
Abstract
Rapidly progressive Alzheimer’s disease (rpAD) is a variant of AD distinguished by a rapid decline in cognition and short disease duration from onset to death. While attempts to identify rpAD based on biomarker profile classifications have been initiated, the mechanisms which contribute to the rapid decline and prion mimicking heterogeneity in clinical signs are still largely unknown. In this study, we characterized prion protein (PrP) expression, localization, and interactome in rpAD, slow progressive AD, and in non-dementia controls. PrP along with its interacting proteins were affinity purified with magnetic Dynabeads Protein-G, and were identified using Q-TOF-ESI/MS analysis. Our data demonstrated a significant 1.2-fold decrease in di-glycosylated PrP isoforms specifically in rpAD patients. Fifteen proteins appeared to interact with PrP and only two proteins3/4histone H2B-type1-B and zinc alpha-2 protein3/4were specifically bound with PrP isoform isolated from rpAD cases. Our data suggest distinct PrP involvement in association with the altered PrP interacting protein in rpAD, though the pathophysiological significance of these interactions remains to be established.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a common form of dementia affecting more than 37 million people worldwide [1, 2]. The prevalence of slow progressive AD (spAD) pathology is well studied [3, 4], however, a subset of rapidly progressive AD (rpAD) cases mimicking prion diseases is emerging [5–9]. These rpAD cases show distinct clinical parameters [5, 9] and pathological features [10], though the risk factors and pathological mechanisms leading to heterogeneous progression rates and phenotypes of AD are still not known [3].
In recent years, neuropathological similarities, genetic acquaintances, and coexistence between AD pathology and prion diseases have been reported [11]. Some studies report coexistence between prion protein (PrP) and amyloid-β (Aβ) in plaques [12, 13]. A genetic correlation and systematic meta-analysis shows PRNP to be a gene potentially related to AD susceptibility [14] with Met/Val 129 polymorphism as a potential risk factor for early onset of AD [13, 16]. Interestingly, the cellular form of PrP also appears to be a high-affinity receptor for Aβ oligomers [17–19], and a population of soluble oligomers (Aβo) was found to interact with PrP in AD and cause behavioral impairment. The coherence between the levels of Aβo interacting with PrP appears to have significance importance in AD progression and PrP plays a pivotal role in AD pathobiology [20]. Despite the fact that such pathological and genetic links between AD and PrP have been identified, there is no evidence of a world-wide PrP-interacting risk factor involved in the course of a slow or a rapid progression of thedisease.
In this study, we demonstrate that post-translationally modified PrP isoforms are reduced specifically in rpAD patients, and that PrP-interacting risk factors may be involved in the slow or rapid progression of AD pathology.
MATERIALS AND METHODS
The brain samples were provided by the Prion Disease Surveillance Units of Germany and Spain including, i.e., spAD, rpAD, sporadic Creutzfeldt–Jakob disease (sCJD), and non-demented control cohorts as described previously [8]. Human samples from Spain were obtained following the Spanish legislation (Ley de la Investigación Biomédica 2013 and Real DecretoBiobancos, 2014) and the approval of the local ethics committees. All experimental protocols were approved and the study conformed to the Code of Ethics of the World Medical Association. All study participants or their legal next of kin gave informed consent and the study was approved by the local ethics committee in Göttingen (No. 24/8/12). All samples were anonymized with regard to at personal data and are summarized in Table 1).
Patient details: Summary of cases used in the present study
Disease duration less than four years (<4) consider as rapid forms and more than four year (>4) considered as slow forms of AD. rpAD, rapid progressive Alzheimer’s disease; spAD, slow progressive classical Alzheimer’s disease; sCJD, Creutzfeldt-Jakob disease; Cont, control (non-demented).
Brain tissue samples were processed as demonstrated previously [8, 21]. Briefly, frontal cortex area 28 was taken from spAD cases (8M/5F) with mean age of 75 years, rpAD cases (5M/4F) with mean age of 69 years, and age-matched non-demented controls (8M/2F). Sucrose velocity gradient ultracentrifugation was performed as described previously with slight modification [7]. Briefly, samples were homogenized by 10% w/v of tissue in 1x PBS containing 2% w/v sarkosyl (pH 7.4). Homogenates were layered onto the top of 10–45% sucrose continuous gradients. Ultracentrifugation was performed at 50,000 rpm for 73 min at 5°C and 20 fractions were collected from top to bottom.
