
Abstract
Alzheimer’s disease (AD) is the most common cause of dementia, and effective therapeutics are lacking. Colivelin (CLN), a novel, strong humanin derivative, is effective in vitro in preventing cell death induced by AD-causative genes and amyloid-β protein (Aβ) even at a low concentration. We recently demonstrated that intrahippocampal injection of CLN prevents Aβ25–35-induced deficits in spatial memory and synaptic plasticity in normal rats. Here, we further observed the effects of chronically intranasally (i.n.) administered CLN on cognitive behaviors and pathological hallmarks in 9-month-old APPswe/PS1dE9 (APP/PS1) AD mice using multiple behavioral tests and immunochemistry. The electrophysiological mechanism of CLN neuroprotection was also investigated by recording in vivo hippocampal long-term potentiation (LTP). CLN pretreatment effectively prevented impairments in new object recognition, working memory, and long-term spatial memory and reversed the depression of in vivo hippocampal LTP in APP/PS1 mice. Additionally, chronic application of CLN obviously reduced Aβ deposition in the hippocampus in APP/PS1 mice. These results indicate that CLN has strong neuroprotective effects on learning and memory behaviors in APP/PS1 mice and that this behavioral improvement is closely associated with the reduction of Aβ deposition and alleviation of LTP suppression in the hippocampus, supporting the potential of CLN for the prevention and treatment of AD.
INTRODUCTION
Alzheimer’s disease (AD), the most common cause of dementia in the elderly, is an incurable, progressive, and devastating neurodegenerative disorder characterized by declining cognitive function, impairments in learning and memory, and deposition of amyloid-β (Aβ) protein in the brain [1]. According to World Alzheimer’s Disease Report 2016, dementia afflicts more than 47 million people worldwide, and this number will increase to 131 million by 2050 [2]. Although the exact pathogenesis of AD remains elusive, mutations of several genes, such as amyloid-β precursor protein (AβPP), presenilin-1 (PS1), and presenilin-2 (PS2), are closely related to AD [3]. In an APP/PS1 double knock-in mouse model, disease progression is accompanied by cognitive deficits in both recognition memory and spatial referencememory [4].
Colivelin (CLN) is a novel Humanin (HN) derivative comprising AGA-(C8R)HNG17 attached to the C-terminus of activity-dependent neurotrophic factor (ADNF) [5]. CLN prevents cell death induced by AD-causative genes and Aβ at concentrations as low as 100 fM and thus is the strongest HN derivative developed to date [6, 7]. In mice, CLN treatment inhibits both working and reference memory impairments caused by scopolamine and Aβ25–35 [5, 8]. We recently demonstrated that CLN effectively prevents Aβ25–35-induced impairments in spatial memory and hippocampal long-term potentiation (LTP) in normal rats [9]. However, evidence supporting neuroprotective effects of CLN in transgenic AD models is lacking. In particular, the ability of chronic application of CLN to ameliorate AD pathological damage and synaptic failure, hallmarks associated with impairments in learning and memory [10], in AD mice remains an open question.
In the present study, we first observed the effects of intranasal (i.n.) administration of CLN on the cognitive behaviors of APPswe/PS1dE9 (APP/PS1) transgenic mice in new object recognition, Y maze, and Morris water maze tests. Then, the possible electrophysiological mechanism was investigated by recording in vivo hippocampal LTP, an electrophysiological neuronal model of learning and memory. In addition, pathologically characteristic Aβ plaques deposited in the hippocampus in AD mice were detected using immunofluorescent and immunohistochemical staining.
MATERIALS AND METHODS
Animals and treatments
Heterozygous male APPswe/PS1dE9 (APP/PS1) mice with a C57BL/6J background and wild-type (WT) littermates were purchased from the Institute of Laboratory Animal Sciences (CAMS & PUMC) and bred at the Research Animal Center of Shanxi Medical University with approval of the Shanxi Committee on Ethics of Animal Research. All mice were housed under controlled room temperature (20–24°C) and humidity (60% –80%) and received food and water ad libitum. The APP/PS1 and WT mice were randomly divided into four groups: WT+Saline, APP/PS1+Saline, WT+CLN, and APP/PS1+CLN (n = 10 for each group). A 10-μl volume of saline or 1 nmol of CLN (Tocris, Bristol, UK) dissolved in 10 μl of saline was successfully transferred to the central nervous system (CNS) via the olfactory bulb by i.n. administration [8]. Drug administration was performed once daily beginning three weeks before the behavioral tests and continued until sacrifice (Fig. 1).
Sequence of events of the experimental procedure. CLN or saline was i.n. administered once daily for 21 days prior to different behavioral tasks, including the open field test (OP), new object recognition task (NOR), Y maze test (YMT), and Morris water maze task (MWM). During the 9 days of behavioral experiments, CLN or saline was administered daily. Then, electrophysiological recordings (LTP), immunofluorescence (IF), and immunohistochemistry (IHC) were performed.
Open field test
The apparatus consisted of an open field arena (length, 55 cm; width, 55 cm; height, 30 cm). Each mouse was acclimatized to the environment for 24 h prior to testing. Then, one mouse was placed in the middle of the open field and allowed to explore freely for 5 min. The total path of each mouse in the open field arena was recorded by an infrared camera to evaluate its motor capability.
New object recognition test
The new object recognition test was performed 24 h after completion of the open field test [11]. First, each mouse was allowed to freely explore the open field for 10 min in the presence of two identical objects (cube or ball) positioned 15 cm from the arena wall (acquisition task). When the 10-min acquisition task was complete, the mouse was returned to its cage for a 5-h delay. Then, the mouse was returned to the open field and exposed to one familiar object and a novel object for 10 min (test task). The objects were placed in the same locations as the objects in the previous acquisition task. The total time spent exploring each object (when the animal’s snout was directly toward the object at a distance ≤2 cm) was recorded, and the recognition index (RI) was calculated as [time exploring the novel object/total time exploring both objects]×100%. A higher RI indicates greater memory retention. In the acquisition and test tasks, if the exploration time was <30 s and <15 s, respectively, the mouse was excluded from analysis. In addition, the arena and objects were cleaned with 70% ethanol after each mouse task to prevent accumulation of olfactory cues.
Y maze spontaneous alternation test
The mice were subjected to the Y maze test 24 h after the new object recognition test. The Y maze comprised three arms connected at angles of 120°; each arm was 30 cm long, 12 cm high, and 5 cm wide. Each mouse was placed at the intersection point of the three arms and allowed to explore the maze freely for 8 min. The order of exploration of the arms and the total arm entries were recorded. The spontaneous alternation percentage, an index of spatial working memory, was measured as the ratio of arm choices differing from the previous two choices (successful choices) to total choices duringthe run [12].
