
Abstract
With increasing survival of patients infected with human immunodeficiency virus type 1 (HIV-1), the manifestation of heterogeneous neurological complications is also increasing alarmingly in these patients. Currently, more than 30% of about 40 million HIV-1 infected people worldwide develop central nervous system (CNS)-associated dysfunction, including dementia, sensory, and motor neuropathy. Furthermore, the highly effective antiretroviral therapy has been shown to increase the prevalence of mild cognitive functions while reducing other HIV-1-associated neurological complications. On the contrary, the presence of neurological disorder frequently affects the outcome of conventional HIV-1 therapy. Although, both the children and adults suffer from the post-HIV treatment-associated cognitive impairment, adults, especially depending on the age of disease onset, are more prone to CNS dysfunction. Thus, addressing neurological complications in an HIV-1-infected patient is a delicate balance of several factors and requires characterization of the molecular signature of associated CNS disorders involving intricate cross-talk with HIV-1-derived neurotoxins and other cellular factors. In this review, we summarize some of the current data supporting both the direct and indirect mechanisms, including neuro-inflammation and genome instability in association with aging, leading to CNS dysfunction after HIV-1 infection, and discuss the potential strategies addressing the treatment or prevention of HIV-1-mediated neurotoxicity.
Keywords
HUMAN IMMUNODEFICIENCY VIRUS 1 EPIDEMIOLOGY
Despite advances in knowledge, treatment, and awareness, human immunodeficiency virus (HIV)/acquired immune deficiency syndrome (AIDS) remains a pandemic with a global prevalence of 0.8%. Since the start of the epidemic, an estimated78 million people have become infected with HIV-1, and 35 million people have died as a result of HIV-1-related illnesses. In 2015, 36.7 million people (including 1.8 million children) were living with HIV-1. Sub-Saharan Africa remains the most affected region, with over 25 million HIV-1 infected individuals. In 2015, 2.1 million new HIV-1 infections were identified, of which 150,000 were children. Most newly infected children live in Sub-Saharan Africa and were infected via their HIV-positive mothers during pregnancy, childbirth, or breastfeeding (http://www.unaids.org/en/resources/fact-sheet).
The global epidemic of HIV-1 infection has changed significantly after the introduction of antiretroviral therapy (ART). As of June 2016, 18.6 million people (46% of all adults and 49% of all children) have had access to combination ART (cART) globally. The global prevalence of HIV-1 infection increased from 31.8 million in 2005 to 36.7 million by the end of 2015, because patients on cART live much longer. The number of new HIV-1 infections in children decreased by 67% between 2005 and 2015, because of increased access to cART, resulting in prevention of mother-to-child HIV-1 transmission. However, despite advances in treatment, HIV-1 remains a major contributor to global deaths. AIDS-related deaths peaked at 2 million in 2005 and decreased to 1.1 million in 2015. Tuberculosis remains the leading cause of death among HIV-1 patients, accounting for one in three AIDS-related deaths. However, some progress has been made, with a 32% drop in tuberculosis-related deaths among people with HIVsince 2004 (http://www.unaids.org/en/resources/fact-sheet).
In 1984, after the first discovery of HIV-1, a second type of the virus was discovered in AIDS patients in West Africa in 1986. Both HIV-1 and HIV-2 use the same modes of transmission and involve similar opportunistic secondary infections upon development of AIDS. However, unlike HIV-1, HIV-2 infections progress slowly. Although, the number of HIV-2-infected patients is growing slowly globally and spreading from Africa to Europe, India, and the United States, HIV-1 infection affects the majority of the population more aggressively, leading to death. In the present review, we will focus on the HIV-1 infection particularly and its disease mechanism and pathology. Hereafter, HIV-1 will be followed as HIV unless and otherwisementioned.
HIV-ASSOCIATED NEUROPATHOLOGICAL COMPLICATIONS
Most HIV-infected patients suffer from neurological dysfunctions collectively known as HIV-associated neurocognitive disorder (HAND). HAND is typically mild in most cases and, thus, is unrecognizable at the beginning. Cognitive impairments in an HIV-affected CNS can be broadly categorized into three groups.
