
Abstract
Mitochondrial dysfunction is proposed to trigger memory deficits and synaptic damage at the onset of Alzheimer’s disease (AD). However, it is unknown how mitochondria dysfunction might trigger synaptotoxicity and if a differential susceptibility of mitochondria located in synapses underlies the greater glutamatergic than GABAergic synaptotoxicity in early AD. Hippocampal synaptosomes (purified synapses) of a rat model of early AD, typified by selective memory deficits two weeks after intracerebroventricular injection of amyloid-β peptides (Aβ1–42, 2 nmol), simultaneously displayed three mitochondria-associated deleterious alterations: 1) hampered metabolism (decreased MTT reduction); 2) increased oxygen radical production (increased hydrogen peroxide production); 3) increased caspase-3 activity. The direct exposure of hippocampal synaptosomes to Aβ1–42 (500 nM) similarly decreased mitochondrial membrane potential (TMRM+ fluorescence) and increased mitochondria-derived oxygen radicals (MitoTraker®red-CM-H2Xros fluorescence) in individual glutamatergic (vesicular glutamate transporter-immunopositive) and GABAergic (vesicular GABA transporter-immunopositive) synaptosomes. However, significantly more glutamatergic than GABAergic synaptosomes were endowed with mitochondria (Tom20-immunopositive). These results indicate that dysfunctional mitochondria located in synapses can trigger synaptotoxicity through multifaceted mechanisms and that it is not the susceptibility of mitochondria to Aβ but more likely a different impact of dysfunctional mitochondria that underlies the greater sensitivity to synaptotoxicity of glutamatergic than GABA synapses in early AD.
Keywords
INTRODUCTION
Mitochondria plays a central role in energy supply, and the dysfunction of mitochondria is associated with different disease conditions as a result of a diminished energy supply, formation of oxygen radical species, or initiation of apoptotic responses [1, 2]. There is abundant evidence for a deterioration of mitochondrial function in Alzheimer’s disease (AD) patients, as well as in animal models of this memory-related disease [3 –5]. Accordingly, oxidative stress is a major contributor to the evolution of AD [6] and mitochondria-targeted antioxidant strategies attenuate memory impairment [7]. This evolving neurodegenerative disorder begins with a dysfunction and damage of synapses in cortical regions [8], namely in the hippocampus [9], prior to the overt damage of neurons. Although alterations of GABAergic transmission have been described in AD models [10], glutamatergic synapses seem more susceptible to damage at the onset of AD [11, 12]. The mechanisms underlying early synaptotoxicity in AD are still unknown and a likely hypothesis is the involvement of mitochondrial dysfunction [4 , 13], since mitochondria located in synapses are critical to sustain synaptic function [14 –16]. In fact, mitochondria located in synapses have a different proteome [17, 18] and lipid composition [19], a different morphology [20, 21], and are more stressed and more prone to deterioration [22 –26] in view of their distance from the nucleus and their effort to meet the energy needs and handle calcium transients associated with synaptic function.
We now aimed to confirm the presence of altered mitochondria in synapses in a rodent model of early AD based on the intracerebroventricular administration of amyloid-β (Aβ) peptides [27], a purported trigger of AD [28]. Furthermore, we also enquired if the exposure to Aβ peptides caused a more robust alteration of mitochondria in glutamatergic than in GABAergic terminals of the rat hippocampus.
MATERIALS AND METHODS
Animals
Adult male Wistar rats (8–10 weeks) were obtained from Charles River (Barcelona, Spain) and were maintained at 23–25°C, under 12/12 h light/dark cycles and with ad libitum water and food. The study was approved by the Ethical Committee of the Center for Neuroscience and Cell Biology (Orbea 78–2013) and conducted in accordance with the principles and procedures outlined as “3 Rs” in the guidelines of European Union (Directive 2010/63/EU).
Exposure to Aβ1–42 and behavioral analysis
The Aβ1–42 peptide fragment (Bachem) was dissolved in water at a concentration of 2.25 mg/ml and 2 nmol in 4 μl were administered intracerebroventricularly (icv), whereas control rats were infused with a similar volume of water, as previously described [27]. The Aβ1–42 solution was constituted by oligomers of up to 4 monomers, as determined by native blue gel analysis of the mixture stained with Coomassie R-250 and an 6E10 antibody (see [27] for details). It takes 15 days to observe memory-related alterations, without mood or locomotor deficits, as well as a hippocampal synaptotoxicity, without gliosis or overt neuronal damage [27].
After 14 days, behavioral experiments were conducted between 9 : 00 AM and 4 : 00 PM (light phase) in a sound-attenuated room under low-intensity light (12 lux), where the rats had been habituated for at least 1 h before the beginning of the tests. Locomotion was evaluated in an open-field arena with a gridded floor, and the number of crossings and rearings was recorded during 10 min, as previously described [29]. On the next day, rats performed the object recognition test in the same arena. Rats first underwent a training session, in which they were exposed to two identical objects in the arena. The test session was performed 90 min after training where two dissimilar objects were presented, a familiar and a novel one, and we measured the time spent exploring each object, as previously described [29]. The discrimination ratio was defined as: TN/(TN + TF), [TN = time spent exploring the novel object; TF = time spent exploring the familiar object]. The rats were sacrificed on the next morning.
Synaptosomal preparation
Rats were anesthetized under halothane atmosphere, killed by decapitation and their hippocampi dissected and homogenized in ice-cold sucrose solution (0.32 M sucrose, 1 mM EDTA, 5 mM Tris, 0.25 mM dithiothreitol, pH 7.4 at 4°C). Synaptosomes (i.e., purified synapses) were obtained as previously described [12, 30], through combined isopicnic centrifugations (2000 g for 3 min followed by 9500 g for 13 min) in sucrose solution followed by a centrifugation (25000 g for 11 min at 4°C, without deceleration) in a discontinuous Percoll gradient in sucrose solution (from bottom to top: 3 ml of a 23%, 5 ml of a 10% and 3 ml of a 3% (v/v) Percoll solution). Synaptosomes with over 95% purity [12, 30] were collected at the interface between 10% and 23% (v/v) Percoll, washed and maintained at 4°C in HEPES-buffered medium (140 mM NaCl, 5 mM KCl, 1.2 mM NaH2PO4, 5 mM NaHCO3, 10 mM glucose, 10 mM HEPES and 1.2 mM MgCl2, pH 7.4).