Morris water maze test
The long-term spatial learning and memory of the mice were evaluated by the Morris water maze (MWM) test [13] 24 h after the Y maze test. The water maze was a circular pool (diameter, 120 cm; height, 50 cm) containing tap water at a temperature of 23±2°C. The water was made opaque with non-toxic white tempera paint. A platform (diameter, 12 cm; height, 29 cm) was submerged approximately 1.0 cm below the water surface at the midpoint of one quadrant of the pool. Various prominent visual cues were positioned on the inner wall of the pool. The swimming activity of each mouse was monitored via a camera mounted overhead, and a video tracking system (Ethovision 3.0, Noldus Information Technology, Netherlands) collected mouse movement information (latency, swim path, and speed). In the hidden platform test, each mouse was trained four times per day for 5 consecutive days. On each training day, a trial was initiated by placing the mouse in the water facing the pool wall in one of the four quadrants, and the mouse was allowed to swim freely to the escape platform. After reaching the platform, the mouse was allowed to remain on the platform for 5 s. The mouse was subsequently returned to the home cage for 20 s before the next trial. If the mouse did not find the platform within the 60 s allowed, it was gently guided to the platform. On the next day after the hidden platform tests, each mouse underwent a 60-s probe trial to evaluate its memory retention ability. During the probe test, the platform was removed, and the searching behavior of the mouse in the target quadrant (where the platform was located during the hidden platform training) was measured. After the probe test, the visual and motor abilities of the mouse were examined via a visible platform test. The time and speed at which each mouse arrived at the target platform were recorded.
In vivo hippocampal LTP recording
Because memory performance is closely associated with hippocampal LTP [14], the same mice used in the behavioral tests were used for the electrophysiological study. Mice were anesthetized with 5% chloral hydrate (0.07 ml/10 g, i.p.) and placed in a stereotaxic device for acute surgery and LTP recording. A bound stimulating/recording electrode was inserted into the left hippocampal CA1 region with the tip of the recording electrode 2.0 mm posterior to the bregma and 1.5 mm lateral to the midline. Baseline field excitatory postsynaptic potentials (fEPSPs) evoked by test stimulation (intensity, 30–50% of maximal EPSPs; frequency, 0.033 Hz) delivered to the Schaffer-collateral/commissural pathway were monitored for 30 min. LTP was induced by a high-frequency stimulation (HFS) protocol consisting of 30 pulses at 200 Hz repeated three times at an interval of 30 s [15]. Test stimuli were used again after HFS for a period of at least 1 h to record any change in slope of the fEPSPs. Paired-pulse facilitation (PPF), a short-term enhancement of synaptic transmission, was also observed in the CA1 region. The change in the PPF ratio (calculated by dividing the slope of the second fEPSP by the slope of the first fEPSP) reflects the alteration of neurotransmitter release from presynaptic terminals.
Immunofluorescent and immunohistochemical staining
Upon completion of LTP recording, the mice were perfused with phosphate-buffered saline (PBS) and 4% paraformaldehyde in PBS. The brains of the mice were rapidly dissected and post-fixed for 24 h at room temperature. For immunofluorescent staining, the brain blocks were sliced into 30-μm-thick coronal sections. The sections were incubated in 5% H2O2 at room temperature for 15 min, followed by washing 3 times in 0.01 M PBS for 5 min. After blocking in 5% bovine serum albumin (BSA) for 30 min, the sections were incubated overnight with anti-Aβ1–16 antibody (6E10, 1 : 500, Biolegend, USA) at 4°C. The next day, the sections were incubated with Alexa Fluor 594-conjugated Goat Anti-Mouse lgG (H+L) (1 : 300, Proteintech, USA) for 2 h and DAPI (10J29C76, Boster, China) for 10 min. An inverted fluorescence microscope was used to obtain immunofluorescence images of Aβ plaques in the right half brain slices. The area of Aβ deposits in each slice section was quantified using Image-Pro Plus, and the area percentage of Aβ deposits in the hippocampus was calculated [16].
Immunohistochemistry was performed on paraffin-embedded sections. In brief, coronal sections (3 μm) were deparaffinized and rehydrated. The sections were incubated in 3% H2O2 for 10 min to block endogenous peroxidases and treated with 0.01 M sodium citrate (pH 6.0) for epitope retrieval. Then, the sections were incubated in 1% BSA for 30 min to eliminate non-specific binding. Subsequently, the sections were incubated with 6E10 (1 : 800, Biolegend, USA) at 4°C overnight. For the negative controls, sections were incubated with PBS instead of antibody. On the second day, the sections were incubated with an anti-mouse HRP-conjugated secondary antibody (Boster, China) for 30 min. Finally, protein localization was visualized using diaminobenzidine tetrahydrochloride (DAB), and the sections were lightly counterstained in Harris’ hematoxylin solution for 40 s. After dehydration and vitrification, the sections were enclosed with a coverslip.
Statistics
All values are expressed as means±standard errors. For MWM tests, the escape latencies were analyzed using three-way repeated measures analysis of variance (ANOVA). The immunofluorescence staining data were analyzed by t test, and all other data were analyzed by two-way ANOVA. Statistical significance was defined as p < 0.05, and all statistical analyses were performed using the statistical software packages SAS 8.0 and SPSS 13.0.
RESULTS
Colivelin treatment reverses the impairment of new object recognition memory in APP/PS1 mice
Before the new object recognition test, the motor capability of the mice was evaluated in the open field test. As shown in Fig. 2A, there was no significant difference in total moving distance among the four groups (APP/PS1: F (1,39) = 0.200, p = 0.658; CLN: F (1,39) = 0.151, p = 0.700; APP/PS1×CLN interaction: F (1,39) = 0.419, p = 0.522). Thus, there is no evidence that APP/PS1 gene mutation and CLN treatment affect the motor capability of the mice. On the second day after the open field test, new object recognition memory was assessed. Two-way ANOVA showed that APP/PS1 gene mutation and CLN treatment had significant main effects on new object recognition memory (APP/PS1: F (1,39) = 67.075, p < 0.001; CLN: F (1,39) = 69.621, p < 0.001; APP/PS1×CLN interaction: F (1,39) = 67.114, p < 0.001). As shown in Fig. 2B, the RI of APP/PS1+Saline mice was 48.4% ±0.7%, significantly lower than the RI of 59.9% ±0.7% in the WT+Saline group (p < 0.001), indicating that the APP/PS1 mice spent less time exploring the novel object. However, the RI of APP/PS1 mice treated with CLN was 60.0% ±0.8%, a statistically significant increase compared with the APP/PS1+Saline group (p < 0.001). Tukey’s post hoc tests showed that the new object recognition memory of APP/PS1 transgenic mice was significantly decreased (p < 0.001) and that CLN reversed this impairment (p < 0.001).