Frozen and paraffin-embedded tissue sections were processed and immunofluorescence and confocal laser scanning microscopy was performed as described previously [8, 22]. Anti-PrP 3F4 (1 : 200), anti-PrP SAF70 (1 : 250), anti- ZAG (1 : 100), Alexa 488/546 conjugated anti-rabbit (1 : 200), and TO-PRO-3 iodide were used. Tissue lysis, immunoblotting co-immunoprecipitation were performed as described previously [8].
Furthermore, we performed in-gel tryptic digestion leading to peptide sequence analysis. Briefly, specific bands were excised from silver-stained 1-DE gel into 1-2 mm2 slices. Slices were then washed in ddH2O, reduced with 10 mM dithiothreitol by incubating for 30 min at 56°C, and alkylated with 55 mM idoacetamide at room temperature in dark for 60 min. Gel slices were then washed in acetonitrile (ACN) for 15 min and dried in a SpeedVac to remove excess solvent. Protease cleavage was done by rehydrating dried gel slices with the minimum amount of porcine trypsin solution (12.5 ng/μl in 0.025 M aqueous ammonium bicarbonate) for overnight at 37°C. After tryptic digestion, gel slices were incubated with 10 μl ddH2O for 15 min at 37°C followed by addition of 80 μl ACN and incubation for 15 min at 37°C. Supernatant was taken after a short spin. Residual peptides were dissolved from gel slices by vortex and incubation in 65 μl 5% formic acid for 15 min at 37°C. ACN 65 μl was added and further incubated for 15 min at 37°C. Supernatant was collected after a short spin and added to supernatant from the previous step. Combined supernatant was evaporated to dryness in a vacuum concentrator. Dried samples were dissolved in 10 μl of 30% ACN and 0.1% triflouroacetic acid.
For mass spectrometric analysis, peptide mixtures were concentrated on a Reversed Phase-C18 precolumn (0.15 mm ID x 20 mm, self-packed with Reprosil-Pur 120 C18-AQ 3 μm material) followed by separation using Reversed Phase-C18 nanoflow chromatography (Picofrit column, 0.07 5 mm ID×200 mm (New Objective, Woburn, USA)) using a 15 min linear gradient on an Easy nLC-1000 nanoflow chromatography system (Thermo Fisher Scientific, Dreieich, Germany). Eluents were analyzed on a Q Exactive hybrid quadrupole/orbitrap mass spectrometry system operated under Excalibur v2.4 software (Thermo Fisher Scientific). Analysis was carried by a Top10 method in the Data Dependent Acquisition mode. Tandem mass spectra were extracted for database searching, using Raw2MSM v1.17 software (Max Planck Institute for Biochemistry, Martinsried, Germany). MS/MS samples were analyzed using Mascot (Matrix Science, London, UK; version 2.4.1) set for searching UniProt/SwissProt database (release 02/14 filtered for Homo sapiens entries) with 5 ppm mass tolerances for precursors and 0.020 Da for fragments, respectively. Methionine oxidation was used as a variable modification and cysteine carbamidomethylation as a fixed modification. Up to two missed cleavages were allowed. Scaffold software v4.4.1.1 (Proteome Software, Portland/OR, USA) was used for validation of MS/MS based peptide and protein identifications. Peptide identifications were accepted, if established at greater than 95.0% confidence. While, a minimum of two confident peptide identifications and a confidence threshold of 99.0% were required for proteinidentifications [23].
RESULTS
Characteristic and distinct PrP isoforms in spAD and rpAD human brains
The distinct PrP isoforms and aberrant localization of PrP in spAD and rpAD brain may be characterized by different phenotypes and binding domains and the interplay of specific mechanisms during the disease progression.