Analysis of alterations related to mitochondrial dysfunction in synaptosomal populations
To evaluate possible consequences of mitochondrial dysfunction in synapses, we compared metabolic activity, the production of H2O2 and the activity of caspase 3 in hippocampal synaptosomes collected from control and Aβ1–42-exposed rats.
Since metabolic dysfunction has been proposed to be a main cause of synaptotoxicity [13, 31], we estimated the metabolic capacity of synaptosomes by the quantification of their ability to reduce 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium (MTT, Sigma-Aldrich) to form a purple formazan product. After 15 min of recovery at 37°C, synaptosomes were incubated with 1 mg of MTT in Krebs-HEPES solution (HEPES medium plus 1.2 mM CaCl2) for 2 h at 37°C to allow MTT reduction to proceed [32]. The synaptosomes were then pelleted and their purple formazan crystals were dissolved in 1 ml isopropanol during 15 min on a shaking table. The absorbance of the resulting purple solution was spectrophotometrically measured at 570 nm.
Since the excessive production of oxygen free radicals by mitochondria has also been proposed to be a main cause of synaptotoxicity [13, 31], the known continuous production of H2O2 by synaptosomes [33] was measured. This was done as previously described [34], with the Amplex® Red fluorescent dye (10-acetyl-3,7-dihydroxy-phenoxazine), which reacts with H2O2 and produces a fluorescent product, resorufin, in the presence of horseradish peroxidase [34]. Synaptosomes (0.5 mg/ml protein) were incubated in Krebs-HEPES solution during 15 min at 37°C, before addition of horseradish peroxidase (0.5 U/ml; Molecular Probes) and Amplex® Red reagent (50 μM; Molecular Probes). After incubation at 37°C for 30 min, fluorescence was measured at 585 nm upon excitation at 550 nm in a VictorTM plate reader (Wallac, Perkin-Elmer). A calibration signal was generated with a known amount of H2O2 (1 μM; Molecular Probes) and the selectivity of the signal was confirmed by its elimination in the presence of catalase (100 U/ml; Sigma-Aldrich).
Since caspase 3 seems to play a role in the development of synaptotoxicity [35], we measured the activity of caspase 3 by following the cleavage of its substrate (Ac-DEVD-AFC peptide; BD-Biosciences) to its fluorescent product, as we have previously described [36]. Synaptosomes were incubated for 15 min at 37°C, before homogenization in ice-cold lysis buffer (25 mM HEPES, 5 mM MgCl2, 5 mM EDTA, 5 mM dithiothreitol, 2 mM phenylmethylsufonyl fluoride, 6.25 μg/ml pepstatin A, 6.25 μg/ml aprotinin, pH 7.4). Aliquots of these samples (50 μl) were mixed with 50 μl of reaction buffer (50 mM HEPES, 2 mM EDTA, 20% glycerol, 10 mM dithiothreitol, pH 7.4) and 4 μl of caspase 3 substrate (final concentration 10 μM from a stock solution of 250 μM in reaction buffer) and added to 96-well plates. Samples were incubated at 37°C for 60 min in the dark, fluorescence was measured in a VictorTM plate reader (excitation 400 nm; emission 505 nm) and enzyme activity was calculated with reference to a standard curve of 7-amino-4-trifluoromethylcoumarin (AFC; 0–10 μM; BD-Biosciences) and values were expressed as nmol AFC/mg protein/min after measuring protein with the BCA assay.
Double immunocytochemical determination of synaptosomes endowed with mitochondria
Since mitochondria dysfunction is hypothesized to be responsible for synaptotoxicity [4 , 13] and glutamatergic terminals are more affected in early AD [11] and in the presently used Aβ1–42 rat model of early AD [12], we first used a double immunocytochemical analysis to determine the number of hippocampal glutamatergic synapses (having vesicular glutamate transporter type I, vGluT1) or GABAergic synapses (having vesicular GABA transporter, vGAT) endowed with mitochondria (stained with its marker Tom20). As previously described [12, 30], synaptosomes were first platted over poly-D-lysine-coated coverslips, fixed during 15 min with 4% of paraformaldehyde in phosphate buffered saline (PBS, composed of 140 mM NaCl, 3 mM KCl, 20 mM NaH2PO4, and 15 mM KH2PO4, pH 7.4 at 4°C) and washed twice with PBS. The synaptosomes were then permeabilized by exposure to 0.2% Triton X-100 in PBS, for 10 min at room temperature (RT), and then blocked with PBS containing 3% bovine serum albumin (BSA) and 5% normal horse serum for 1 h to prevent non-specific binding. After washing twice in PBS, the synaptosomes were incubated with primary antibodies against either vGluT1 (1 : 2000; Cat.n° 135304 from Synaptic Systems) or vGAT (1 : 1000; Cat.n° 274104 from Synaptic Systems) and against the mitochondrial preprotein translocases of the outer membrane (Tom20, 1 : 1000; Cat.n° sc-11415 from Santa Cruz) in PBS with 3% BSA for 1 h at RT. After washing three times with PBS containing 3% BSA, the synaptosomes were stained with the secondary antibodies Alexa Fluor® 594-conjugated goat anti-guinea pig (1 : 1000, Invitrogen) and either Alexa Fluor® 647-conjugated goat anti-rabbit (1 : 2000, Invitrogen) or Alexa Fluor® 350-conjugated goat anti-rabbit (1 : 2000, Invitrogen) for 1 h at RT. After washing, the preparations were mounted on glass slides with ProLong® Antifade and visualized by fluorescence microscopy (Zeiss Axiovert 200 microscope with a HXP 120V lluminator and images collected through a Plan Apochromat 63× oil 1.4 DIC M27 objective with a high resolution Axiocam HRc digital camera with a total magnification of 630×). The co-localization of the two emitting Alexa Fluor® tags was carried out as previously described [12, 30], using the ImageJ software (NIH), and counting a minimum of 200 individual nerve terminals in at least 3 different fields in each coverslip. It was confirmed that none of the secondary antibodies produced any signal in preparations to which the addition of the corresponding primary antibody was omitted.
