
Abstract
Background:
The first consensus criteria for dementia with Lewy bodies (DLB) published in 1996 were revised in 2005, partly because the original clinical criteria had suboptimal sensitivity. Few studies have assessed the accuracy of the 2005 criteria applied prospectively in newly diagnosed patients who have been followed longitudinally.
Objective:
To explore the correlation between clinical and pathological diagnoses in patients with DLB and Parkinson’s disease with dementia (PDD).
Methods:
From a prospective referral cohort study with enriched recruitment of patients with DLB and PDD, we included the first 56 patients coming to autopsy. Patients had mild dementia at inclusion and were followed annually until death with standardized clinical assessments. Pathological assessment was performed blind to clinical information according to standardized protocols and consensus criteria for DLB.
Results:
20 patients received a pathological diagnosis of Lewy body disease; the corresponding clinical diagnoses were probable DLB (n = 11), PDD (n = 5), probable (n = 2) or possible (n = 2) Alzheimer’s disease (AD). Of 14 patients with a clinical diagnosis of probable DLB, 11 had DLB/PDD and 3 had AD at pathology. One patient with clinically possible DLB fulfilled criteria for pathological AD. Sensitivity, specificity, positive predictive value, and negative predictive values for probable DLB were 73%, 93%, 79%, and 90%.
Conclusion:
Our findings suggest that the international clinical consensus criteria for DLB perform reasonably well. However, false positive and false negative diagnoses still occur, indicating that the criteria need to be improved, that biomarkers may be needed, and that neuropathological feedback is vital to improve accuracy.
INTRODUCTION
Dementia with Lewy bodies (DLB) is a common form of neurodegenerative dementia. The first consensus criteria for the disease were published in 1996 [1], and later revised in 2005 [2], partly because the original clinical criteria were satisfyingly specific, but not sensitive enough. A large study of 2,861 patients found a sensitivity of 32% and a specificity of 98% using the 1996 clinical criteria [3]. The pathological criteria were sensitive, but without acceptable specificity [2]. In the 2005, clinical criteria REM sleep behavior disorder (RBD), severe neuroleptic sensitivity, and reduced striatal dopaminergic transporter activity (DAT) imaging were given more weight, in addition to several changes in the neuropathological classification. Inclusion of RBD has been shown to improve the sensitivity of the criteria [4]. A study with prospectively diagnosed patients found a sensitivity of 88% and specificity of 100% for DAT imaging versus autopsy, thus performing better than the clinical 1996 criteria which had sensitivity of 75% and a specificity of 42% versus autopsy in the same study [5]. DLB symptoms may develop late in the course of the disease, and may also occur in patients with Alzheimer’s disease (AD), thus diagnosing DLB can be difficult. Relatively few studies have assessed the accuracy of the 2005 criteria applied prospectively in newly diagnosed patients who have been followed longitudinally with a comprehensive and standardized clinical battery.
Our objective was to examine the accuracy of the 2005 McKeith clinical criteria [2] for DLB versus neuropathology in a referral cohort of dementia patients followed prospectively as part of the Dementia study in western Norway (DemVest). We also wanted to describe the clinical and pathological details of cases with a mismatch between clinical and pathological diagnosis.
METHODS
Cohort selection procedures
All patients with newly diagnosed mild dementia at specialist clinics in Geriatric Medicine and Old Age Psychiatry in the counties Hordaland and Rogaland between 2005 and 2007 were asked to participate. Specialist clinics in Neurology were also invited to refer patients. Inclusion criteria were Mini-Mental State Examination (MMSE) scores ≥20 and/or Clinical Dementia Rating score ≤ 1, no acute delirium, terminal illness, major somatic or psychiatric illness with effects on cognition. Between 2007 and 2013, DLB and Parkinson’s disease dementia (PDD) patients only were included to increase samplesize.
The study originally included 265 patients: 18 patients withdrew because they did not want toparticipate, 2 proved not to have dementia with follow-up, and 3 withdrew for other reasons (see Fig. 1). Of the remaining 242 patients 205 had died by November 14, 2016. Of these, 56 patients had come to autopsy by January 1, 2017, and constitute the autopsy group presented in this paper. This included one case who was later excluded since the cognitive impairment did not progress. The autopsy cohort did not differ significantly from the non-autopsy cohort for gender, education, and baseline MMSE score, but was slightly younger and had longer survival (Table 1).