Asymptomatic neurocognitive impairment (ANI)
ANI, which was first recognized as a key neuropathological disorder in HIV-infected patients in 2007 [1], is more prevalent than HIV-associated dementia (HAD) among infected individuals. As it is not an overt pathology, it is often difficult to clinically distinguish ANI from mild neurocognitive disorder (MND). According to the criteria for HAND diagnosis, ANI is characterized by cognitive impairment involving at least two cognitive domains without any functional deficits in everyday performance. To date, it remains controversial whether ANI can be considered an early sign of developing MND or HAD among HIV-infected patients. However, a longitudinal study led by Grant et al. for the CNS HIV Anti-Retroviral Therapy Effects Research (CHARTER) Group concluded that individuals suffering from ANI exhibit a 2- to 6-fold higher risk of developing early symptomatic cognitive decline, suggesting an urgent need to identify treatment avenues for those at highest risk of MND [2]. Furthermore, about 16–19% of HIV-negative individuals over 35 years of age were characterized as having ANI based on American Academy of Neurology criteria, suggesting that further scrutiny of the parameters of test batteries for diagnosing ANI is required [3].
The major bottleneck in finding predictive biomarkers and/or definitive neuropsychological parameters for characterizing ANI is the identification of actual disease onset and progression. ANI can develop from multiple secondary factors, such as genetic susceptibility toward neurodegeneration, substance abuse, other infectious diseases, and neurotoxicity of ART. Moreover, at the initial stage of highly active ART (HAART), most cognitive deficits (both asymptomatic and symptomatic) are reversed, making it difficult to pinpoint the natural history of disease origin [4, 5]. Furthermore, the symptoms of ANI may fluctuate over time depending on the age of the infected individual, rendering it difficult to directly correlate progression of HIV infection with MND or HAD.
To date, ANI diagnosis as a disease is challenging, requiring a very stringent approach for characterizing the disease. However, recent advances in brain imaging techniques may have much higher potential to identify the disease than other previously employed criteria. Because asymptomatic decline is not associated with anatomic alterations in brain architecture, it is difficult to utilize structural imaging tools. However, molecular imaging techniques, such as proton magnetic resonance spectroscopy (MRS), show increases in the levels of choline, a cell proliferation and inflammation marker, and myo-inositol, a tissue glial marker, in almost all cases of HIV infection, even in totally asymptomatic individuals, N-acetyl-aspartate (NAA), a neuronal marker of injury and an indirect measure for brain metabolism, is found to be closely related to the degree of cognitive dysfunction. Also, blood oxygen level-dependent functional magnetic resonance imaging, which can measure the blood flow rate in the brain associated with specific cognitive functions to correlate and understand the extent of damage due to HIV infection in comparison with age-matched healthy individuals [6], could be a promising tool in the diagnosis and treatmentof ANI.
Mild Neurocognitive Disorder (MND)
MND is defined as an impairment of at least two cognitive domains associated with learning, memory, or thinking with detectable impairment/aberration in motor skills and other daily life activities[7, 8]. Unlike ANI, MND involves mild to moderate anatomic changes associated with the affected cognitive domains, including substantial volumetric reductions in cortical and subcortical brain regions [9]. As mild to moderate neurocognitive disorder can even occur in HIV-negative individuals depending on their age, genetic factors, dementia, delirium, alcohol or drug abuse, or unattended infectious diseases, extremely cautious and stringent diagnostic approaches should be taken to confirm that MND is only attributable to HIV infection. Although still under debate, a growing body of evidence shows that early onset of MND increases the risk of developing HAD by several fold, specifically fueled by chronic HIV and persistent immunosuppression, in the later stages of HIV infection predominantly amongindividuals genetically predisposed to neurodegenerative diseases [10, 11].
