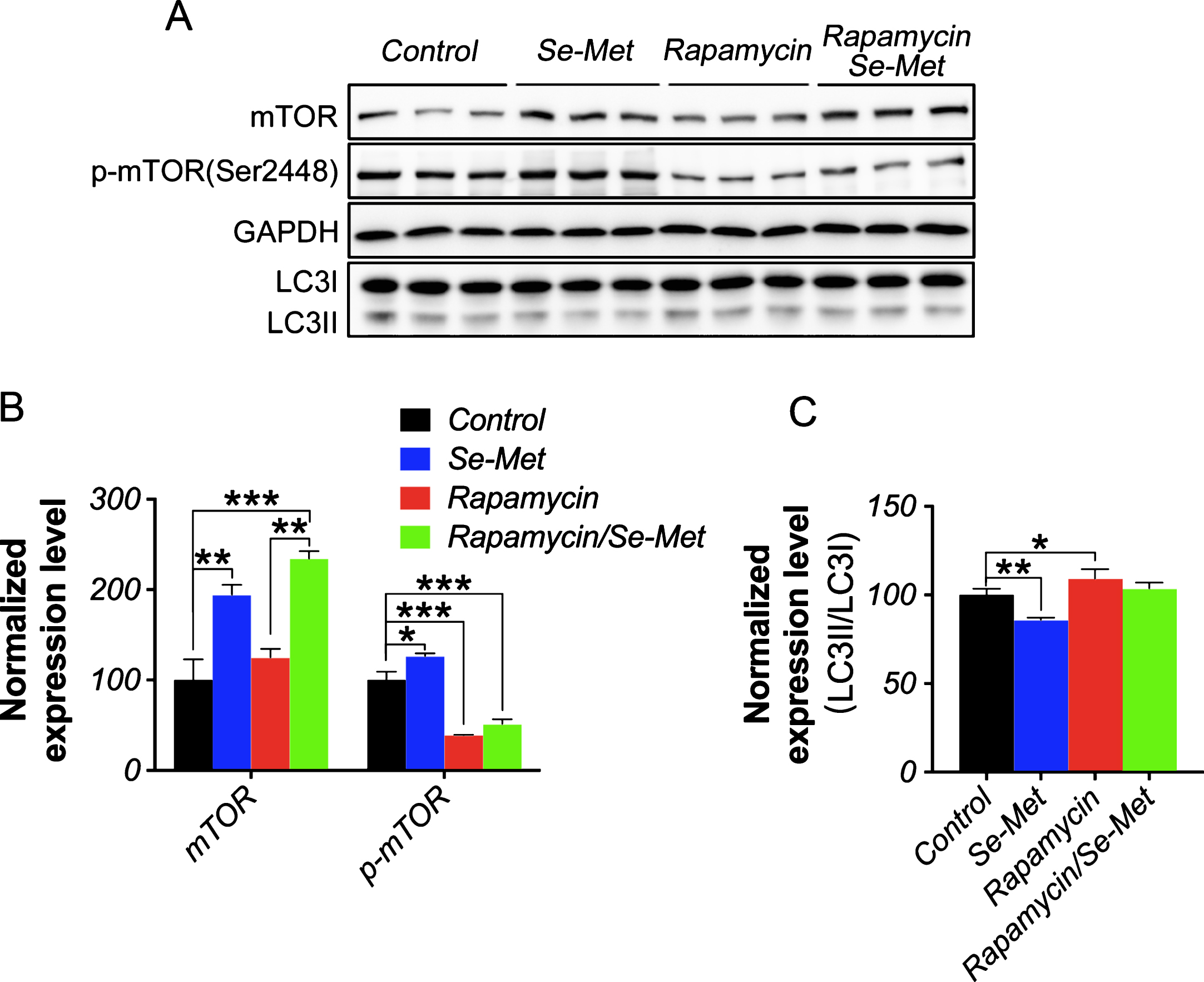
Rapamycin suppressed the Se-Met-induced inhibition of autophagy in N2asw cells. A, B, C) Representative western blot images and densitometric analysis of mTOR, p-mTOR (Ser2448), LC3II, LC3I levels in control, Se-Met-treated, rapamycin-treated, and rapamycin-Se-Met-treated N2asw cells. The quantitative data were normalized to the GAPDH levels. The values are expressed as percentages of the control (set to 100%) and are presented as the group mean±SEM (n = 6). * p < 0.05, ** p < 0.01, *** p < 0.001.