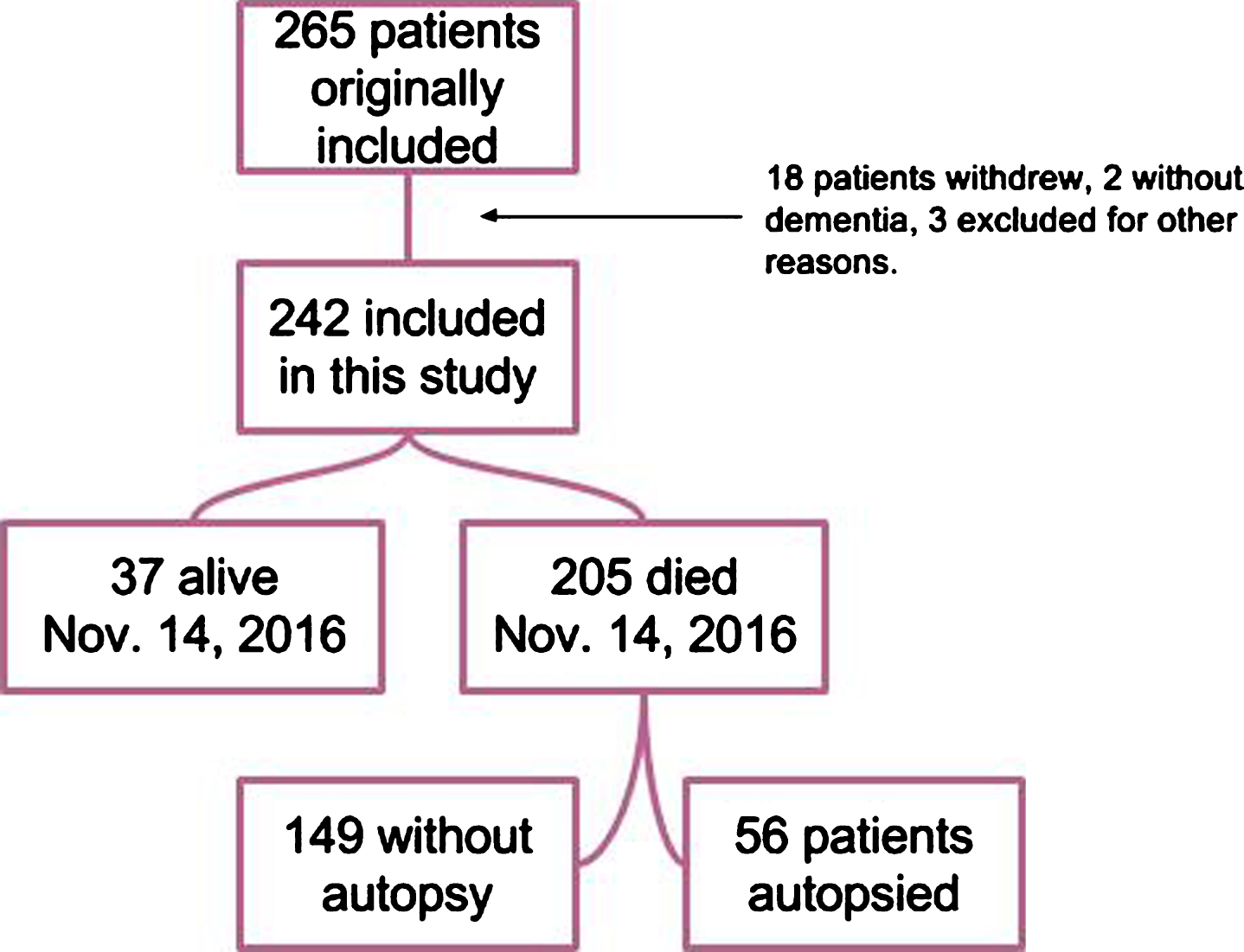
Flowchart included patients in the study.
Baseline demographics
*The autopsy group is compared to the deceased who did not go to autopsy; by November 14, 2016, 37 of the 242 were still alive. π Groups were compared using the Chi-Square test with continuity correction. #Groups were compared using the Mann Whitney test. Missing data is annotated. p < 0.05 is highlighted.