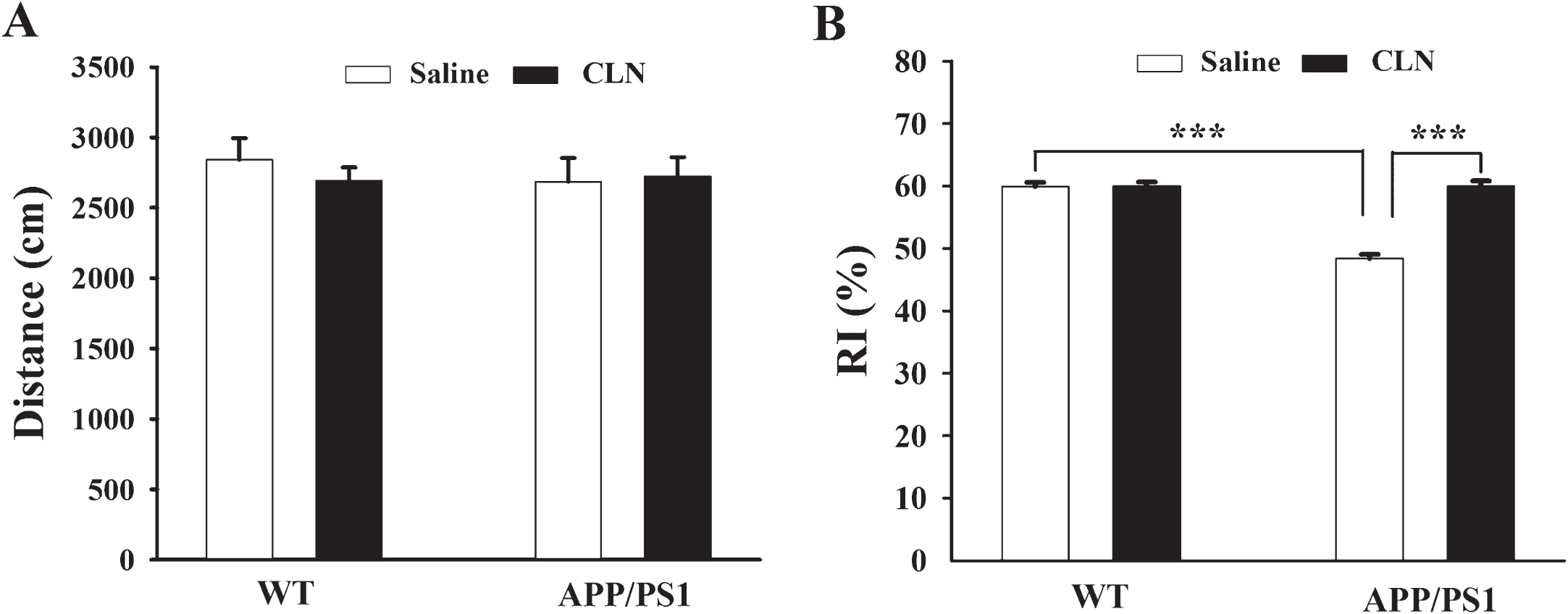
CLN treatment reversed the impairment of new object recognition memory in APP/PS1 mice. A) Histograms showing no significant difference in the total distance moved in the open field test (n = 10 for each group). B) Histograms showing that CLN treatment reversed the decrease in RI in APP/PS1 mice (***p < 0.001, n = 10 for each group).
Colivelin treatment prevents impairment of working memory in APP/PS1 mice
The Y maze spontaneous alternation test was performed to examine the spatial working memory of the mice. Two-way ANOVA showed that APP/PS1 gene mutation and CLN treatment had significant main effects on the spontaneous alternation of the mice (APP/PS1: F (1,39) = 4.686, p = 0.037; CLN: F (1,39) = 2.898, p = 0.026; APP/PS1×CLN interaction: F (1,39) = 7.399, p = 0.010). The percentage of right alternation of mice in the WT+Saline, APP/PS1+Saline, and APP/PS1+CLN groups was 60.7% ±3.1%, 46.4% ±2.8%, and 59.3% ±3.3%, respectively. According to Tukey’s post hoc tests, compared with the WT+Saline group, the percentage of right alternation was significantly lower (p = 0.002) in the APP/PS1+Saline group, and CLN treatment reversed this detrimental effect in APP/PS1 mice (Fig. 3A, p = 0.004). However, the total arm entries did not differ significantly among these groups (APP/PS1: F (1,39) = 0.000622, p = 0.980; CLN: F (1,39) = 0.0156, p = 0.901; APP/PS1×CLN interaction: F (1,39) = 0.762, p = 0.388; Fig. 3B), suggesting that the differences in spontaneous alternation among these groups were due to impairment of spatial working memory rather than a disability of locomotor activity.