Se-Met promoted autophagosome–lysosome fusion and increased protein clearance
Defects in autophagosome maturation at the stage of autophagosome-lysosome fusion have been reported to be a general feature of AD pathology [31]. Therefore, the expression levels of p62/SQSTM1, an autophagy adaptor protein that is degraded upon autophagosome-lysosome fusion [32], and cathepsin D (CatD), the principal aspartate protease responsible for degrading engulfed proteins in the lysosome [33], were detected to determine whether Se-Met can promote autophagic maturation. As shown in the western blot analysis, the p62 levels were significantly decreased in the Se-Met-treated cells (p < 0.05, Fig. 6A, B). The CatD precursor (46–52 kD) is activated by proteolysis in the acidified lysosome to produce a mature proteolytic product (mCatD) (32 kD). The expression level of mCatD was significantly increased in Se-Met-treated cells compared to the control cells (p < 0.01, Fig. 6A, B). Se-Met enhanced the fusion of autophagosomes and lysosomes and thus promoted autophagic degradation.

Se-Met decreased the p62 levels and increased the cathepsin D levels. A, B) Representative western blots, showing the p62 and cathepsin D (Cat D) levels in N2asw cells. The quantitative data were normalized to the beta-actin levels. The values are expressed as percentages of the control (set to 100%) and are presented as the group mean±SEM (n = 6). * p < 0.05, ** p < 0.01. C) Representative fluorescent images of N2asw cells that had been transiently transfected with mTagRFP-mWasabi-LC3 and treated with Se-Met. Scale bars, 10 μm.
A tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) plasmid was transfected into N2asw cells to monitor autophagic flux using the different pH stabilities of green and red fluorescent proteins to ascertain the effect of Se-Met on promoting autophagosome-lysosome fusion. As described above, the numbers of both yellow and red puncta increased when autophagic flux was increased in cells, and only the number of yellow puncta increased when autophagic flux was blocked in cells [34]. Se-Met notably increased the number of yellow and red puncta in the cells compared with the control group (Fig. 6C), which is further evidence that Se-Met increases the fusion of autophagosomes andlysosomes.
Se-Met increased Aβ clearance by regulating autophagic flux
Finally, we examined whether inhibition of the autophagic pathway influenced the degradation of amyloid peptides in N2asw cells. Bafilomycin A1, an autophagic flux inhibitor, blocks the fusion of autophagosomes with lysosomes and thus inhibits the autophagic degradation of proteins. The Se-Met treatment significantly reduced the levels of both AβPP and Aβ oligomers, whereas bafilomycin A1 obviously increased the levels of Aβ oligomer in N2asw cells (p < 0.01, Fig. 7A, B). Se-Met significantly ameliorated the bafilomycin A1-induced increase in the Aβ oligomer levels (p < 0.05, Fig. 7A, B). Importantly, based on these data, Se-Met modulated autophagic flux to promote autophagic proteolysis, thereby eliminating Aβ in N2asw cells.