Baseline clinical evaluation
The baseline evaluation was conducted by experienced specialists in geriatric medicine or psychiatry aided by a research nurse. It consisted of a semi-structured interview of patients and caregivers, general clinical and neurological examination, a neuropsychological test battery including MMSE, neuropsychiatric evaluation, blood tests, as well as MRI scan and cerebrospinal fluid sampling for those who consented to this as described previously [6]. Specifically the core and suggestive DLB features were ascertained using the following clinical rating scales: the Neuropsychiatric Inventory (NPI) [7], the motor examination of Unified Parkinson’s Disease Rating Scale (UPDRS) [8], the Mayo Fluctuations Questionnaire [9] or the Clinician Assessment of Cognitive Fluctuations [10], and the Mayo Sleep Questionnaire [11] for visual hallucinations, parkinsonism, fluctuating cognition, and RBD, respectively. The semi-structured interview contained a specific question about antipsychotic hypersensitivity. After inclusion, patients were followed annually with a similar examination program [12]. Bi-annual training meetings with the study clinicians were held to ensure intra- and inter-rater reliability and harmonization of procedures.
Clinical diagnostic procedures
Dementia was diagnosed according to DSM IV criteria. For differential diagnosis, we applied commonly used diagnostic criteria; for AD the criteria from the National Institute of Neurological and Communicative Disorders and Stroke – Alzheimer’s and Related Disorders Association [13], vascular dementia (VaD) according to the National Institute of Neurological Diseases and Stroke – Association Internationale pour la Recherche et l’Enseignement en Neuroscience criteria [14], DLB according to the 2005 consensus criteria [2], and Parkinson's disease with dementia according to the Movement Disorder Task Force [15]. Lund-Manchester criteria for frontotemporal dementia [16] and DSM IV criteria for alcoholic dementia were also used. Details regarding how the clinical scales were used and their cut-offs are provided elsewhere [17]. The protocol recommended that patients with suspect DLB should be referred for (123I)FP-CIT SPECT, as described in detail previously [18]. However, due to availability and local factors, only a relatively small proportion (n = 50) were scanned.
Two experienced old age psychiatrists (DA, AR) independently applied diagnostic criteria based on all available information including the case note from the baseline visit. In cases of disagreement and in patients fulfilling more than one set of diagnostic criteria, patients were discussed until consensus was reached. The diagnoses were re-evaluated twice, after two and five years, the last consensus also included an experienced geriatrician (HS). The re-evaluation focused on patients with a previous “possible” (AD/DLB/VaD) diagnosis, and those with an unexpected course, for example slow cognitive decline. All available information was used in order to arrive at the most likely diagnosis, including clinical records and neuroimaging, but without any pathological information.
Ethical approval
The study was approved by the regional committee for medical and health research ethics in western Norway (REK 2010/633). As the patients all had mild dementia at inclusion, they were able to provide written informed consent, including consent to autopsy.
Neuropathological examination
Sampling and characterization of postmortem brain samples
Brain dissection, macroscopic description, regional sampling, tissue processing, and staining were done following standard protocols including BrainNet Europe and Brains for Dementia Research UK [19–22]. Block taking for histological and immunohistochemical studies and neuropathological assessment of neurodegenerative and control cases was performed in accordance with published guidelines [19–23]. For histology, 7 μm thick sections were cut and stained with hematoxylin and eosin (H&E) and selected blocks also with Luxol fast blue/Nissl (LFB/Nissl), for first stage neuropathological assessment of cytoarchitecture and basic cytopathology (for example the presence of Lewy bodies), extent and neuroanatomical distribution of neuronal and myelin loss, and selection of tissue blocks for detailed immunohistochemicalanalysis.
Immunohistochemistry
Briefly, 7 μm-thick paraffin sections were routinely de-waxed, blocked for endogenic peroxidase activities in ethanol containing 1.5% (v/v) H2O2, and heat-treated in appropriate antigen retrieval buffer solutions using a household electronic pressure cooker. After protein blocking in 50 mM TRIS-buffered saline (TBS pH 7.4) containing 5% (w/v) low fat milk powder, the sections were incubated with the primary antibodies at room temperature for 70 min. Detection was performed using Novolink polymer kit (Leica Biosystems/Novocastra), and nuclear staining was carried out with Mayer’s hematoxylin. For primary goat antibody, rabbit anti-goat linker IgG (GenWay Biotech), for primary rat antibody, rabbit anti-rat linker IgG (Vector Laboratories) were used.
Immunostaining (location and intensity) was examined by a consultant neuropathologist (TH) and the intensity of cytoplasmic staining assessed using the following standard semiquantitative scale: 0 – no staining, 1 – mild staining, 2 – moderate staining, and 3 – intense staining.