HIV-Associated Dementia (HAD)
HAD, or AIDS dementia complex, is a late-onset neurological complication observed in nearly 30% of HIV-infected individuals and diagnosed based on changes in neuropsychological behaviors of the infected person [12, 13]. Continuing viral load, combined with immunosuppression and prolonged neurotoxicity of ART drugs, causes severe anatomic deformity in the brain architecture, leading to HAD. Although it is beyond the scope of this review, it is worth mentioning that secondary pathogenic infections in the CNS can lead to HAD in AIDS patients with longer lifespans following HAART. HAD symptoms and severity widely vary from patient to patient, depending most importantly on patient age at onset of the disease. HAD is primarily characterized by cognitive impairments involving short-term memory loss combined with lack of concentration, difficulty in learning new things, difficulty in finding words, confusion in following instructions, longer response time, inability to perform daily tasks, and/or abrupt psychological changes such as social withdrawal, personality changes, and depression. Advanced stages of HAD are associated with more severe systemic motor dysfunctions, such as movement disorder, muscle weakness, vision problems, speech problems, and balance disturbances. The severity and stage of dementia are usually assessed by the International HIV Dementia Scale (IHDS), for which a score of ≤10 out of 12 is considered the upper limit for further evaluation of possible dementia [14]. Despite the well-established IHDS screening methods routinely used for American and African populations, recent evaluations of the clinical utility of the IHDS, compared with the HIV Dementia Scale (HDS), reveal divergent outcomes. In one study among Spanish-speaking adults, the HDS showed more appropriate screening results and clinical utility in terms of time and cost compared with the IHDS [15]. Conversely, a separate study among German-speaking people recommended the IHDS as a useful screening tool [16], suggesting that the pattern of HAD symptoms and their severity could be linked to the genetic background and lifestyle of individual patients.
Due to the lack of predictive biomarkers and early physical diagnosis, HAD diagnosis largely relies on the exclusion of other diseases. Although earlier studies in primate models suggest the possibilityof separating simian immunodeficiency viruses (SIV)/AIDS from SIV encephalitis by identifying novel metabolites using MRS scanning, its clinical application in human patients has not been tested [17]. However, recent advances in in vivo molecular and structural brain imaging techniques for patients with dementia, or dementia-associated diseases like Alzheimer’s disease (AD) or Parkinson’s disease (PD), offer a great opportunity to design precise therapeutic strategies to combat the disease from its early onset [18].
NEURO-MUSCULAR COMPLICATIONS IN HIV-ASSOCIATED SENSORY NEUROPATHY
HIV-associated sensory neuropathy (HIV-SN), which involves both myelinated and unmyelinated axons of distal nerves, is another common neurological complication in the majority of HIV-infected elderly people. HIV-SN is thought to be the outcome of chronic HIV-infection along with opportunistic secondary infections and/or antiretroviral drug toxicity. Peripheral sensory neuropathy can be categorized into four groups based on their possible origins: (1) Painful Sensory Neuropathy (PSN) includes distal sensory polyneuropathy and toxic neuropathy from antiretroviral drugs, with undistinguishable neuropathological symptoms; (2) Inflammatory demyelinating polyradiculoneuropathies (IDP) including both acute and chronic forms occur with high frequency at the early stage of HIV-infection, possibly due to autoimmune phenomena. Clinically and electrophysiologically, IDPs in HIV-infected people are indistinguishable than that of non-HIV people [19]; (3) Mononeuropathy multiplex can occur at both early stage due to immune dysfunction and advanced stage due to secondary opportunistic pathogens like cytomegalovirus, Varicella-Zoster virus, and hepatitis B or C virus in immunocompromised conditions [20, 21]; (4) Progressive polyradiculopathy causes due to progressive loss of sensory and motor neurons involving the lumbar and sacral roots, characterized by lumbosacral pain, saddle anesthesia, and impairment of urinary retention and rectal sphincter control as early signs. Cytomegalovirus and Varicella-Zoster virus are considered to be primary pathogens to develop this disease at the advanced stage of AIDS [22–24].