In this study, we found differential PrP immunoreactivity in the neuronal cell bodies of the frontal cortex region of spAD brains, rpAD, and control brain tissues. PrP expression was significantly increased in spAD and rpAD cases as compared to controls. rpAD cases also showed significantly weaker immunoreactivity as compared to spAD cases (Fig. 1A). Notably, PrP showed aberrant localization toward the nuclear region specifically in rpAD as compared to age-matched controls (Fig. 1A). The differential expression of PrP by one- and two-dimensional immunoblot reveals a 1.2-fold (p≤0.05) decrease in rpAD as compared to the characteristic isoforms of PrP in the post-translationally modified di-glycosylated PrP isoforms (Fig. 1B-F). Colocalization and the FRET index suggest that the rpAD cases showed the significant decrease of PrP isoforms at the size of 37 kDa (Fig. 1F).

Characteristically distinct PrP isoforms in spAD and rpAD human brains. A) Localization of PrP and nuclear immunoreactivity in the frontal cortex brain region in spAD, rpAD, and control brain tissues using SAF70 anti-PrP antibody. Zoom shows overlapping of nucleus with PrP. ICA and scatter plots illustrate the colocalization pixel intensities between PrP and nucleus. B) PrP expression in spAD and rpAD by using SAF70 anti-PrP antibody and immunoblotting analysis and beta actin as loading control. DG, di-glycosylated isoforms; MG, mono-glycosylated isoforms; UG, un-glycosylated isoforms. C-E) Densitometry analysis from four independent (±SD) immunoblotting experiments using 15 controls (Con), 15 spAD, and 8 rpAD cases. F) Immunoblots from 2-DE gels of controls, spAD and rpAD proteins, using SAF70 anti-PrP antibody. Linear 7 cm IPG strips (pH 4–8) were used and loaded with 50 μg of protein. Upper panels represent co-localization points (ImageJ WCIF plugin) and lower panel represents co-localization and FRET index (ImageJ FRET plugin) of silver-stained 2-DE gels in spAD, rpAD, and controls. Pearson’s co-localization correlation co-efficient rp (–1≤rp≤1) experiments and FRET index were generated by ImageJ (WCIF plugin) software. The significance was calculated with the one-way ANOVA Friedman test (*p < 0.05) and AU arbitrary units.
To further characterize the hydrodynamic information on the size and shape of PrP molecules in rpAD as compared to spAD and controls, we used a high-speed ultra-centrifugation and sucrose gradient to evaluate the sedimentation velocity. We also used sCJD cases as PrP controls in this sedimentation assay to check for a possible overlap of PrP isoforms between rpAD and CJD cases. The PrP in the brain tissue of controls, spAD, rpAD, and sCJD cases remained in the top (1–6) fractions, as expected for monomers or dimers of PrP (Fig. 2). However, a broad range of densities and a variable fraction (7–12) remained floating in spAD and rpAD cases (Fig. 2C, D). Interestingly, rpAD showed a distinct species of PrP in the fraction (13, 14, and 16), similar to sCJD cases (Fig. 2A, D, E). However, these fractions in rpAD were not proteinase K-resistant as compared to sCJD (data not shown here).