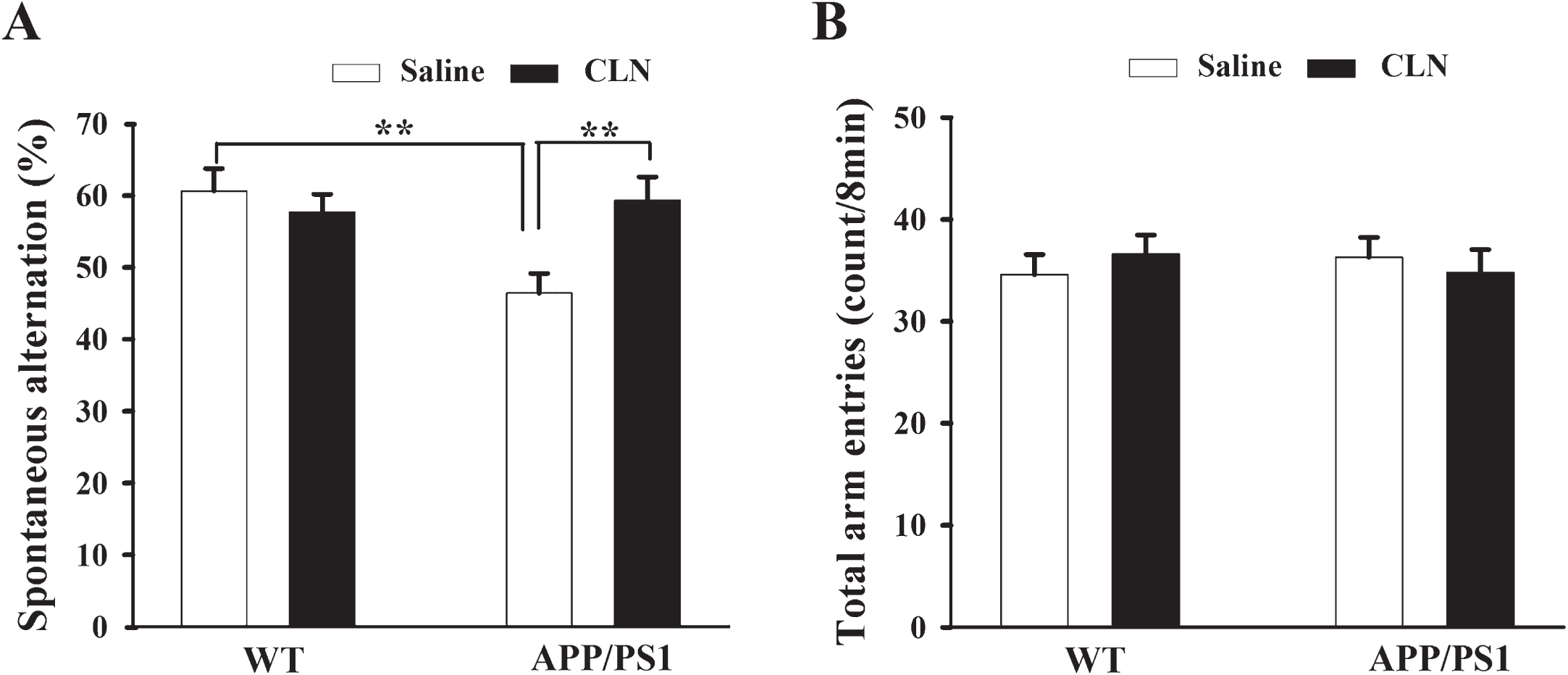
CLN treatment prevented working memory impairment of APP/PS1 mice in the Y maze spontaneous alternation test. A) Histograms showing lower spontaneous alternation in the APP/PS1+Saline group and significant recovery in the APP/PS1+CLN group (**p < 0.01, n = 10 for each group). B) Histograms showing no significant difference in total arm entries among the different groups.
Colivelin treatment protects long-term spatial memory in APP/PS1 mice
The MWM test was used to assess the ability of CLN treatment to prevent impairment of long-term spatial memory in APP/PS1 mice. In the MWM test, the learning ability of the mice to acquire spatial information was first assessed by the hidden platform test on five consecutive days. As expected, during the 5 training days, the average escape latency of the mice to find the hidden platform gradually decreased (F (4,160) = 208.15; p < 0.001). APP/PS1 gene mutation and CLN treatment had significant main effects on escape latency (APP/PS1: F (1,39) = 15.41, p < 0.001; CLN: F (1,39) = 17.27, p < 0.001; APP/PS1×CLN interaction: F (1,39) = 13.39, p < 0.001). According to the least squares means test, there was no statistically significant difference in escape latency among the four groups on day 1. However, on days 2–5, as shown in Table 1 and Fig. 4A and 4B, the escape latency was increased significantly in the APP/PS1+Saline group compared with the WT+Saline group. However, the escape latency was significantly decreased in the APP/PS1+CLN group compared with the APP/PS1+Saline group. These results indicated that the spatial learning ability of APP/PS1 mice was significantly impaired and that chronic CLN treatment effectively reversed this impairment.
Mean escape latencies during training days 1–5 (s, mean±S.E.M.)
Compared to APP/PS1+Saline group, *p < 0.05, **p < 0.01.
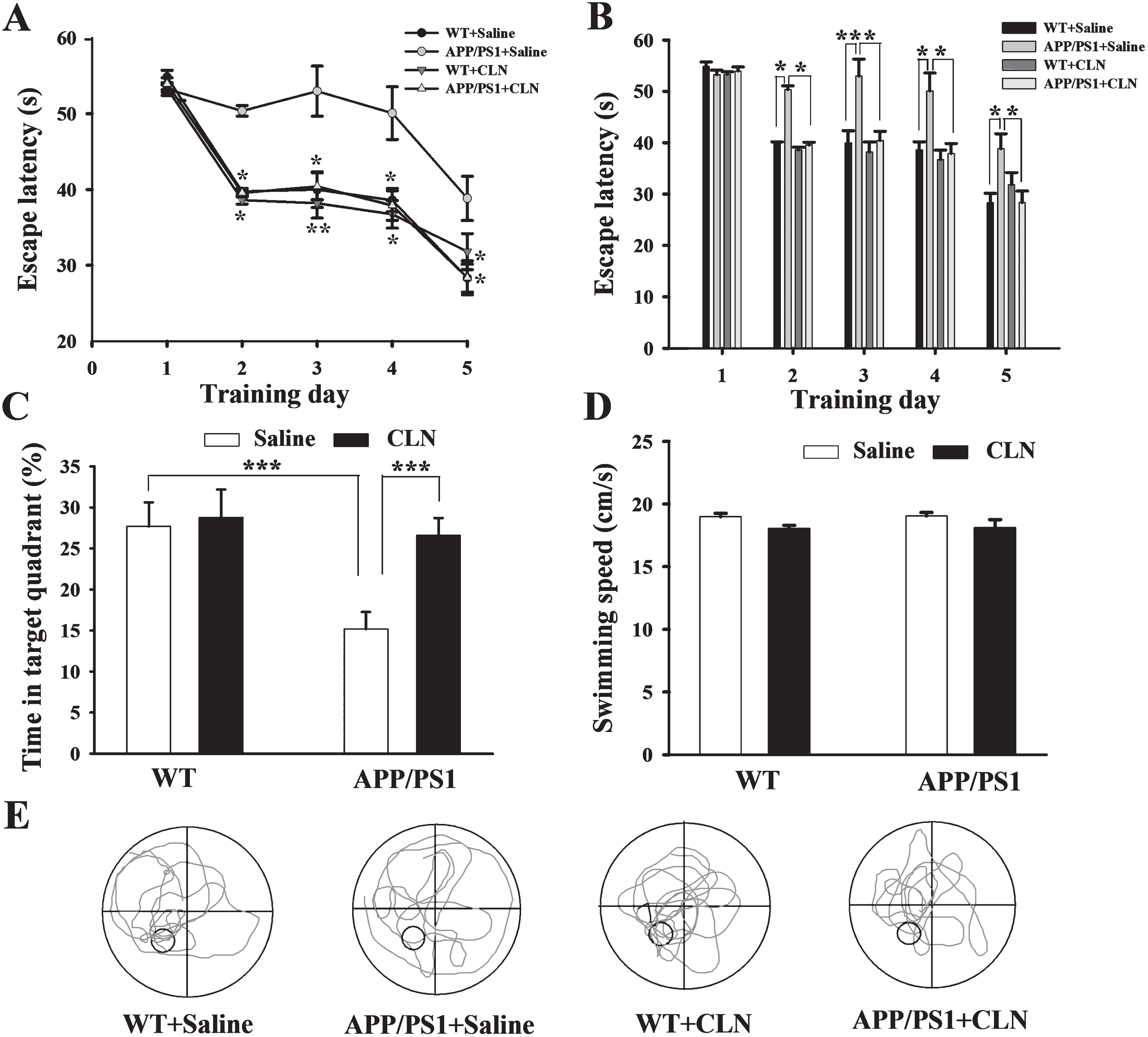
CLN treatment protected against the impairment of spatial learning and memory in APP/PS1 mice. A) Plots showing the average escape latencies of mice when searching for the hidden platform over five consecutive training days (compared to the APP/PS1+Saline group, *p < 0.05, **p < 0.01, n = 10 for each group). B) Histograms showing that the escape latency was significantly increased in the APP/PS1+Saline group on training days 2–5 and that this increase was reversed by CLN treatment (*p < 0.05, **p < 0.01). C) Histograms showing that the decrease in swimming time in the target quadrant in the APP/PS1+Saline group during the probe trial test was reversed by CLN treatment (***p < 0.001). D) Histograms showing no significant difference in the swimming speed of mice among the groups in the visible platform test. E) Representative swimming traces of mice in the four groups during the probe trial. The large circle represents the water maze pool, and the small circle represents the platform.