Western blot analysis
To confirm that there was a selective decrease of glutamatergic rather than GABAergic markers accompanying early memory dysfunction after Aβ1–42 icv treatment, we prepared extracts from hippocampal synaptosomes of vehicle- and Aβ1–42-treated rats, by homogenizing a sample of synaptosomes in SDS-PAGE buffer containing 30% (v/v) glycerol, 0.6 M dithiothreitol, 10% (w/v) sodium dodecylsulfate (SDS) and 375 mM Tris-HCl pH 6.8 and boiling at 95°C for 5 min. After diluting and determining the amount of protein (bicinchoninic acid, BCA, protein assay; Thermo Scientific), these samples (50 μg protein) were analyzed by western blot to determine the immunodensity of vGluT1 and vGAT using primary antibodies against either vGluT1 (1 : 5000; Synaptic Systems) or vGAT (1 : 1000; Synaptic Systems), as previously described [12, 37]. Membranes were then re-probed for α-tubulin (1 : 10,000; Sigma) as a loading control. We randomly paired a sample from vehicle-treated and from Aβ1–42-treated rats in each western blot analysis and expressed the changes of immunodensity as a percentage of the immunodensity in the vehicle sample, after normalization with tubulin immunodensity.
Single synaptosome live imaging analysis
This group of experiments aimed at determining if different types of synaptosomes from control (i.e., non-treated) animals displayed a different susceptibility to the in vitro acute exposure to Aβ1–42. Aβ1–42 was directly dissolved in water and used immediately (within 90 min) after preparation, when the mixture was mainly constituted by monomers and oligomer containing up to four monomers, as previously described [27]. We previously carried out a concentration-response curve showing the ability of acute Aβ1–42 exposure to modify the reducing capacity of hippocampal synaptosomes (MTT assay) in the range of 0.1–5 μM, and we selected to use the single concentration of 500 nM of Aβ1–42 because it caused the maximal modifications of metabolic reduction without damage of the synaptosomes (release of lactate dehydrogenase) within a period of incubation of 2 h [27].
Hippocampal synaptosomes were diluted to an optical density between 0.055–0.057 at 600 nm in Krebs-HEPES solution, and plated over poly-D-lysine-coated 12 mm gridded coverslips (Bellco Biotechnology) during 45 min at RT. Synaptosomes were then exposed during 30 min to Krebs-HEPES solution in the absence (control) or presence of soluble Aβ1–42 (500 nM), prepared as previously described [27]. Synaptosomes were then incubated with 7.5 nM tetramethylrhodamine methyl ester (TMRM+; Invitrogen), a mitochondrial membrane-permeable fluorescent dye, to determine mitochondrial membrane potential (Δ ψm) and 1 μl/ml of the plasma membrane potential indicator (PMPI; 1 : 2000 dilution of a R-8042 Component A vial from Molecular Devices, reconstituted in 1 ml water) or with 5 μM MitoTraker® red CM-H2Xros (Thermo Fisher Scientific), a reduced, non-fluorescent dye that fluoresces upon oxidation. Coverslips were then placed in a superfusion chamber (RC-20; Warner Instruments), with 500 μl of Krebs-HEPES solution, on the stage of an inverted fluorescence microscope (Axiovert 200; Carl Zeiss). Synaptosomes were alternately excited at 490 and 550 nm using an optical splitter (Lambda DG4; Sutter Instruments), and the emitted fluorescence was captured at 600 nm through a 100×oil objective connected to a digital camera (CoolSNAP; Roper Scientific). Image acquisition was performed during 500 ms every 30 s for a total of 10 min. Acquired images were processed using MetaFluor software (Universal Imaging). The masks of synaptosomes were drawn, and the average value of pixel intensities was evaluated at each time point.
Mitochondrial membrane potential (Δ ψm) was estimated using a cell permeant, cationic, fluorescent dye that is rapidly sequestered by active mitochondria, TMRM+. Plasma membrane potential (Δ ψp) was estimated using PMPI, a lipophilic anion, bis-oxonol-type probe that is excluded from the cytosol by membrane potential. We first established a baseline for 2.5 min before the addition of 5 μl of cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP, 1 mM; Sigma-Aldrich) and oligomycin (1 mg/ml; Sigma-Aldrich) or 30 mM KCl both made up in Krebs-HEPES solution and added directly to the bath. Δ ψm was estimated as the difference between TMRM+ fluorescence values before and after addition of FCCP+oligomycin, controlling for the photobleaching of the dye, as well as for the effect of solvents (ethanol), both assessed in parallel experiments. Reactive oxygen species was estimated by the change in fluorescence of the mitochondrial selective probe, Mitotraker® Red CM-H2Xros, before and after the addition of antimycin A (5 μM; from Sigma-Aldrich).
After live imaging, the synaptosomes were processed for the immunocytochemical detection of markers of glutamatergic and GABAergic synapses, namely vGluT1 and vGAT, as described above [12, 30]. The initial collection of a transmission image together with the gridded marks in the coverslip allowed drawing a mask to delineate each of the individual synaptosomes, which were then allocated as GABAergic or glutamatergic in nature, thus enabling to distinguish the alterations of Δ ψm and of the production of reactive oxygen species in each type of hippocampal synaptosomes.