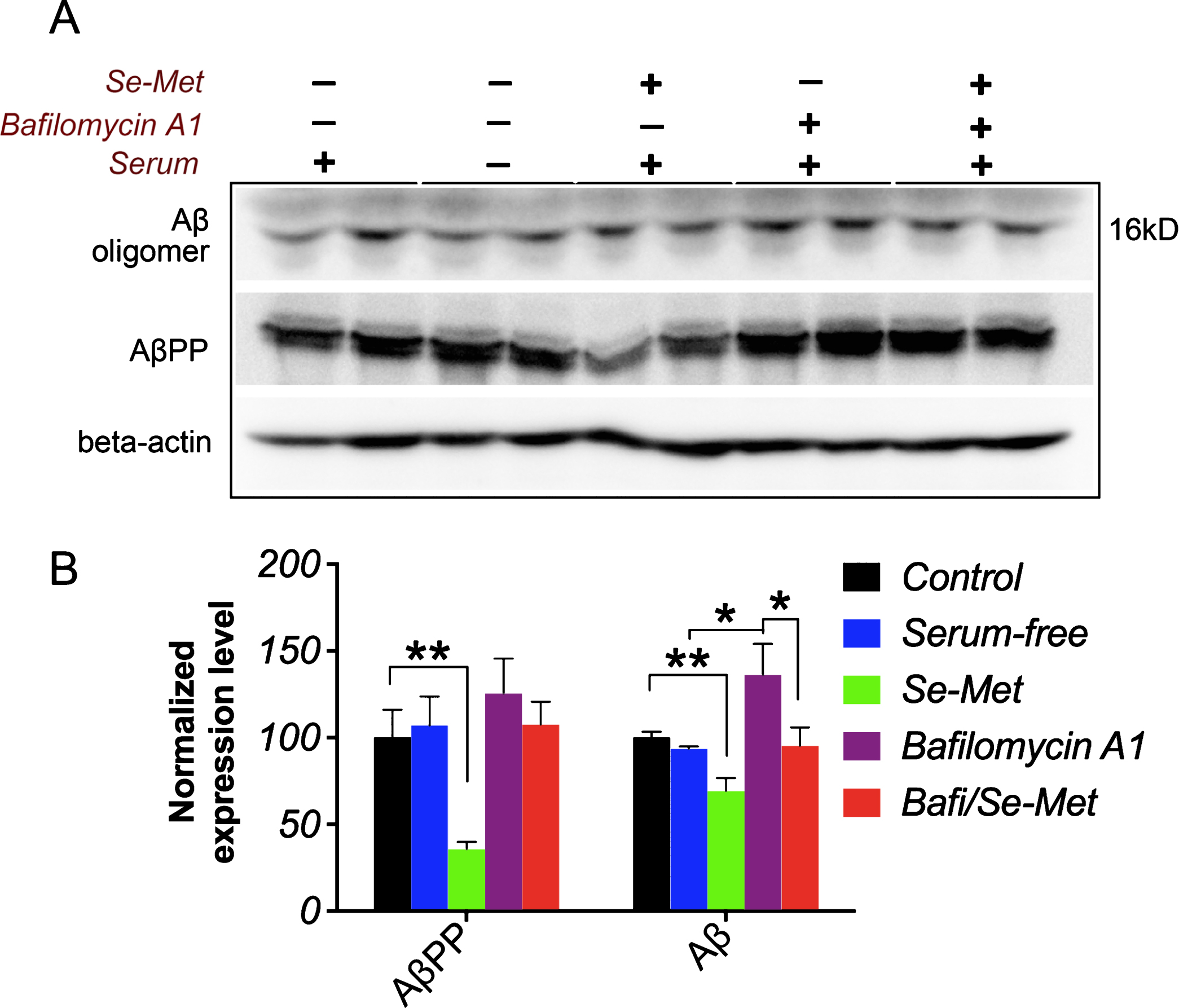
Se-Met reduced the bafilomycin A1-induced increase in the Aβ oligomer levels. A, B) Representative western blots showing the levels of AβPP and Aβ oligomers in N2asw cells. The quantitative data were normalized to the beta-actin levels. Values are expressed as percentages of the control (set to 100%) and are presented as the group mean±SEM (n = 4). * p < 0.05, ** p < 0.01.
DISCUSSION
Although many factors have been reported to contribute to the pathogenesis of AD, Aβ is still the most extensively validated therapeutic target. The imbalance between the production and clearance of Aβ and related peptides is understood to be responsible for initiating the pathological cascade that eventually leads to AD. Agents that can decrease Aβ production and/or increase Aβ clearance have been confirmed to be effective in retarding AD progression [35, 36]. In our previous study, Se-Met ameliorated the decline in cognitive function and reduced Aβ deposition and tau hyperphosphorylation in AD mice and might thus be a promising therapeutic option for AD [20, 21]. In the present study, Se-Met was a suitable agent for ameliorating Aβ pathology in vitro and we tried to clarify the underlying mechanism by investigating both the inhibition of Aβ production and the promotion of Aβ clearance.
Aβ is generated from AβPP by the successive actions of BACE-1 and γ-secretase. As a rate-limiting enzyme in the amyloidogenic pathway, the expression level and activity of BACE1 are elevated in the brains of patients and mice with AD, and blocking the proteolytic activity of BACE1 suppresses Aβ generation [37, 38]. Downregulation of the BACE1 levels has been regarded as an important strategy for anti-AD therapy [39]. Over the past few decades, extensive efforts have been directed toward potent inhibitors of BACE1 activity for AD therapy. GSK3β is currently regarded as a critical molecular link between the two major pathological pathways (amyloid and tau) in AD. A similar increase in the activity of GSK3β has also been observed in the brains of patients with AD [40]. Activated GSK3β was strongly associated with the increased levels of BACE1 observed in AD [27 , 42]. GSK3β inhibition reduced BACE-1-mediated cleavage of AβPP and thus reduced Aβ pathology by decreasing the transcription and the expression of the BACE1 gene [27]. Here, in addition to downregulating the AβPP levels, Se-Met effectively inhibited GSK3β activity by activating Akt, thus decreasing the BACE1 expression level and activity in N2asw cells to inhibit Aβ production.