Among these different subtypes, PSN is the most commonly occurring form, affecting ∼60% ofHIV-infected population. The clinical symptoms of PSN includes reduced sharp sensation for vibration in the legs and feet, and delayed ankle reflexes in a distal symmetrical pattern. Pain symptoms are characterized by gradual onset of bilateral pain, tingling, and numbness with a burning or aching sensation. Pain generally starts in toes, gradually spreads to proximal regions, and worsens in soles. In advanced stages, pain spreads to hands.
Distal sensory polyneuropathy has been found to occur at the early stage of primary HIV infection due to high rate of activated macrophage, cytokines, and chemokines infiltration in the CNS resulting from systemic and nervous system immune responses [25]. Studies in the SIV-infected primate models have shown that primary HIV-infection induces sensory neuropathy by damaging dorsal root ganglia nerve fibers and intra-epidermal nerve fiber density [26, 27]. It has been found that sCD133 and regulated on activation normal T cell expressed and secreted (RANTES) can serve as potential biomarkers for SN. Additionally, CD137 signaling regulates trafficking of activated macrophage to dorsal root ganglia, leading to severe loss of intra-epidermal nerve fiber density [28]. Recently, as a potential therapeutic approach, treatment with α4-integrin antibody has been found to rescue dorsal root ganglia fiber damage by blocking the trafficking of activated monocyte/macrophage, but not T-lymphocyte [29]. Similar antibody treatment in another study has been found to decrease cardiac pathology by regulating monocyte/macrophage trafficking to the heart in SIV-infected primate model of HIV-AIDS [30]. CHARTER Group studies on a large cohort of HIV-infected people receiving cART have shown that ∼50% of the patients suffer from PSN, causing physical disability and reduced quality of life despite successful cART treatment [31]. A cross-sectional deep profiling study revealed that patients with HIV-SN presented metabolic dysfunction (higher plasma triglyceride), lack of concentrations, depression and anxiety, higher pain catastrophizing scores, and insomnia compared to HIV participants without peripheral sensory neuropathy [32].
In this context, it is important to mention that although majority of the HIV-infected patients suffer from PSN, the extent of severity differs among different races and genders, even some HIV-infected patients showing resistance to PSN. This suggests the underlying roles of human genetic variations toward disease susceptibility. A study on large cohort of US women has revealed that HIV-PSN has been less prevalent in women than previously reported. They have also found that African-Americans are highly predisposed to HIV-PSN than other racial groups, and particularly patients with comorbid hepatitis C virus infection, older age and diabetes mellitus type 2, and metabolic syndrome in terms of higher triglyceride levels are at highest risk [33, 34]. In the context of genetic variations, mitochondrial DNA (mtDNA) undergoes spontaneous mutagenesis generating different mitochondrial haplogroups, leading to variable disease susceptibility across the races. It has been observed from the very beginning of the implementation of HAART treatment that the HAART receiving HIV-infected patients develop similar neuromuscular diseases like people with inherited mitochondrial dysfunction. Following that observation, it was discovered that the most common clinically effective nucleoside-analogue reverse transcriptase inhibitors (NRTI) used to block the activity of mitochondria specific DNA polymerase γ, required for mtDNA replication, led to mitochondrial dysfunction [35–37]. In recent years, the association between HIV-sensory neuropathy and mtDNA variation has further been established by the CHARTER Group Cohort Study, where they identified the two most significant single-nucleotide polymorphisms, namely A12810G and T489C corresponding to the African haplogroup L1c and European haplogroup J, respectively, with decreased occurrences of HIV-linked sensory neuropathy compared to all other haplogroups [38]. However, these incidences of PSN have been inevitable because of the widespread clinical applications of NRTIs. Recent findings have established a link between genetic variation in iron-metabolism and PSN in HIV +ve patients on cART treatment [39]. They have found that polymorphisms in transferrin, transferrin receptor, bone morphogenetic protein 6, aconitase 1, solute carrier family 11 member 2, and frataxin genes confer reduced risk; other variants of transferrin, aconitase 1, bone morphogenetic protein 6, beta-2-microglobulin, and ceruloplasmin genes confer increased risk to peripheral neuropathy.