Antibodies
Primary antibodies dilutions used in this study are as follows: mouse monoclonal anti-amyloid-β (Agilent/DAKO)(1 : 100), rat monoclonal anti-phospho-TDP-43 (Ser 409/410)(Merck/Millipore)(1 : 100), rabbit polyoclonal anti-phospho TDP-43 (pSer409/410-1)(Cosmo-Bio)(1 : 1000), mouse monoclonal anti-phospho-tau (Ser 202/Thr205) (ThermoFisher/Invitrogen) (1 : 100), goat polyclonal anti-α-synuclein (R&D Systems)(1 : 1.000), mouse monoclonal anti α-synuclein (clone 5G4)(Roboscreen)(1 : 2.000), mouse monoclonal anti-p62 (lck ligand)(1 : 200)(BD Biosciences), rabbit polyclonal anti-ubiquitin (DAKO)(1 : 1.000). Fixation time varied considerably in our cohort which may have affected immunoreactivity, especially regarding α-synuclein [24]. Therefore, we applied two α-synuclein antibodies when immunoreactivity was unequivocal or inconsistent with findings on HE and p62 (which was applied when primary immunohistochemical work-up was negative or immunoreactivity questionable).
Neuropathological diagnosis
Specific stains for identification of AD-type and Lewy body pathologies and immune histochemical procedures were used for detection of hyperphosphorylated tau (pretangles, tangles, dystrophic neurites, and neuropil threads), amyloid-β (diffuse and classical plaques and amyloid angiopathy), and α-synuclein (Lewy bodies and Lewy neurites), according to standard immunohistochemical protocols (see above). Each case was assessed by an experienced neuropathologist (TH) who was blinded to clinical data. Pathological diagnosis was made according to international consensus criteria for DLB [2, 25] and AD [22, 26–28]. The presence of possible co-existing TDP-43 proteinopathy was assessed according to guidelines [29], and microscopic vascular lesions considered and recorded [30]. The neuropathological syndrome of DLB or PDD is in the following referred to as DLB/PDD. Patients were classified as having DLB/PDD if the likelihood of a DLB syndrome according to McKeith et al. [2] was “intermediate” or “high”. Patients with DLB/PDD pathology at autopsy were classified as PDD or DLB if they had a clinical diagnosis of PDD or DLB, respectively. The unclassifiable case (Patient 3) with ‘skipping’ of neuroanatomical regions typically affected in DLB was labelled as ‘predominantly limbic’ and was described in detail. Regarding vascular pathology and its contribution to dementia we applied the concepts outlined our recent consensus papers [31, 32] for neuropathological correlates of VaD.
After the evaluation, the neuropathologist was ‘unblinded’ (i.e., clinical diagnosis and data were disclosed). All false positive and false negative cases were reviewed. The initial diagnosis was upheld.
Statistics
Data were analyzed using SPSS version 23. Age, MMSE total, education, and disease duration were not normally distributed and thus non parametric statistics were applied for comparisons between groups. Sensitivity and specificity values were calculated using the appropriate formulas from standard 2×2 frequency tables. Since DLB and PDD have clinical and pathological similarities [33], sensitivity and specificity were calculated for both DLB and PDD both combined and DLB separately.
RESULTS
The pathological diagnoses among the 56 autopsy cases were AD (n = 31), DLB/PDD (n = 20), VaD (n = 2), and one case each of progressive supranuclear palsy, TDP-43 positive frontotemporal dementia, and one without significant pathology. The patient without significant pathology did not demonstrate progression of cognitive impairment. One patient with “low” probability of the DLB-syndrome and Braak α-synuclein stage of III [25] was in the absence of other significant pathology (Braak tau stage I, only mild small vessel disease) classified as DLB/PDD, with a clinical diagnosis of PDD. 10 AD patients had some degree of concomitant Lewy body pathology including three with α-synuclein pathology in the amygdala only, and nearly all DLB/PDD patients had some degree of AD pathology. Nine AD patients had TDP-43 pathology, two mild in amygdala only, two mild, four moderate, and one patient severe pathology in amygdala and hippocampus; one of these patients also had mild TDP-43 proteinopathy in the neocortex. Six DLB/PDD patients had TDP-43 pathology, five had moderate changes in the amygdala and hippocampus, and one had severe pathology in the amygdala and moderate in the hippocampus and neocortex. Vascular pathology, such as amyloid angiopathy and small vessel disease, was common in both AD and DLB/PDD, and some patients also had infarctions.