Statistical analysis
Results are mean±Standard Error of the Mean (S.E.M.) of the indicated number of experiments (n). To test the significance of the differences between two groups, a Student’s t-test was used (unpaired for behavioral analysis and paired for biochemical assays). The level of significance of the differences is indicated in the text.
RESULTS
Rats injected with 2 nmol Aβ1–42 icv suffered a deterioration of memory performance in the object recognition test when evaluated 15 days after Aβ1–42 administration (Fig. 1A), whereas they displayed a locomotion similar to that of control rats (Fig. 1B). We then enquired at this time point, representative of early AD, if hippocampal synaptosomes displayed alterations compatible with a modified function of mitochondria in synapses. Fig. 1C shows that the metabolic redox capacity was hampered in hippocampal synaptosomes from Aβ1–42-treated rats, as testified by their lower ability to reduce MTT (absorbance of formed formazan = 0.362±0.023, n = 8; p = 0.0039) compared to synaptosomes from vehicle-treated rats (absorbance of formed formazan = 0.427±0.009, n = 8). Likewise, the generation of oxygen radicals, another consequence of mitochondrial dysfunction that can trigger the loss of synapses [4 , 31], was also increased, as heralded by the increased production of H2O2 in hippocampal synaptosomes from Aβ1–42-treated (Amplex® red fluorescence = 0.0081±0.0011, n = 7; p = 0.0031) compared to control rats (Amplex® red fluorescence = 0.0018±0.0002, n = 7) (Fig. 1D). Finally, we also report that the activity of caspase 3, which is involved in synaptotoxicity in animal models of AD as a result of mitochondrial dysfunction [35], was also enhanced in hippocampal synaptosomes from Aβ1–42-treated (0.441±0.075 nmol AFC/mg protein/min, n = 6; p = 0.025) compared to control rats (0.187±0.027 nmol AFC/mg protein/min, n = 6) (Fig. 1E).
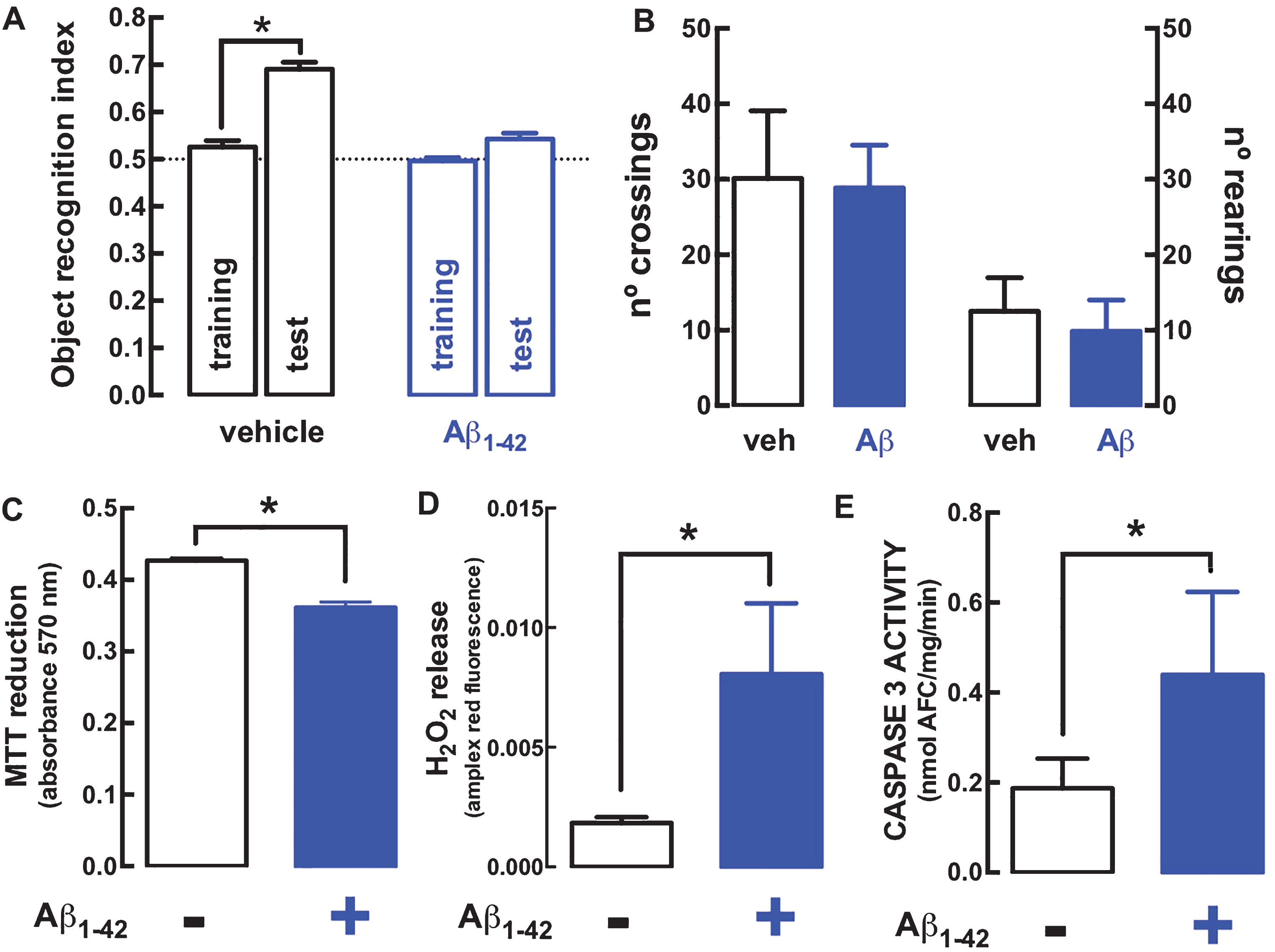
Deterioration of different mitochondria-related parameters in rat hippocampal synaptosomes prepared 15 days after exposure to amyloid-β peptides (Aβ1–42) to model early AD as typified by an impaired performance in a reference memory test. A) Fifteen days after an intracerebroventricular (icv) administration of Aβ1–42 (2 nmol), rats displayed a lower ability to recognize a novel object compared to control rats injected icv with vehicle. B) In contrast, there was no difference in locomotion assessed in an open field arena. The behavioral data are mean±SEM of n = 8-9 rats per group. Hippocampal synaptosomes from Aβ1–42-treated rats displayed a lower metabolic capacity, as measured by a lower ability to reduce MTT (C), a greater production of oxygen radicals measured with the Amplex® red hydrogen peroxide assay (D) and a larger activity of caspase 3, measured by the cleavage of its selective substrate (E). The biochemical data are mean±SEM of n = 6– 8 rats per group. * p < 0.05 using a Student’s t test (unpaired in A, paired in C-E).