To further assess the spatial memory of the mice in the MWM test, probe trials without the platform were performed on day 6. The swimming time percentage of the mice in the WT+Saline, APP/PS1+Saline, WT+CLN, and APP/PS1+CLN groups was 27.7% ±2.9%, 15.2% ±2.1%, 28.8% ±3.4%, and 26.6% ±2.1%, respectively. Two-way ANOVA indicated that APP/PS1 gene mutation and CLN treatment had significant main effects (APP/PS1: F (1,39) = 109.897, p < 0.001; CLN: F (1,39) = 117.979, p < 0.001; APP/PS1×CLN interaction: F (1,39) = 76.245, p < 0.001, Fig. 4C, E) on the swimming time percentage of the mice in the target quadrant. Tukey’s post hoc test indicated a significant difference in memory retrieval between the APP/PS1+Saline and WT+Saline groups (p < 0.001). Similarly, CLN treatment reversed the APP/PS1 gene mutation-induced reduction of swimming time in the target quadrant (p < 0.001). These results indicated that pretreatment with CLN protected against the deficit of spatial memory in APP/PS1 mice.
To exclude the possibility that the results above were due to impairment of visual or motor ability, the swimming speed of the mice was tested with a visible platform after the probe trials. As shown in Fig. 4D, there were no statistically significant differences in average swimming speed among the groups (APP/PS1: F (1,39) = 2.880, p = 0.098; CLN: F (1,39) = 0.422, p = 0.520; APP/PS1×CLN interaction: F (1,39) = 0.153, p = 0.698), indicating that the changes in escape latency and swimming time percentage resulted from impairments of cognition and not changes in the visual or motor ability of the mice.
Colivelin treatment reverses in vivo hippocampal LTP suppression in APP/PS1 mice
After the MWM tests, in vivo hippocampal fEPSPs and LTP in the CA1 region in APP/PS1 mice were recorded. Synaptic efficacy was represented by the change in fEPSP slope. After stable baseline recordings of fEPSPs for 30 min, HFS was applied to induce LTP. Immediately after HFS, the slopes of the fEPSPs (percentage of baseline) abruptly increased from 100% to 159.9% ±2.7%, 159.0% ±2.6%, 161.9% ±2.9%, and 157.6% ±3.8% in the WT+Saline, APP/PS1+Saline, WT+CLN, and APP/PS1+CLN groups, respectively, indicating that LTP was successfully induced in the four groups (Fig. 5A). However, the LTP values in the APP/PS1+Saline group began to significantly decrease beginning 20 min post-HFS. As shown in Fig. 5A and 5C, the LTP values were 157.5% ±1.7%, 136.1% ±2.8%, 156.1% ±3.2%, and 154.6% ±2.7% at 30 min post-HFS and 151.3% ±3.5%, 120.3% ±2.6%, 150.1% ±3.6%, and 148.2% ±3.6% at 60 min post-HFS in the WT+Saline, APP/PS1+Saline, WT+CLN, and APP/PS1+CLN groups, respectively. Two-way ANOVA demonstrated that APP/PS1 gene mutation and CLN treatment had significant main effects on the fEPSP slope at 30 min (APP/PS1: F (1,23) = 266.054, p < 0.001; CLN: F (1,23) = 137.300, p < 0.001; APP/PS1×CLN interaction: F (1,23) = 192.342, p < 0.001; Fig. 5C) and 60 min post-HFS (APP/PS1: F (1,23) = 1119.703, p < 0.001; CLN: F (1,23) = 801.891, p < 0.001; APP/PS1×CLN interaction: F (1,23) = 880.524, p < 0.001; Fig. 5C). Tukey’s post hoc test showed that the LTP value was significantly suppressed (p < 0.001) in APP/PS1 mice and that the depression of LTP was significantly prevented by CLN treatment (p < 0.001).