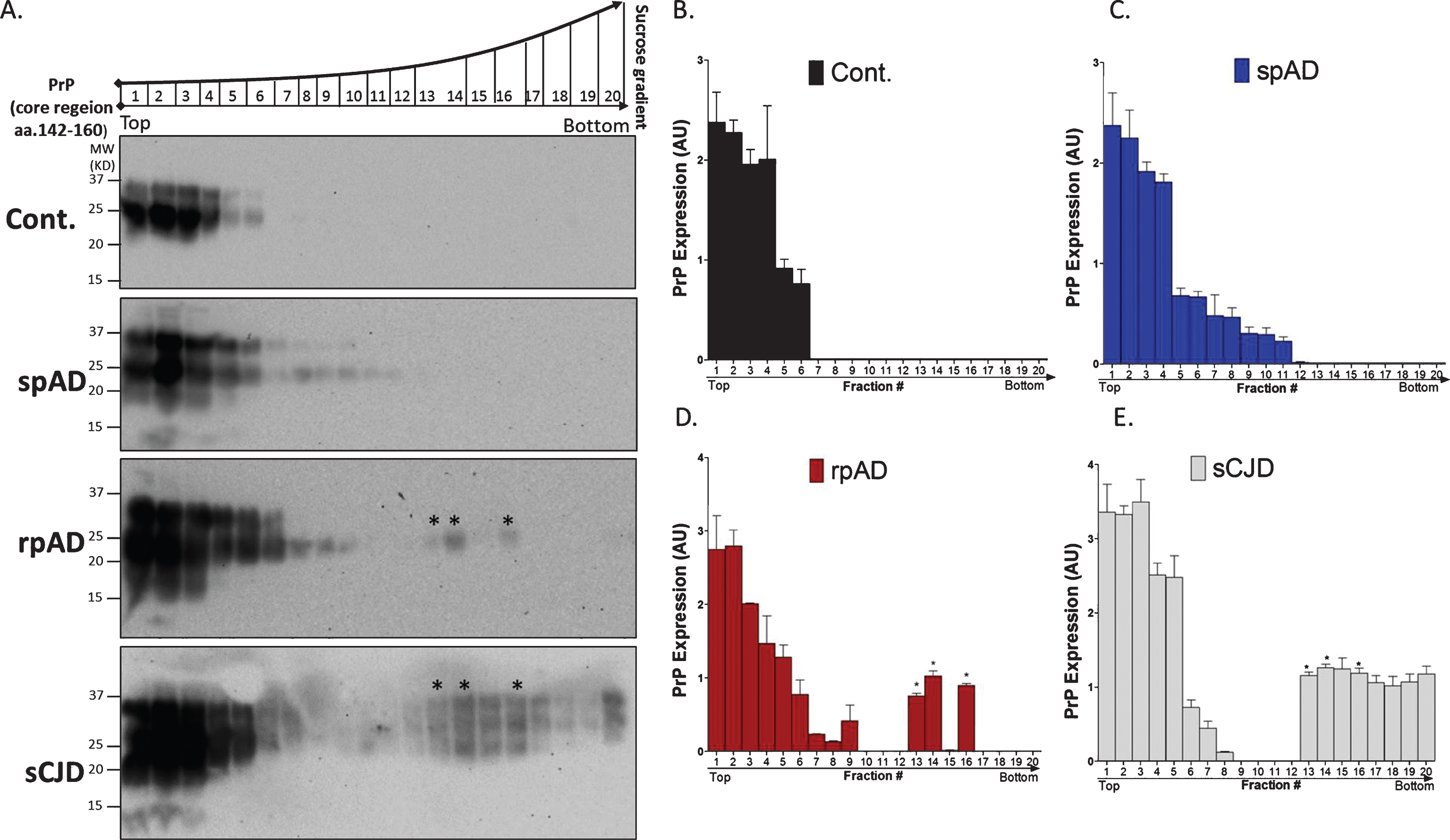
Separation of characteristically distinct PrP isoforms by sedimentation velocity in sucrose gradient in spAD and rpAD human brains. A) Separation of PrP isoforms by sedimentation velocity in sucrose gradient. The gradient fractionation profile of PrP particles from spAD, rpAD, sCJD, and controls after separation by ultracentrifugation in sucrose gradient. Fractions collected from the bottom of the tubes were immunoblotted by anti-PrP mAb SAF70. B-E) Densitometry analysis from four independent (±SD) immunoblotting experiments in 15 controls (Con), 15 spAD, 8 rpAD cases, 15 sCJD, and 15 non-demented controls and graph was generated by Prism 5 software. The significance was calculated with the one-way ANOVA Friedman test (*p < 0.05) and AU arbitrary units.