Based on accumulating evidence, Aβ aggregates, particularly the oligomeric states, are more toxic than fibrils in causing memory loss and synaptic dysfunction in AD [43, 44]. Modulation of autophagy has been proposed as a reasonable strategy to help neurons clear abnormal protein aggregates, as autophagy dysfunction contributes substantially to the abnormal protein generation and accumulation (such as Aβ) observed in AD [45, 46]. Strategies that promoted autophagy by regulating several key signaling pathways, including the Akt-mTOR-p70S6K pathway, protected neurons from Aβ-induced cytotoxicity and enhanced the clearance of protein aggregates in vitro and in vivo [47 –50]. The kinase mTOR is a major modulator of autophagy and a downstream target of Akt kinase. Activation of mTOR by Akt leads to phosphorylation of its substrate protein p70S6K, thus inhibiting the initiation of autophagy. As shown in recent studies, mTORC1 (mammalian target of rapamycin complex 1), a complex that mediates the classic functions of mTOR, inhibits lysosome function, thereby revealing a dual suppressive effect of mTOR on autophagy [51]. In our study, Se-Met inhibited the formation of autophagosomes by activating the Akt-mTOR-p70S6K pathway to decrease the stress of autophagic clearance.
Autophagy begins with the formation of autophagosomes, followed by the fusion of these vesicles with lysosomes and their subsequent degradation by enzymes in the lysosome [52]. Based on accumulating evidence, deficits in autophagosome-lysosome fusion and lysosomal clearance are more common in advanced stages of AD [50]. Genetically enhanced lysosomal activity and the restoration of autophagic flux in the brains of TgCRND8 AD mice significantly reduced the number of Aβ deposits [11]. The adaptor protein p62 is selectively sequestered in autophagosomes and then degraded after autophagosome-lysosome fusion; therefore, p62 levels are typically inversely correlated with autophagic degradation. As one of the major lysosomal acidic proteases, cathepsin D directly participates in lysosomal substrate clearance by degrading unfolded or oxidized protein aggregates that are delivered to the lysosomes by the autophagosomes [32, 53]. Strong evidence supports a role for cathepsin D in the autophagic clearance of Aβ, and Aβ accumulation in the brain is increased by inhibiting lysosomal activity and decreased by increasing cathepsin activity [54]. Se-Met significantly decreased p62 expression and increased cathepsin D expression, suggesting that Se-Met promotes autophagosome-lysosome fusion and the clearance of autophagic substrates in N2asw cells. Transfection with the mTagRFP-mWasabi-LC3 plasmids and the subsequent bafilomycin A1 treatment further verified that the effects of Se-Met on Aβ clearance are mediated by enhancing autophagic flux in AD.
As the study progressed, more details regarding the pathogenesis of AD were revealed. The autophagic process is associated with Aβ generation and localization; autophagic vesicles are highly enriched with AβPP, β-cleaved AβPP, and the components of the γ-secretase complex [4]. Dysfunctional autophagy accelerated the accumulation of autophagic vesicles and promoted Aβ overproduction [54, 55]. Abnormal autophagy also regulated the expression level of BACE1 in N2a/AβPP695 cells [56, 57] and upregulated the Aβ peptide levels by increasing the activity of the γ-secretase complex [58]. However, accumulation of these amyloidogenic species, including Aβ and β-cleaved AβPP, within autophagic organelles disturbed normal autophagic functions and thus formed a vicious cycle in which the accumulation of these proteins may be both the cause and a consequence of an endolysosomal-autophagic dysfunction in AD [59, 60]. In addition, GSK3 might also play a role as a “positive regulator” of the mTOR pathway, as inhibition of GSK3 promotes the clearance of Aβ burdens and the reactivation of mTOR [33]. All of these data suggested a sophisticated cross-talk mechanism between autophagy and Aβ generation, as well as the multifaceted and comprehensive effects of Se-Met on Aβreduction.
In summary, Se-Met modulated the activity of the Akt-GSK3β pathway to reduce Aβ production by inhibiting AβPP processing. Se-Met also promoted Aβ clearance by modulating the Akt-mTOR-p70S6K pathway and increasing autophagosome-lysosome fusion and autophagic degradation. Taken together, these results highlight the pleiotropic effects of Se-Met as an AD treatment.
Footnotes
ACKNOWLEDGMENTS
We are extremely thankful to Dr. Cuihong Zhou (Beijing Nuclear Magnetic Resonance Center, Peking University) for providing the mTagRFP-mWasabi-LC3 plasmids. We also thank Professor Yunwu Zhang (Institute for Biomedical Research, Xiamen University) for providing the N2a-sw cells.
This study was financially supported by the National Natural Sciences Foundation of China (Nos. 81400847, 31470804) and the Shenzhen Bureau of Science, Technology and Information (JCYJ20150529164656093 and JSGG20140703163838793).