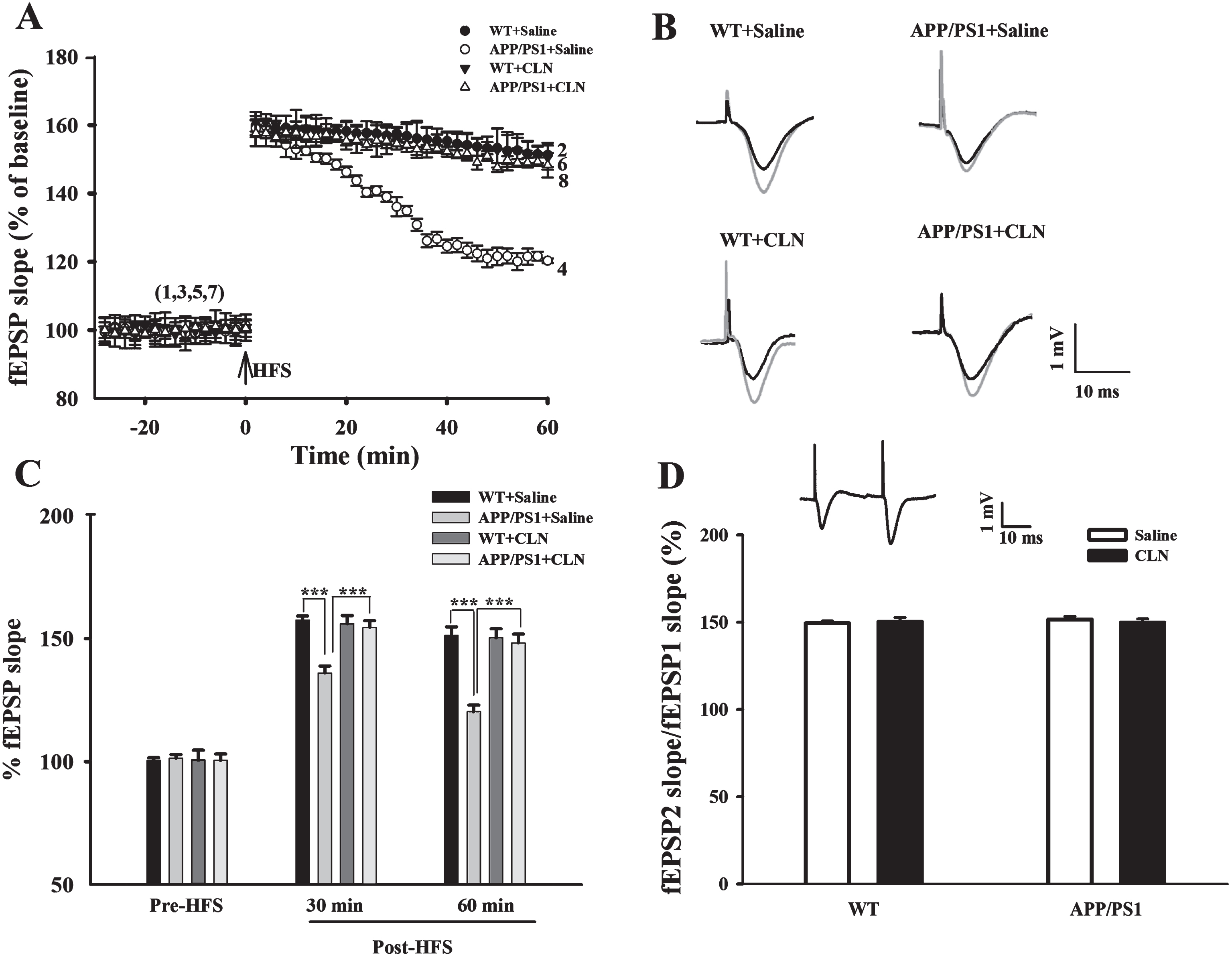
CLN treatment reversed the depression of hippocampal LTP in APP/PS1 mice. A) Time course of fEPSPs and LTP induced in the hippocampal CA1 region of mice in the four groups (n = 6 for each group). B) Typical fEPSP traces before and after HFS recorded from mice in the four groups. Scale bars, 1 mV and 10 ms. C) Histograms showing that the depression of LTP 30 min and 60 min after HFS in the APP/PS1+Saline group was reversed by CLN treatment (***p < 0.001). D) APP/PS1 gene mutation and CLN treatment did not affect PPF (fEPSP2/fEPSP1). Inset, representative paired fEPSPs.
To clarify the potential involvement of the presynaptic mechanism in the effects of APP/PS1 gene mutation and CLN treatment, paired pulse facilitation (PPF) was examined. Application of paired pulses to the Schaffer collaterals always induced PPF. Two-way ANOVA showed that APP/PS1 gene mutation and CLN treatment had no significant main effects on PPF (APP/PS1: F (1,23) = 1.419, p = 0.247; CLN: F (1,23) = 0.257, p = 0.617; APP/PS1×CLN interaction: F (1,23) = 2.734, p = 0.114; Fig. 5D). Thus, there is no evidence that APP/PS1 gene mutation and CLN treatment affected presynaptic neurotransmitter release in the hippocampal CA1 region in mice.
Colivelin treatment reduces the deposition of Aβ in the hippocampus in APP/PS1 mice
An abnormal increase in Aβ and deposition of senile plaques are characteristic pathological hallmarks in AD patients. Changes in Aβ deposition in the hippocampus in APP/PS1 mice were detected using immunofluorescence (Fig. 6A) and immunohistochemistry (Fig. 6B). As shown in Fig. 6A and 6B, no Aβ deposition was detected in the hippocampus in the WT+Saline and WT+CLN groups, whereas many Aβ immunopositive plaques were detected in the hippocampus in the APP/PS1+Saline group. Importantly, CLN pretreatment (APP/PS1+CLN) significantly reduced Aβ deposition. The area percentage of Aβ deposits in the hippocampus calculated from the immunofluorescence images decreased significantly (p < 0.001) from 10.2% ±2.5% in the APP/PS1+Saline group to 5.5% ±1.8% in the APP/PS1+CLN group (Fig. 6C).