HAND IN THE ERA OF c ART
The introduction of cART in the 1990 s dramatically improved the clinical outcome of HIV-infected patients in terms of morbidity and mortality. cART significantly reduced the incidence of CNS-associated opportunistic infections in patients. Also, the prevalence of HAD, the most severe form of HAND, declined from 20% to 5% [40, 41]. However, progress toward eliminating HAND has been discouraging, as 20–50% of patients continue to suffer from a milder form of HAND, namely ANI and MND [40, 42]. In a cross-sectional study conducted in Switzerland, HAND was present in 84% of patients with evidence of cognitive decline and in 64% of patients without cognitive decline. In the first group of patients, 24% had ANI, 52% had MND, and 8% had HAD, whereas in the second group, 60% had ANI, 4% had MND, and 0% had HAD [43]. In another cross-sectional study performed by the CHARTER Group including 1,555 HIV patients, HAND was prevalent in 52% of patients, of which 33% had ANI, 12% had MND, and 2% had HAD [3]. These milder forms of HAND not only impact quality of life[44, 45] but also affect cART adherence [46]. There have been three characteristic changes in HAND in the cART era. First, as previously mentioned, HAND has become less severe, with ANI and MND becoming the predominant subtypes among patients[42, 48]. Second, cortical deficits can develop in HAND patients, with executive function and learning ability being the most frequently affected domains [47–49]. The development of vascular cognitive impairment also poses a substantial concern, but the evidence for this is preliminary and largely obtained from neuroimaging studies [50–52]. Third, HAND can be associated with extrapyramidal features that overlap with PD [53, 54]. Emerging evidence shows that HAND shares some common features with other neurodegenerative diseases in terms of pathogenesis, including dysfunction in autophagy [55, 56] and the ubiquitin-proteasome system [57, 58].
ART-induced neurotoxicity
Various mechanisms have been implicated in the pathogenesis of HAND, including ongoing viral replication [59, 60], the presence of activated CD8-positive T cells in the brain [61–64], and indirect neurotoxicity caused by viral proteins such as Gp120 [65–71] and Tat [72–77]. As several ART drugs, including NRTIs and protease inhibitors (PIs), can attain therapeutically effective concentrations in the CNS, a decline in HAND should be expected. However, increasing therapeutically effective drug concentrations in the CNS by cART does not always result in improved cognition and, in fact, can increase the risk of HAND by more than 50% [78]. Emerging evidence now demonstrates direct neurotoxic effects of ART drugs. Nucleoside reverse transcriptase inhibitors have been reported to reduce neuronal axon length [79, 80] and mitochondrial DNA content [81, 82]. Recently, efavirenz which was found to promote amyloid-β (Aβ) production in vitro and in vivo [83], abrogate neural stem cell proliferation [84], and alter mitochondrial dynamics (i.e., increased mitochondrial depolarization, decreased mitochondrial DNA, and altered mitochondrial respiratory function) [81, 86]. In addition, raltegravir was shown to enhance IL-8 production, providing further evidence for cART-mediated neurotoxicity [87]. ART drugs can also compromise the structural integrity of the corpus callosum [88] and worsen neurocognitive function in clinical [89] and experimental [90] models. Moreover, PIs (i.e., saquinavir, indinavir, and ritonavir), efavirenz, and zidovudine disrupt the blood-brain barrier (BBB) by decreasing the expression of tight junction proteins (TJPs), causing oxidative and endoplasmic reticulum (ER) stress as well as mitochondrial dysfunction [91–93]. The regulation of TJPs is controlled by signaling factors originating from endothelial cells and is also influenced by astrocytes and microglia. Dysfunction in the BBB and increased BBB permeability are hallmarks of several acute and chronic CNS pathologies, including HIV-infection [94]. ART drugs significantly diminish HIV burden but fail to restore damage to BBB integrity caused by HIV proteins. Therefore, it can be safely assumed that in the presence of ongoing HIV-infection, the neurotoxic effects of ART drugs on the BBB could be further worsened, as both HIV proteins and ART drugs induce oxidative stress, disrupt normal mitochondrial function, alter gene expression, and activate various cell signaling cascades [95]. The studies described above highlight the possible role of cART-induced neurotoxicity in HAND. Further studies examining the effects of ART drugs, with or without HIV or the role of viral proteins in combination with ART drugs, may provide novel insights into the underlying mechanisms. The outcomes of these studies will be of vital importance to developing better HIV treatmentapproaches.