Characteristic PrP interactome identification in spAD and rpAD human brains
We isolated PrP-interacting partners from spAD, rpAD, and controls by co-purification of PrP, using Protein G coupled to super magnetic Dynabeads® (Fig. 3A). Eluates from this co-immunoprecipitation with SAF32 and SAF70 PrP were stained silver (Fig. 3A) and whole lanes from spAD, rpAD, and control eluate were excised and recovered with tryptic in-gel/in-solution digestion. Proteins were identified with ESI/Q-TOF MS/MS. Both known and novel PrP-interacting partners were among the proteins identified in this study (Table 2). The ESI/Q-TOF MS/MS identified proteins based on protein identification probability, the exclusive unique peptide count, and total spectrum count; the spectrum and sequence are summarized in Supplementary Table 1 listing fifteen proteins as interacting partners of PrP. Of these fifteen proteins, two— histone H2B type 1-B (H2B-1B) and zinc alpha-2 protein (ZAG)— were specifically bound with PrP isolated from rpAD cases. Furthermore, three proteins, i.e., tubulin beta 2C, synaptojanin-1, and synaptopodin, showed no potential interaction with the PrP isoform from spAD and rpAD cases, and appeared to bind with controls. Interestingly, one protein, myelin P2, was alone isolated as the binding partner with the PrP isoform in AD samples. Four interacting proteins, i.e., peroxiredoxin–1, four-and-half LIM domains protein 1, transketolase variant (fragment), and the basic myelin protein, were commonly found in AD and rpAD cases. Four interacting proteins, namely ribonuclease UK114, fructose-bisphosphate aldolase A and lysozyme C, were found in common in AD and non-demented healthy controls; and lastly, synapsin-1 and myelin proteolipid proteins were found to interact with PrP in all cases.
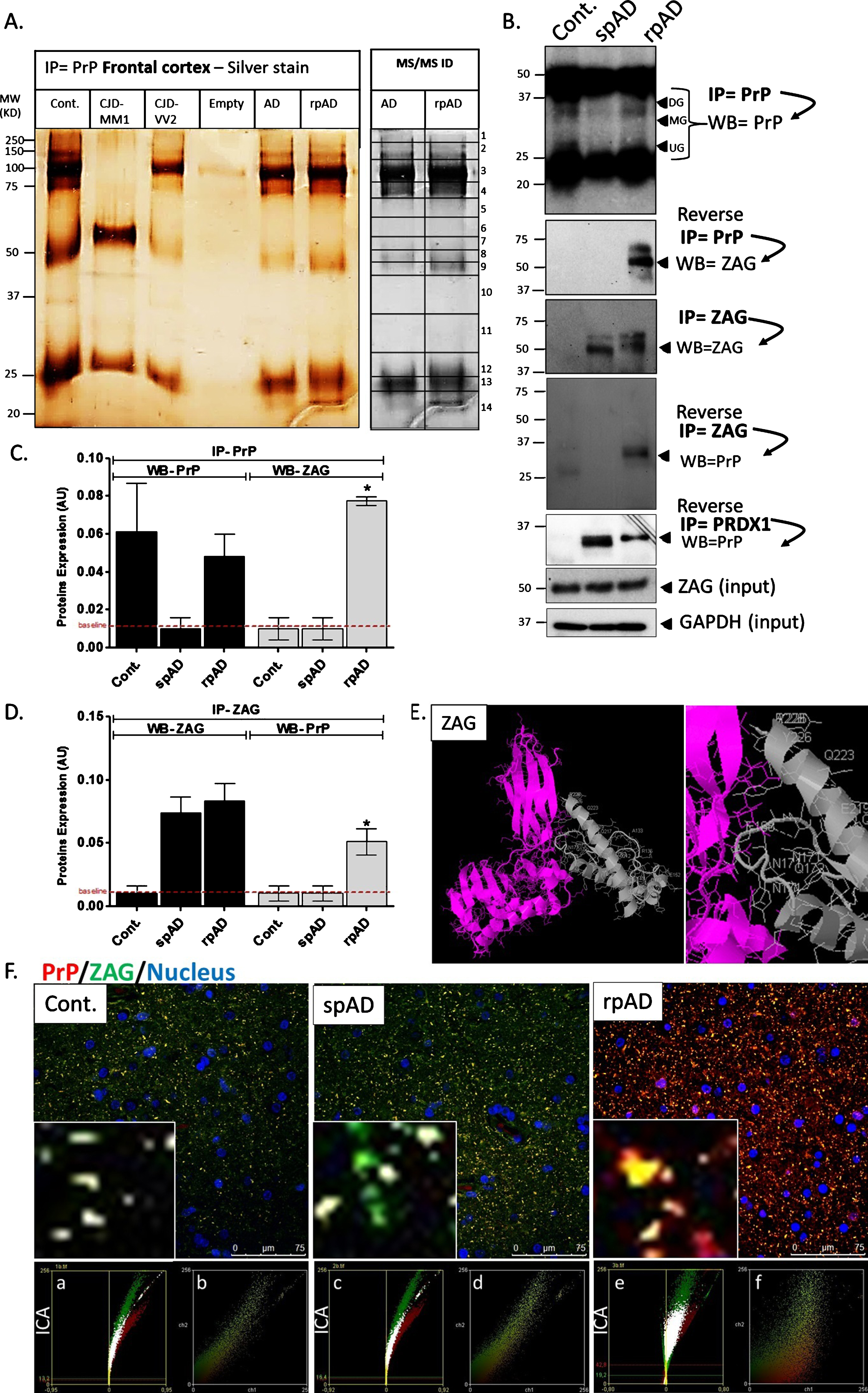
Characteristic PrP interactome identification in spAD and rpAD human brains. PrP-interacting partners isolated from spAD, rpAD, and control cases by using SAF32 and SAF70 PrP Protein G coupled to super magnetic Dynabeads. The bands were excised, in-gel digested, and proteins were identified by ESI/Q-TOF MS/MS analysis. A) IP eluates were stained silver, illustrating the PrP-interacting proteins in spAD, rpAD, and controls. B) IP eluates were immunoblotted against PrP and ZAG by using specific monoclonal antibodies. ZAG and GAPDH immunoblotting analysis in spAD, rpAD, and control samples were used as