We next aimed determining the number of synaptosomes endowed with mitochondria. This was assessed first in the hippocampus of control rats by quantifying the number of excitatory synaptosomes (i.e., glutamatergic synaptosomes endowed with their selective marker, vesicular glutamate transporter type 1, vGluT1) or of inhibitory synaptosomes (i.e., GABAergic synaptosomes endowed with their selective marker, vesicular GABA transporter, vGAT) endowed with a generic marker of mitochondria (Tom20) [38]. Figure 2A shows representative double labelling immunocytochemical staining of hippocampal synaptosomes with vGluT1 (i - red) or Tom20 (ii - green) and their co-localization (iii - yellow), that allowed concluding that 65.1±3.8% (n = 5) of glutamatergic synapses are endowed with mitochondria (Fig. 2C). A similar analysis of the co-localization of vGAT (i - red) and Tom20 (ii - blue) (Fig. 2B) allowed concluding that 43.3±3.4% (n = 6) of GABAergic synapses are endowed with mitochondria (iii - co-localization in white) (Fig. 2C).
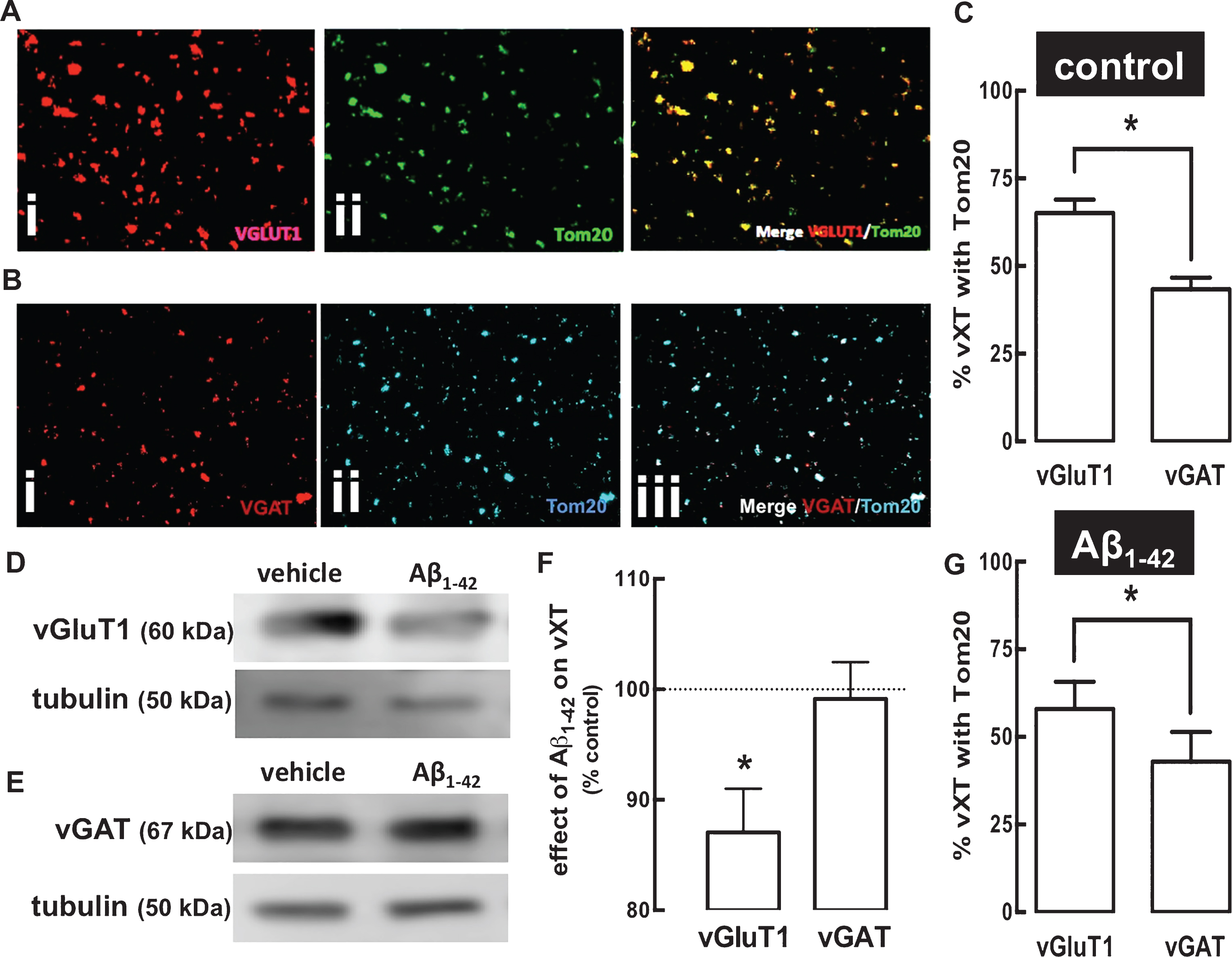
A greater percentage of glutamatergic compared to GABAergic terminals are endowed with a mitochondrial marker and this is unaffected in rat hippocampal synaptosomes prepared 15 days after exposure to amyloid-β peptides (Aβ1–42) to model early AD. A, B) Immunocytochemical analysis in hippocampal nerve terminals from control rats of a marker of glutamatergic nerve terminals, vesicular glutamate transporters type 1 (vGluT1 stained with Alexa Fluor® 594 – red; i in A) or of a marker of GABAergic nerve terminals, vesicular GABA transporters (vGAT stained with Alexa Fluor® 594 – red; i in B) and of a marker of mitochondria, Tom20 (stained with Alexa Fluor® 647 – green; ii in A; or with Alexa Fluor® 350 – blue; ii in B), shows the extent of their co-localization in the merged image (yellow; iii in A; white; iii in B). C) This enabled determining that a larger number of glutamatergic than GABAergic synaptosomes of control rats are endowed with mitochondria. D, E) Western blot comparison of the density of vGluT1 and vGAT in synaptosomal extracts from the hippocampus of control rats and from rats 15 days after exposure to Aβ1–42 (2 nmol, icv), showing that there is a reduction of the density of vGluT1, but not of vGAT after Aβ1–42 treatment (F). In spite of this purported reduction of glutamatergic terminals after Aβ1–42 treatment, the percentage of the remaining glutamatergic and GABAergic terminals endowed with Tom-20 remained unchanged after Aβ1–42 treatment (G). Immunocytochemistry data are mean±SEM of n = 4–12 rats per group and western blot analysis are mean±SEM of n = 6 rats per group. * p < 0.05 using an unpaired Student’s t test in A and G and a t-test comparing with 100% in F. The magnification used to obtain the images was 630×.