Clinico-pathological correlation
The associations between clinical and neuropathology diagnoses are shown in Table 2. 14 patients had a clinical diagnosis of probable DLB; three of these had a pathology diagnosis of AD and 11 had DLB/PDD. One patient with clinically possible DLB had AD. Among the 20 patients with a pathology diagnosis of DLB/PDD, the corresponding clinical diagnoses were probable DLB (n = 11), PDD (n = 5), probable (n = 2) and possible (n = 2) AD. Sensitivity, specificity, positive predictive value, and negative predictive value was 80%, 92%, 84%, and 89% respectively for DLB and PDD combined, for probable DLB alone 73%, 93%, 79%, and 90%.
Correlation between clinical and pathological diagnosis
#The clinical diagnosis was alcohol related dementia. $The pathological diagnosis was progressive supranuclear palsy. +One of these patients had clinical and pathological FTD, the other clinical MCI, and a neuropathological diagnosis of aging and mild AD.
Overview of severity of AD and DLB/PDD pathology in patients with concomitant disease at autopsy, with the McKeith et al. [2] classification of likelihood of the DLB syndrome
#3 of these were AD patients with α-synuclein pathology in the amygdala only.
Sensitivity, specificity, positive predictive value, and negative predictive value for a clinical diagnosis of probable AD, including the two mixed cases, was 81%, 88%, 89%, and 79%, respectively.
Patients with discrepancy between clinical and neuropathological diagnosis
In 7 patients, the clinical and pathological diagnosis of DLB did not match. Three patients were “false positives”, i.e., the clinical diagnosis of probable DLB was not verified at autopsy, and four were “false negatives”, i.e., the pathological DLB diagnosis was missed and the patients diagnosed as having AD. While 12 of the cohort had performed DAT imaging, none of these seven patients had a scan. A summary of the clinical and pathological details are provided here. For a more detailed overview, please see Table 4 and patient data in the Supplementary Material.
Characteristics of false positive and false negative DLB/PDD patients
*Two scales were used to assess fluctuations, the Clinicians Assessment of Cognitive Fluctuations (FI) and The Mayo Fluctuation Questionnaire (MFQ) using recommended cut-off scores, one example given from the last month from question a) or b) and a total of 5 or more from the Clinician Fluctuation scale and 3 or 4 features from the Mayo Fluctuation Questionnaire. #The presence of visual hallucinations based on the NPI hallucinations item, the clinical interview and comments in the medical records. currenRem-sleep behavior disorder was diagnosed if there was a history of recurrent nocturnal enactment behavior, assessed by the Mayo Sleep Scale. CAA = cerebral amyloid angiopathy.
Patients with a “false negative” diagnosis of DLB
At baseline, no core features were present among the four false negative cases. However, three had no bed-partners and thus RBD could not be assessed, and two had borderline UPDRS motor scores (11 and 9, see Rongve et al. [17]) which progressed significantly, and one also developed cognitive fluctuations and visual hallucinations. All cases had some degree of hippocampal atrophy on MRI. Of the two cases without parkinsonism, one developed visual hallucinations and fluctuations from year 3 which persisted, whereas the other was entirely without core features during the first 4 years of follow-up; only at year 5 were cognitive fluctuations and visual hallucinations reported. The pathological assessment showed severe AD with Braak stage 6 in all four cases; in addition to severe Lewy body pathology (all had neocortical Lewy body disease).
Patients with a “false positive” diagnosis of DLB
At baseline, all 3 false positives had visual hallucinations and cognitive fluctuations which persisted for several years as well as relatively preserved hippocampi on MRI. Of note, 2 of the cases had eye disease and visual impairment, one in both eyes. Parkinsonism tended to slowly increase and UPDRS-III scores were high from 3 years after baseline in all cases. However, two patients had received treatment with antipsychotics. One patient received olanzapine, although this was withdrawn prior to first follow-up and well before UPDRS motor score started to increase. The other patient received risperidone from before his first follow-up until death. Both these patients, however, had visual hallucinations and fluctuating cognition and thus fulfilled criteria for probable DLB independent of parkinsonism. The neuropathological assessment showed that all cases had severe AD with Braak tau stage 6. Two cases were without any α-synuclein pathology; both showed tau-pathology in substantia nigra, one severe, and one also TDP-43 pathology in hippocampus and amygdala. The third case had limbic Lewy pathology in addition to some small vessel pathology and amyloid angiopathy. This case, which did not fit into the McKeith criteria and was at the initial neuropathological assessment diagnosed as limbic DLB, had atypical neuroanatomical distribution (with α-synuclein pathology skipping the substantia nigra and presence of cortical involvement of variable severity). The clinico-pathological context of this case is discussed in more details in the Supplementary Material.