ROLE OF HIV-1 PROTEINS IN NEUROTOXICITY
HIV crosses the BBB soon after infection, leading to CNS infection and eventually causing HAND. Neurotoxicity is mediated by the direct effects of the virus and/or viral particles on neurons and glial cells through the release of a myriad of toxic factors. Viral particles shed from the virus include Gp120, HIV-1 Tat, Nef, and Vpr. Fig. 1 provides an illustration of multifaceted nature of HIV mediated neurotoxicity.
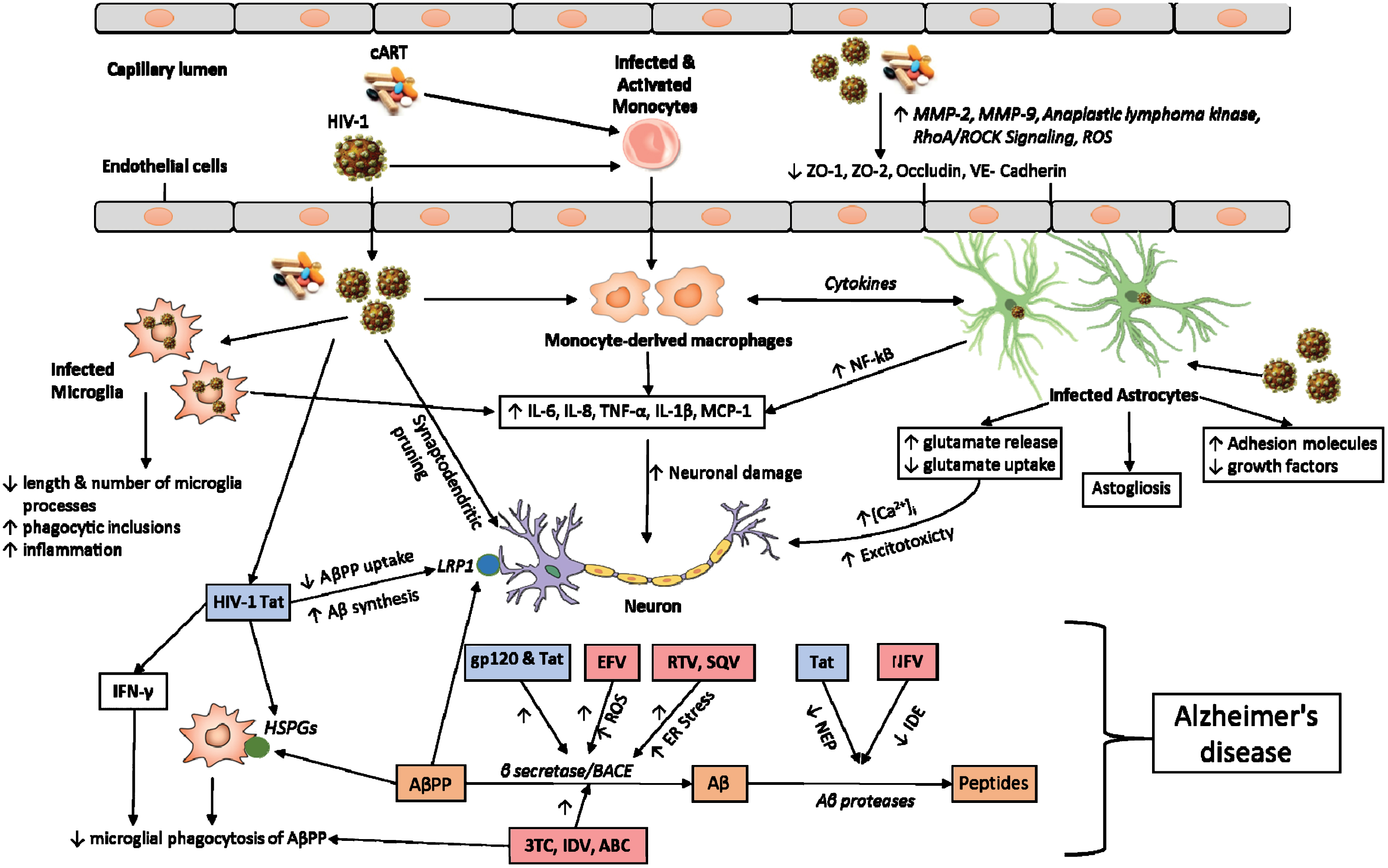
Schematic overview of HIV-1 effects in CNS. The figure illustrates the mechanisms by which HIV-1, HIV-1 proteins, and cART mediate neurotoxicity; act on endothelial cells to decrease the expression of tight junction proteins; act on microglia to promote the release of various inflammatory factors and increasing phagocytic inclusions; act on monocyte derived macrophages and astrocytes to promote the release of various cytokines that act on neurons directly. HIV-1 infected astrocytes also promote neurotoxicity by decreasing the production of growth factors and altering the glutamate homeostasis. HIV-1 viral proteins (HIV-1 Tat and gp120) and cART promote the formation of Aβ by decreasing AβPP uptake or increasing the activity of BACE or decreasing the activity of NEP and IDE. HIV-1 viral proteins are indicated in blue boxes and various antiretroviral drugs are indicated in light pink boxes. Also included are cART, combination antiretroviral therapy; MMP, matrix metalloproteinase; ROS, reactive oxygen species; ZO, zona occludens; RhoA, Ras homolog gene family, member A; ROCK, Rho-associated protein kinase; IL, interleukin; TNF, tumor necrosis factor; MCP-1, monocyte chemotactic protein 1; AβPP, amyloid-β protein precursor; LRP1, low density lipoprotein receptor-related protein 1; Aβ, amyloid-β; IFN, interferon; HSPGs, heparan sulfate proteoglycans; BACE, beta-site amyloid precursor protein cleaving enzyme; ER, endoplasmic reticulum; EFV, Efavirenz; RTV, Ritonavir; SQV, Saquinavir; 3TC, Lamivudine; IDV, Indinavir; ABC, Abacavir; NFV, Nelfinavir; NEP, Neprilysin; IDE, Insulin degrading enzyme.