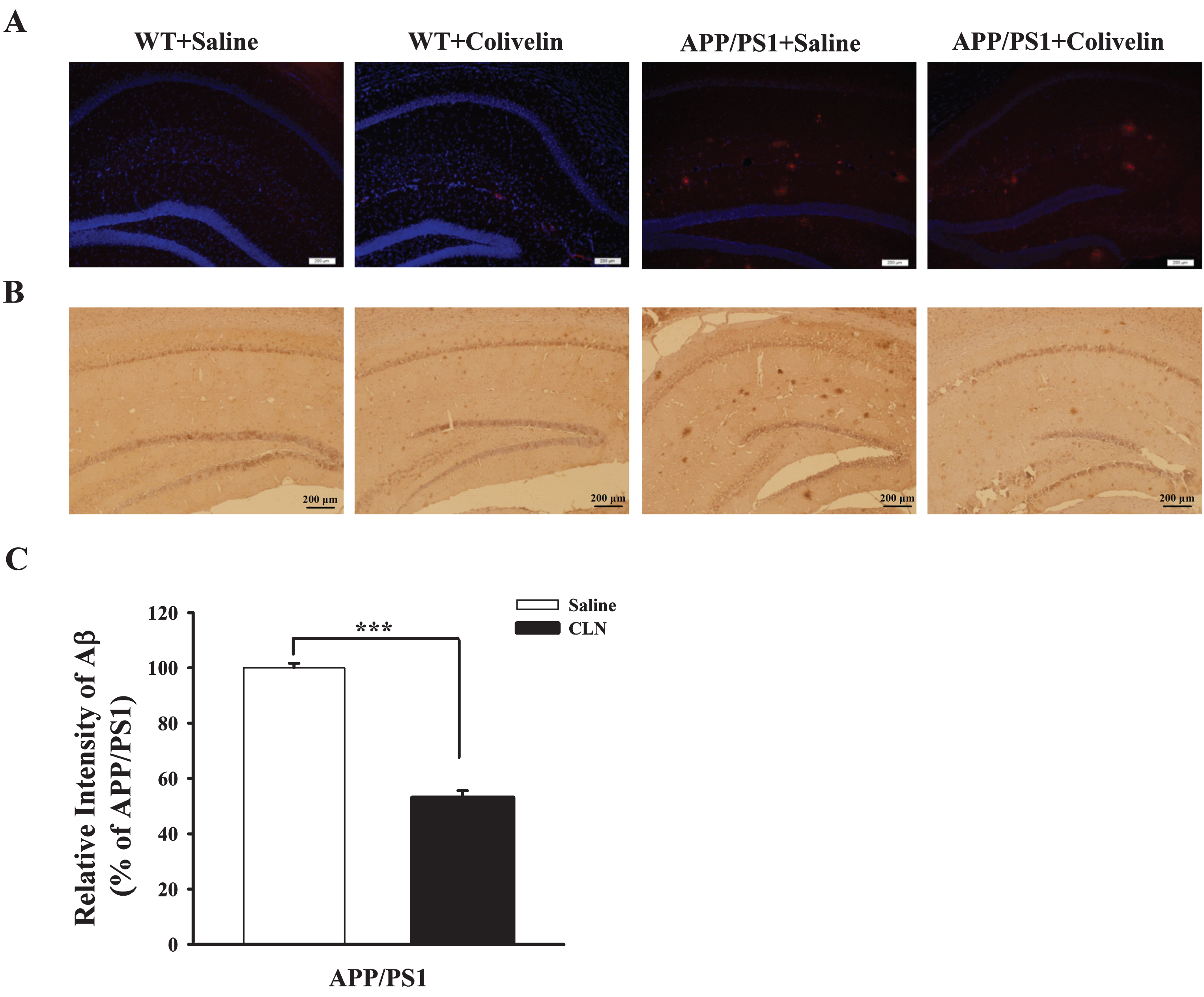
CLN treatment reduced Aβ deposition in the hippocampus in APP/PS1 mice. A) Immunofluorescent imaging of Aβ plaques (red color) deposited in the hippocampus in APP/PS1 mice. Tissue sections from the four groups were incubated with DAPI (blue channel) and 6E10 (red channel) antibodies (scale bar: 200 μm). B) Photographs showing immunohistochemical staining of Aβ deposition in WT and APP/PS1 mice treated with saline or CLN. The number of Aβ plaques in the hippocampal CA1 region was obviously increased in the APP/PS1+Saline group, and the Aβ load was reduced after treatment with CLN. Bar = 200 μm. C) Histograms showing a significant decrease in the area percentage of Aβ deposits in the hippocampus in APP/PS1 mice after CLN treatment (***p < 0.001).
DISCUSSION
In the present study, we investigated the ability of CLN pretreatment to improve cognitive function impairment and LTP suppression in APP/PS1 mice and detected the effect of CLN on Aβ deposition. We found that chronic i.n. administration of CLN improved new object recognition, working memory, and long-term spatial memory and reversed in vivo hippocampal LTP depression in 9-month-old APP/PS1 mice. In addition, CLN effectively reduced Aβ deposition in the hippocampus in APP/PS1 mice.
Memory dysfunction, including short-term and long-term spatial memory, is an important clinical performance in AD patients [17]. In the present study, 9-month-old APP/PS1 mice were first subjected to multiple behavioral tests. We did not observe significant differences in total moving distance (in the open field test), total arm entries (in the Y maze test), and swimming speed (in the MWM test) between APP/PS1 mice and WT mice. Therefore, the decreased RI (new object recognition test), decreased percentage of spontaneous alternation (Y maze test), increased escape latency and decreased swimming time percentage in the target quadrant (MWM tests) of APP/PS1 mice reflect multiple cognitive impairments, including impaired short-term and long-term memory. These results are consistent with previous reports for the APP/PS1 mouse model of AD [4, 19].
Due to the close association between spatial memory and hippocampal synaptic plasticity, hippocampal LTP is widely viewed as a major cellular mechanism underlying learning and memory [20]. In a previous study, application of Aβ42 to C57BL/6J mice brain slices significantly blocked LTP induced by HFS [21]. We previously demonstrated that intracerebroventricular (i.c.v.) injection of Aβ40 suppressed in vivo hippocampal LTP in normal rats [22]. Although basic synaptic transmission was not affected in APP/PS1 mice, HFS-induced LTP was impaired in hippocampal slices from 7- to 12-month-old and even 3-month-old APP/PS1 mice [23–25]. In the present study, we confirmed that in vivo LTP in the hippocampal CA1 region was also seriously impaired in APP/PS1 mice. Importantly, the LTP recording experiments in the present study were conducted on behaviorally trained mice, providing further insight on the electrophysiological mechanisms underlying the impairments of spatial memory in APP/PS1 mice. After performing behavioral tests and LTP recording, Aβ deposition in the hippocampus, a classic hallmark of AD [26], was detected to clarify the possible internal relationship among the behavioral deficits, electrophysiological changes, and pathological damage observed in APP/PS1 mice. As expected, obvious deposition of Aβ plaques was observed in the hippocampus in 9-month-old APP/PS1 mice. Therefore, we propose that this abnormal increase in hippocampal Aβ is directly involved in the synaptic plasticity impairment and multiple behavioral deficits in APP/PS1 mice.