DISCUSSION
Here we present the results of a prospective cohort study assessing the accuracy of the international consensus criteria for clinical DLB versus autopsy in patients followed annually from dementia diagnosis to death with standardized clinical procedures in a research setting. We were able to correctly identify and diagnose 16 of 20 patients with neuropathological DLB/PDD, resulting in a sensitivity and a specificity for probable DLB alone of 73% and 93%, respectively. All PDD patients were correctly classified. With a sensitivity of 73% compared to 81% for AD, it could be argued that the current clinical DLB criteria are not yet sufficiently sensitive; the specificity, however, is satisfactory. Notably, all the 4 false negative patients had severe concomitant neuritic plaque AD pathology (Braak stage V-VI) and intermediate likelihood of the DLB syndrome [2], which has been shown to decrease the frequency of core clinical DLB features [34] and thus make the clinical diagnosis of DLB more difficult.
Few studies have examined the accuracy of the 2005 clinical DLB criteria against autopsy in prospectively diagnosed patients. In a pivotal study by McKeith et al. based on the 1996 criteria including 50 patients, 24 of 29 autopsy-confirmed DLB cases were correctly identified clinically, resulting in a sensitivity of 83% and a specificity of 95% [35]. The 2005-criteria, where RBD was added as a suggestive feature, were validated in 76 patients with dementia [34]. The criteria performed “reasonably well”; 40 of 43 patients with a clinical diagnosis of probable DLB had intermediate or high likelihood DLB pathology. Another prospective study found the results of DAT imaging to be closely linked to neuropathology, as positive DAT imaging alone had a sensitivity of 80% and a specificity of 92% against DLB at autopsy, while clinical diagnosis alone had a sensitivity of 87% and specificity of 72% [36].
The three false positive cases with a diagnosis of probable DLB but AD at autopsy had visual hallucinations and cognitive fluctuations at baseline. Of note, two of them had visual impairment due to eye disease. The interaction between eye disease, cognitive decline, and visual hallucinations is poorly understood but the presence of eye disease in patients with early cognitive decline is thought to lower the threshold for visual hallucinations [37]. A patient with AD and coincidental eye disease may thus present with visual hallucinations early in the disease course, resulting in a potential misdiagnosis of AD as DLB. False positive patients 1 and 2 are likely to fall into this category. Patient 3 did not have eye disease but limbic DLB pathology that has been linked to visual hallucinations in the absence of cortical Lewy body pathology or dementia [38]. Patient 3 also had evidence on MRI of posterior cortical atrophy (Table 4), an imaging phenotype linked neuropathologically to both AD and DLB and associated with visual hallucinations in up to 25% of cases [39]. Occipital atrophy may also be caused by eye disease [40] and the posterior cortical changes found in patient 2 may be related to bilateral AMD. All false positive DLB cases had severe AD pathology also affecting the primary visual cortex and para- and pericalcarine areas.
Although one might speculate these pathological changes are directly responsible for visual hallucinations in these cases, most patients with AD do not experience visual hallucinations despite severe pathology in the primary visual cortex and adjacent areas. It seems likely in the false positive DLB cases that concurrent eye disease or limbic pathology combined with AD pathology in visual cortex led to visual hallucinations early in the course of AD.
Four patients who had Lewy body pathology at autopsy were clinically diagnosed as AD. All four were without any core feature at baseline, but three of them subsequently developed several features after 2-3 years of follow-up. This suggests that although both parkinsonism and visual hallucinations are not uncommon in AD, even when they occur several years after the dementia diagnosis they may indicate Lewy body disease.