We next tested if the percentage of glutamatergic and GABAergic synapses endowed with Tom20 was affected after an in vivo treatment with Aβ1–42. In accordance with the different susceptibility of these two main types of hippocampal nerve terminals in early AD [11, 12], we first confirmed that 15 days after exposure to Aβ1–42-association there was a reduction of vGluT1-immunoractivity (to 87.1±3.9% of control, n = 6; p = 0.022; t5 = 3.276, t-test compared to 100%) (Fig. 2D, F) but not of vGAT-immunoreactivity (to 99.1±3.3% of control, n = 6; p = 0.807; t5 = 0.257, t-test compared to 100%) (Fig. 2E, F). In contrast, the percentage of the remaining glutamatergic or GABAergic synapses endowed with mitochondria was not altered in synaptosomes collected from Aβ1–42-treated rats (58.0±6.7% of vGluT1-positive synapses, n = 4, and 42.9±6.8% of vGAT-positive synapses, n = 6, were Tom20-positive) (Fig. 2G).
These findings of lack of alteration of the association of mitochondria markers with these two types of synapses in contrast to the alterations caused by in vivo Aβ1–42 exposure selectively on glutamatergic rather than GABAergic markers, lead us to test in an in vitro assay if the known direct impact of Aβ1–42 on mitochondria located in synapses [27 , 40] was different in glutamatergic and GABAergic synapses. For that purpose, we exposed hippocampal synaptosomes from naïve rats directly to Aβ1–42 and monitored on-line its impact on TMRM+ fluorescence (more depolarized mitochondria will have their interior less negative and will accumulate less dye) to record alterations of mitochondrial membrane potential (Δ ψm) in individual synaptosomes; then, the tested synaptosomes were fixed and stained against vGluT1 or vGAT to ascribe each synaptosome as glutamatergic or GABAergic. As shown in Fig. 3, Aβ1–42 decreased TMRM+ fluorescence (corrected for background fluorescence, as obtained through subsequent exposure of the synaptosomes to FCCP+olygomicin, which blunts the mitochondrial membrane potential) in both glutamatergic (Fig. 3A, C) and GABAergic synaptosomes (Fig. 3B, D); Δ ψm was reduced by 28±6% (p = 0.0065; n = 6) in glutamatergic synaptosomes and by 25±9% (p = 0.0533, n = 5) in GABAergic synaptosomes. Importantly, we controlled that this different modification of TMRM+ responses was indeed reflecting a different alteration of Δ ψm, by showing that Aβ1–42 was devoid of effects on the plasma membrane potential, which can affect TMRM+ responses [41], as assessed by the lack of modification of PMPI fluorescence in both glutamatergic and GABAergic synaptosomes upon challenging with FCCP and olygomycin (data not shown). As a positive control, we confirmed that PMPI fluorescence displayed a transient and robust increase upon addition of 30 mM KCl (peak value of 93±19% over baseline, n = 6) in the whole population of hippocampal synaptosomes.