loading controls. C, D) Densitometry analysis from four independent (±SD) IP eluates immunoblotting experiments with15 controls (Con), 15 spAD, and 8 rpAD cases each. The significance was calculated with the one-way ANOVA Friedman test (*p < 0.05) and AU arbitrary units. E) Bioinformatic protein-protein prediction shows PrP (grey) and ZAG (pink) interacting sites by ZDOCK 3.0, and the molecule represents its corresponding centered orientation. F) Co-localization of PrP and ZAG in the frontal cortex brain region of spAD, rpAD, and control brain tissues using SAF70 anti-PrP antibody (red signal) and ZAG antibody (green signal). Zoom shows overlapping (yellow signal) of PrP and ZAG in the frontal cortex brain region of spAD, rpAD, and control brains. ICA (a, c, e) and scatter plots (b, d, f) illustrate the interaction between PrP and ZAG in rpAD brain samples.
List of prion protein interacting proteins in rpAD and spAD by using ESI/MS analysis
rpAD, rapid progressive Alzheimer’s disease; AD, slow progressive classical Alzheimer’s disease; Cont, control (non-demented); Nu, nucleus; Pm, plasma membrane; Ee, extracellular exosome; C, cytoplasm; Ms, myelin sheath; Fa, focal adhesion; Mm, myelin membrane; Cs, cytoplasmic side; Cm, cell membrane; Cp, clathrin coat of coated pit; Cj, cell junction, Ck, cytoskeleton; S, secreted; Ga, Golgi apparatus; Sv, synaptic vesicle.
The localization of proteins and accession number is assigned as in ExPASy protein database and Uniprot data base. Relevance with AD, prion and PrP ligand were established by deep NCBI literature database search. Tandem mass spectra were extracted and all samples were analyzed using Mascot (Matrix Science, London, UK; version 2.3.02). Mascot was set up to search the uniprot_human_database and used trypsin enzyme for protein digestion. Mascot was searched with a fragment ion mass tolerance of 0.60 Da and a parent ion tolerance of 10.0 PPM. Methyl of lysine and arginine, oxidation of Methionine, dimethyl of lysine and arginine, acetyl of lysine, trimethyl of lysine were specified as variable modifications, whereas carbamidomethyl of cysteine was specified as fixed modification in Mascot database search. For protein identification Scaffold (version Scaffold_4.4.7, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if probability by the Peptide Prophet algorithm showed greater than 96.0% [31], greater than 95.0% probability and at least 2 identified peptides.