Previous studies have reported important neuroprotective activities of CLN in multiple neurodegenerative diseases. For example, CLN suppressed amyotrophic lateral sclerosis (ALS)-related neurotoxicity, dose-dependently improved motor performance and prolonged the survival of ALS mice [27]; enhanced the viability of primary cortical neurons exposed to alcohol and prevented alcohol-induced apoptosis in the fetal brain in C57BL/6 mice [28]; and elicited full neuroprotection at a concentration of 100 fM against neuronal cell death caused by V642I-APP and M146L-PS1 in vitro and by Aβ42 i.c.v. injection in vivo. In the present study, we further assessed the neuroprotective effects of CLN pretreatment on multiple cognitive behaviors, hippocampal Aβ deposition, and synaptic plasticity in APP/PS1 mice. In the Y maze test, CLN pretreatment did not affect total arm entries but completely reversed the decrease in spontaneous alternation of APP/PS1 mice, suggesting that CLN effectively ameliorated the impairment of spatial working memory caused by APP/PS1 gene mutation without affecting basic motor activity. Similarly, a previous study showed that i.c.v. and intraperitoneal injection of CLN almost completely suppressed Aβ25–35-,Aβ42-, scopolamine-, and cholinergic blocker-induced spatial working memory impairment in CD1 mice [5, 8]. Our results also support those of Chiba et al., who observed that i.n. CLN treatment improved the memory of Tg2576 mice in Y maze, elevated plus maze, and water-finding tasks [7]. Deficits in visual short-term memory binding, a test used to assess recognition of changes in shapes or shape-color bindings, have recently been proposed as an early and specific marker of AD [29]. As another evaluation of short-term memory, the new object recognition test was performed in our study. The decrease in RI of 9-month-old APP/PS1 mice for the new object was obviously reversed in the CLN treatment group, indicating that CLN effectively improved new object recognition memory. In the MWM tests, CLN did not change the swimming speed but effectively decreased the escape latency of APP/PS1 mice in the hidden platform tests and increased swimming time in the probe trials, clearly indicating the neuroprotective effects of CLN on long-term spatial learning and memory. These results for transgenic AD mice are consistent with our previous findings in normal rats after Aβ25–35 injection [9]. Similarly, recent research showed that intraperitoneal injection of CLN improved long-term spatial memory in PDAPPV717I transgenic AD model mice, and the effect of CLN was significantly stronger than that of synchronous administration of AGA-HNG and ADNF [30]. These behavioral experimental results indicate that CLN has important positive effects on the new object recognition memory, spatial working memory, and long-term reference memory of APP/PS1 mice, and the present study provides further evidence that CLN may be a novel, effective alternative strategy for the prevention and treatment of AD.
Although behaviorally based neuroprotective effects of CLN have been reported, the in vivo electrophysiological mechanism and pathological improvement remain to be confirmed. Given the close relationship between spatial memory and hippocampal synaptic plasticity [31, 32], the present study investigated the effect of CLN on in vivo hippocampal LTP in behaviorally trained mice. We observed that CLN alone did not affect the induction and maintenance of LTP in WT mice but effectively protected against the depression of LTP in APP/PS1 mice. In addition, PPF, an electrophysiological indicator reflecting changes in presynaptic neurotransmitter release, was not affected by CLN. These in vivo LTP experimental results in transgenic mice are supported by recent in vivo and in vitro non-transgenic studies using Aβ25–35 and Aβ31–35 [9, 34]. In addition, the present study attempted to associate cognitive behaviors with pathological changes in the brain. We observed that chronic i.n. application of CLN not only improved multiple cognitive behaviors and in vivo hippocampal synaptic plasticity but also significantly reduced Aβ deposition in the hippocampus in APP/PS1 mice. As supporting evidence, Zhang et al. reported that chronic administration of HNG attenuated cognitive deficits and Aβ loads in middle-aged APP/PS1 mice, including mice with pre-existing substantial Aβ neuropathology [35]. Similarly, Yin et al. reported that CLN significantly reduced elevated Aβ42 and Aβ40 levels in the brains of AD mice [30].
The molecular and synaptic mechanisms underlying the neuroprotective effects of CLN against AD are poorly understood. Previous reports suggest that CLN plays neuroprotective roles by binding to an unidentified cell-surface receptor linked to Tyr kinase activity, followed by upregulation of phosphorylated STAT3 (p-STAT3) levels [7, 36] and maintenance of intracellular calcium homeostasis in AD-like animals [9]. In addition, STAT3 can bind to the 5’ upstream region of the PS1 gene and regulate its transcription [37]. PS1 is a major component of the γ-secretase that cleaves AβPP and contributes to the generation of Aβ [38], and thus CLN might decrease Aβ levels in the brain by increasing p-STAT3 levels and consequently reducing PS1 gene transcription.
Interestingly, STAT3 is activated in a variety of solid tumors, including brain tumors and hematological malignancies [39, 40]. Thus, the potential induction of cancer by long-term CLN application should be considered. Favorably, Chiba et al. reported that 3-week i.n. administration of CLN alone did not affect p-STAT3 levels in WT mice and only restored the reduced p-STAT3 levels in Tg2576 mice to normal levels [7]. Moreover, CLN alone did not increase the viability of primary cortical neurons [28] and did not activate the JAK2/STAT3 axis in glial cells in the CNS, which might promote gliosis in AD models [36]. However, the translation of a novel drug from basic research to clinical application is a complicated and lengthy process. In addition to ascertaining reliable effectiveness, the side effects, half-life, application pathway, and application scope of CLN must be established. Promising preliminary data from subacute safety/toxicity tests in mice indicated no obvious side effects in the liver and kidney [8].
In summary, the present study indicates that CLN has strong neuroprotective effects on learning and memory in APP/PS1 mice. The behavioral improvement induced by CLN, as well as the alleviation of Aβ deposition and LTP suppression in the hippocampus, suggest that CLN could be significant in the prevention and treatment of AD.
Footnotes
ACKNOWLEDGMENTS
This project was supported by the following: 1) Fund Program for “Sanjin Scholars” of Shanxi Province; 2) National Natural Science Foundation of China, 31471080 and 31300968; 3) Shanxi Scholarship Council of China, 2013-054; 4) Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shanxi Province; 5) Fund for Shanxi Key Subjects Construction.