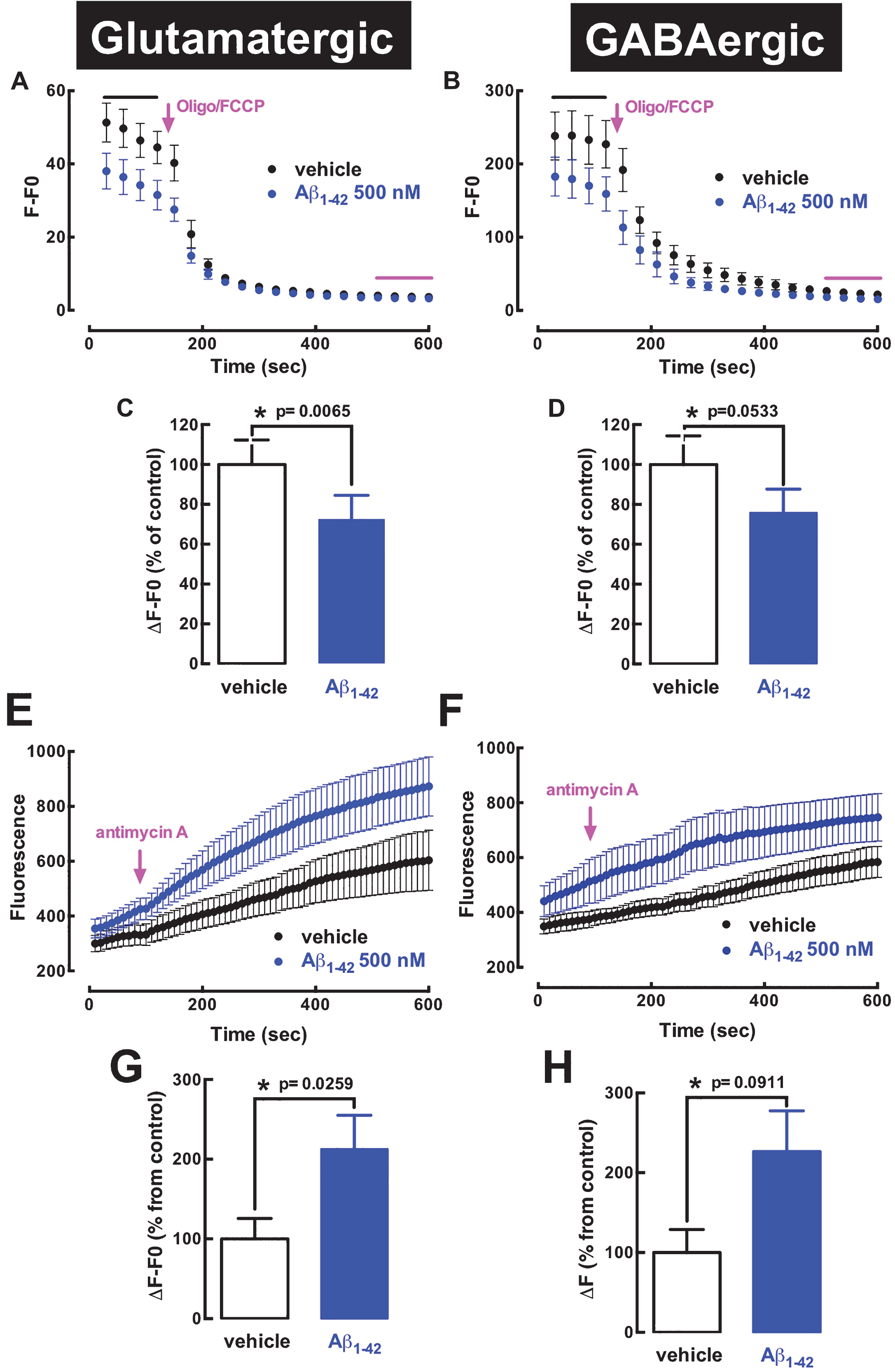
Direct in vitro exposure to Aβ peptides similarly affect mitochondria in glutamatergic and GABAergic hippocampal synapses. Hippocampal synaptosomes were obtained from naïve rats, platted in gridded coverslips and exposed to 500 nM Aβ1–42, following a 30-min pre-incubation with TMRM+ to measure mitochondria membrane potential (Δ ψ m) or with MitoTraker® Red CM-H2Xros to determine the production of mitochondria-derived oxygen radicals in individual synaptosomes. After the in situ on-line recordings of Aβ1–42-induced alteration of mitochondria located in synapses, the synaptosomes were immunocytochemically stained for vesicular glutamate transporters type 1 (vGluT1) or for vesicular GABA transporters (vGAT) to ascribe them as glutamatergic or GABAergic synapses, respectively. The alteration of TMRM+ fluorescence before and after the addition of FCCP+olygomycin (F0) allowed estimating Δ ψ m in glutamatergic (A) and GABAergic hippocampal synapses (B), showing that the exposure to Aβ1–42 similarly affected Δ ψ m in glutamatergic (C) and GABAergic synapses (D). The alteration of MitoTraker® Red CM-H2Xros fluorescence before and after the addition of antimycin A (to block complex III of mitochondrial chain, inducing maximal reactive oxygen species generation) allowed estimating the production of oxygen radicals in glutamatergic (E) and GABAergic hippocampal synapses (F), showing that the exposure to Aβ1–42 similarly affected mitochondria-derived oxygen radicals in glutamatergic (G) and GABAergic synapses (H). Data are mean±SEM of n = 5-6 rats per group. *Significance with the indicated p value, using a Student’s t test.
We then aimed determining if the alteration of mitochondrial function resulted in deleterious consequences that were different in glutamatergic and GABAergic synaptosomes. We were limited to testing the formation of mitochondria-derived oxygen radicals, which is the only endpoint that can currently be followed by live imaging in single synaptosomes, with the use of the Mitotraker® Red CM-H2Xros. Upon addition of antimycin A, an inhibitor of the electron transport chain, Aβ1–42 (500 nM) increased the production of mitochondria-derived oxygen radicals in both glutamatergic (Fig. 3E, G) and GABAergic synaptosomes (Fig. 3F, H), with similar amplitudes (110±43% increase, p = 0.0259; n = 6; and 127±61% increase, p = 0.0911, n = 5, respectively).
DISCUSSION
The present study confirms in a rat model of early AD, the occurrence in hippocampal synapses of different deleterious consequences resulting from mitochondrial dysfunction, namely lower metabolic reducing capacity, higher formation of oxygen radicals, and increased activity of caspase 3. In an attempt to understand the different susceptibility of different hippocampal synapses, we unexpectedly concluded that the exposure to Aβ peptides triggered a similar alteration in glutamatergic and GABAergic terminals of mitochondria membrane potential as well as mitochondrial production of oxygen radicals. However, more glutamatergic than GABAergic synaptosomes contained mitochondria, raising the hypothesis that mitochondrial dysfunction might cause a larger impact on the function and viability of glutamatergic than GABAergic synapses of the hippocampus.