ZAG and PRDX1 were further validated by immunoblotting after reverse and co-immunopre-cipitation. We used ZAG and PRDX1 proteins as bait protein under non-denaturing conditions and were able to isolate (prey) PrP protein only in rpAD brain samples from specifically ZAG–binding complex (Fig. 3B-D). This immunoprecipitation and reverse co-immunoprecipitation were performed under physiological conditions and proteins were post-translationally modified and conformationally natural. Additionally, we used PrDX1 as bait and managed to isolate PrP from both AD disease subtypes (spAD and rpAD). Isolated PrP and immunoblotting showed no signal from control brain samples (Fig. 3B).
Furthermore, we performed co-localization analysis by using confocal laser scanning microscopy and demonstrated that ZAG specifically and significantly showed increased co-localization with PrP in rpAD in the cortex region of the brain in contrast to spAD and controls groups (Fig. 3F). Use of ICA and scatter plots highlight the interaction between PrP and ZAG in rpAD brain samples (Fig. 3F a-f). We could also predict the molecular interactions by using the computational protein docking program ZDOCK to look for possible interaction sites between PrP and ZAG, which we indeed found (Fig. 3E).
DISCUSSION
The emerging field of proteomics and protein-protein interaction complexes can provide important insights into disease mechanisms. Recent studies using genome-wide protein expression by microarrays successfully predicted the disease outcome of a patient by correlating human interactome data [24].
In this study, we were able to successfully isolate PrP-interacting proteins from patients with heterogeneous AD during disease progression. This study clearly demonstrates a differential expression and localization of PrP in the rpAD brain as compared to the brains of spAD patients.
In spAD and rpAD cases, the differences in PrP expression, localization, and particle size may influence tissue-specific complexes and their associated cellular signaling pathways. Lately it has also been demonstrated that PrP binds with Aβ in AD patients, and exosomal PrP neutralizes the neurotoxicity caused by Aβ through the fibrillization mechanism [19, 25]. We were able to isolate PrP-interacting protein complexes, demonstrating a number of novel interacting proteins involved in AD pathology. We also identified several proteins already known to contribute to the disease pathology, which confirms the validity of our data. Other novel proteins were identified with a disease-specific contribution.
Interestingly, two proteins— H2B-1B and ZAG— showed potential interaction with PrP, specifically in rpAD cases. H2B-1B has already been studied in AD cases in relation to damage to DNA and cell cycle activation, and it is critical for rapid p53-mediated cell cycle inhibition, caspase-3 activation, and for inducing the phosphorylation of histone leading to cell death [26]. ZAG is a 40 kDa glycoprotein; one study shows that the relevance of ZAG in dementia is the differentially regulated cerebrospinal fluid protein in frontotemporal dementia patients [27]. It is interesting to note that one protein, myelin P2, was isolated as a binding partner with the PrP isoform in spAD samples, suggesting a possible link with spAD cases.
Furthermore, our data set demonstrates that three proteins, ribonuclease UK114, fructose-bisphosphate aldolase A, and lysozyme C, specifically interact with PrP isolated from spAD and controls. Fructose-bisphosphate aldolase A is known as binding partner of the cellular form of PrP [28] and has also been reported to be significantly upregulated in the cerebrospinal fluid of sCJD (human prion disease), but not in AD [29]. There was no regulatory response observed in the brains of CJD and AD cases [29]. The involvement of fructose-bisphosphate aldolase A in CJD and rpAD cases demonstrates a similarly rapidly progressive mechanism in both diseases.
Our interactomic approach includes a list of known and novel PrP-interacting proteins. Several proteins identified in this study are unique in relation to AD and its various forms, and are crucial for various cellular pathways. Determining this translational aspect of disease progression is of importance in further understanding the slow or fast progression of AD.
Footnotes
ACKNOWLEDGMENTS
This work was supported by a grant from Helmholtz-Alberta Initiative-Infectious Diseases Research (HAI-IDR) and APRI-Human prions–distinguishing sporadic from familial forms via structure and function as well as from the DZNE clinical project (Helmholtz). The study was performed within the recently established Clinical Dementia Center at the University Medical Hospital Göttingen and was partly supported by grants from the EU Joint Program–Neurodegenerative Disease Research (JPND – DEMTEST (Biomarker based diagnosis of rapid progressive dementias-optimization of diagnostic protocols, 01ED1201A).