The presence and interactions of multiple pathologies in brains of patients with dementia, as observed in this cohort, is well known [41]. Although the hallmark protein in DLB/PDD is α- synuclein, amyloid-β deposition is virtually always present and tau pathology is also common [42, 43]. Cases which show neocortical Lewy bodies and full-blown AD can be neuropathologically diagnosed as having mixed dementia (mixed AD/DLB/PDD), since the severity of either AD or Lewy body pathology alone would be sufficient to represent a robust neuropathological correlate for clinical dementia. This was the case in several of our cases, including those with false negative DLB. Although AD is the most frequent coexisting pathology in DLB/PDD, TDP-43 pathology is present (usually limited to the amygdala and hippocampal formation) in around 30% of cases [44], although the clinical relevance is unknown. Cerebrovascular disease is also frequent, reported up to 94% [45].
The time course and sequence of development of co-existing pathologies in dementia have implications for both pathogenesis and clinical presentation. Tau and amyloid-β pathology can occur at an early age [46], whereas there is no data suggesting similarly early onset of α-synuclein pathology in DLB/PDD. Possibly, in the cases with mixed AD/DLB, the α-synuclein pathology might have developed later during the disease course which could explain why the clinical presentation at early stages of the disease was more consistent with AD. This notion is supported by findings of higher hyperphosphorylated tau loads in those who clinically presented with AD than those with clinical DLB [47]. In contrast, in cases clinically manifesting as DLB, α-synuclein could initially be the major abnormal hallmark neurodegenerative protein, in later stages of the disease followed by AD type pathologies of tau and amyloid-β. The synergistic and aggravating effects of co-existing pathologies are supported by neuropathological studies, showing that mixed cases had more rapid disease progression (i.e., shorter survival fromdiagnosis) [43].
How the clinical manifestations (i.e., dementia phenotype, for example AD versus DLB) relate to the individual proteinopathies remains to be established and our study provides some insight into this problem. The dominant pathology will likely dominate the clinical presentation, with the pathological neurodegenerative proteins having an additive effect. However, our findings with some false negative and false positive cases with mixed pathologies where the clinical presentation was not determined by the predominant AD versus DLB type pathology contradict this notion and suggest a more complex mechanism. This is in concert with a previous report that severe cortical tau pathology can lead to DLB clinical presentation even in the absence of cortical Lewy bodies (false positive case), particularly in elderly cases [48]. False negative diagnosis of DLB appears more frequently at early stages of dementia [3] and our false negative cases showed pronounced clinical DLB symptoms 2-3 years after baseline. Improved diagnostic accuracy of DLB remains a challenge, particularly in cases when there is neuropathological overlap with AD and possibly other pathologies.
Strengths of our study include that patients were diagnosed prospectively, and since all dementia clinics in the region were involved, 80% of the cohort had died and only age and duration differed between autopsied and non-autopsied patients, our cohort is likely representative of patients referred to specialist clinics for dementia work-ups in western Norway. The thorough clinical protocol, including standardized assessments for core and suggestive DLB symptoms is also a strength. The long follow-up from time of diagnosis is vital, allowing us to detect both early symptoms and those that developed later during the disease course. Limitations include the relatively small sample number, and the use of DAT imaging in only a subgroup of patients.
To conclude, our findings suggest that although the current clinical DLB criteria are reasonably accurate, some patients presented the core symptoms late and were therefore missed, while other patients with the DLB features proved not to have DLB at autopsy. Visual hallucinations in the presence of visual impairment can be misleading and core DLB features developing 2-3 years after diagnosis may also indicate DLB. Biomarkers such as DAT imaging, MIBG scintigraphy, and polysomnography may improve diagnostic accuracy, and quantitative EEG might also prove to be useful [49]. Neuropathological verification of clinical diagnosis remains essential for diagnostic quality. More research is needed to improve the accuracy of clinical diagnosis of DLB, and to increase our understanding of clinico-pathological relationships and the role ofnon α-synuclein pathologies, such as amyloid, tau, cerebrovascular disease, and TDP-43.
Footnotes
ACKNOWLEDGMENTS
We would like to express our deep gratitude to the patients and caregivers for their time, efforts, and cooperation. We are also indebted to all study nurses and doctors. Thanks to the departments of Pathology at Haugesund Hospital, Stavanger University Hospital, and Haukeland University Hospital and the involved nursing home staff. The project was funded by the Western Norway Regional Authority (Helse Vest RHF). We would like to thank the NIHR Biomedical Research Centre for Mental Health and the NIHR Biomedical Research Unit for Dementia at King’s College London for supporting the involvement of Clive Ballard and Paul Francis in the study and the Hungarian Brain Research Program forsupporting the participation of Tibor Hortobágyi.