Several studies have reported the presence of altered mitochondria in AD patients [42, 43], as typified by an increased free radical production and oxidative damage, decreased density and activity of Krebs cycle enzymes, decreased ATP production, abnormal expression of mitochondria-encoded genes, and alterations of mitochondrial ultrastructure and trafficking [4, 5]. These modifications are largely recapitulated in cell lines expressing mutant forms of AβPP or treated with Aβ peptides, and in transgenic AD mouse models, contributing to the growing body of evidence suggesting that the accumulation of Aβ peptides plays a central role in mediating mitochondrial toxicity [4, 5]. This hypothesis of a key role of mitochondrial dysfunction to trigger AD-related memory impairment was consolidated by the parallel observations that synaptic dysfunction is an early feature in AD [8, 9] and that mitochondria within synapses are critical to sustain synaptic activity [22, 44]. In fact, hampering the trafficking to synapses and/or the dynamics within synapses of mitochondria impacts on single pulse responses at individual synapses and disrupts synaptic plasticity [22, 44]. Accordingly, mitochondria located in synapses are modified in AD patients and AD animal models [3–5 , 13]. The mechanism by which mitochondrial dysfunction might lead to synaptotoxicity is still unclear. One possibility is an energetic deficit [31], since synaptic activity [45] and in particular vesicle cycling [46] are metabolically expensive processes. Accordingly, glucose hypometabolism is an early feature in AD, emerging before the occurrence of morphological or phenotypic signs of the disease [47, 48]. Another possibility is the excessive production of free radicals [5, 31], in accordance with the ability of mitochondria-targeted antioxidant strategies to attenuate memory impairment [3 , 7]. Still another possibility would be a local recruitment of caspase 3 to trigger synaptotoxicity, as was concluded in a mouse AD model [35]. We now report that all three mitochondria-related alterations are simultaneously present in hippocampal synaptosomes from an Aβ1–42-induced rat model of early AD, re-enforcing the multifaceted contribution of the dysfunction of mitochondria located in synapses [49] to the synaptotoxicity known to occur at the onset of memory impairment in this model of early AD [12, 27].
In spite of these observations linking mitochondria deterioration with synaptotoxicity in early AD, we were surprised to conclude that glutamatergic and GABAergic hippocampal synapses display mitochondria with similar susceptibility to damage upon exposure to Aβ1–42. In fact, it has previously been reported that the exposure of synaptosomes to Aβ1–42 was sufficient to alter the morphology and function of mitochondria located in synapses [27 , 40]. Since different synapses are differentially affected in the course of AD [50], with a greater susceptibility of glutamatergic synapses at the onset of AD [11 , 51–53] whereas GABAergic synapses seem less affected [27 , 55], we posited that Aβ1–42 might cause a greater alteration of mitochondria in glutamatergic compared to GABAergic synapses. However, our findings negated this working hypothesis, showing that both the Aβ1–42-induced decrease of Δ ψm as well as the Aβ1–42-induced generation of mitochondria-derived oxygen radicals were similar in both types of synapses. Although this does not invalidate the purported critical contribution of mitochondrial dysfunction to synaptotoxicity in early AD, it adds a new dimension to the different susceptibility of different synapses to excitotoxicity. In fact, the difference between glutamatergic and GABAergic synapses might not be due to a different susceptibility of their mitochondria, but rather to the different impact of defective mitochondria on the viability of the two different types of synapses. In accordance with this new hypothesis, it is interesting to note that the association of mitochondrial dysfunction with altered synaptic transmission has been defined in glutamatergic synapses [14 , 35], whereas the specific impact of mitochondrial dysfunction in GABA-mediated inhibitory transmission is comparatively less explored [56]. Furthermore, we now report that fewer GABAergic than glutamatergic synaptosomes were endowed with mitochondria, as assessed by Tom20 immunoreactivity. Overall this is suggestive of a greater dependence of glutamatergic than GABAergic synapses on the presence and function of mitochondria. It is worth noting that we observed a surprisingly low number of synapses endowed with mitochondria (circa 50%), which is clearly at odds with the expected energy burden of the process of synaptic transmission [57]. One obvious possibility is that Tom-20, which is expected to be present in all mitochondria [38], might not be accessible to antibody recognition in mitochondria located in synapses. Another possibility resides in the rapid trafficking of mitochondria (reviewed in [58]), possibly moving rapidly in and out of synapses more rapidly than their time of residence in synapses, so that a snapshot would only show a few synapses with resident mitochondria. However, elegant dynamic studies in cultured hippocampal neurons showed that less than 10% dendrites (actually as low as 2%) are endowed with mitochondria [59] and synaptic scaling has a far greater impact on morphology and activity compared to the association of mitochondria with dendrites [59]. It is striking to realize that we know far more about the process of neurotransmission than about its energetic support. We do not know if synapses without mitochondria correspond to silent synapses, if mitochondria rely on energy produced nearby but outside synapses [60], how fast mitochondria move in and out of synapses, how many mitochondria might be present in central mammalian synapses and what substrates are oxidized to generate the energy required to maintain synaptic function (e.g., [61, 62]).
In spite of these limitations, the present study provides the first evidence that the susceptibility of mitochondria located in synapses to Aβ peptides is similar in different types of nerve terminals. This suggests that it is not the sensitivity of mitochondria in excitatory and inhibitory terminals, but rather the impact of modified mitochondria that can explain the different susceptibility to synaptotoxicity of excitatory and inhibitory synapses of the hippocampus in animal models of AD. Thus, future studies should address if GABAergic terminals are more resistant than glutamatergic synapses to mitochondria-related dysfunction or if other parameters [63], such as increased activity [64, 65] or a different density and/or impact of tau [4] selectively at glutamatergic synapses, need to be present together with dysfunctional mitochondria to bolster the selective damage of glutamatergic terminals in early AD [11, 12]. Clearly, the identification of the factor responsible for the selective destruction of some particular synapses in early AD will be paramount to design effective strategies to counteract this key process at the onset of AD.
Footnotes
ACKNOWLEDGMENTS
We thank Carla G. Silva for helping in preparing some of synaptosomal preparations. This study was supported by Maratona da Saúde and Santa Casa da Misericórdia; support was also obtained from ERDF, through Centro 2020 (project CENTRO-01-0145-FEDER-000008:BrainHealth 2020), and through FCT (project POCI-01-0145-FEDER-007440).