Role of HIV-1 Tat
HIV-Tat is a functional protein that is produced early during virus replication. Depending on the viral subtype, it consists of 86–101 amino acids. The cysteine-rich region of Tat is responsible for the transcriptional activity of the virus (reviewed in [96]). Tat is secreted from HIV-infected cells and acts on surrounding cells, affecting their function and causing toxicity. Various in vitro and in vivo studies show that Tat administration increases the apoptosis of neurons [97, 98]. Various mechanisms have been implicated in Tat-mediated neurotoxicity. Tat toxicity affects the integrity of the BBB by inducing oxidative stress and lowering the expression of various TJPs and adhesion proteins, including occludin, ZO-1, and VE-cadherin, in brain endothelial cells [99–101]. Disruption of BBB integrity occurs through activation of matrix metalloproteinase (MMP)-9 and the RhoA/ROCK signaling pathway, resulting in the buildup of various immune cells in the CNS. Tat acts on different cell types in the CNS to promote the release of various soluble mediators. Tat increases the expression of various pro-inflammatory cytokines such as IL-6,IL-8, TNF-α, IL-1β, and MCP-1 from astrocytes, microglia, and monocytes [102–106]. Tat-mediated induction of cytokines is mediated through activation of the NF-κB pathway and different MAPKs as well as elevated levels of intracellular calcium. Mice receiving stereotactic Tat injections exhibit shorter and fewer microglial processes than vehicle-treated mice. Furthermore, the number of dendritic spines is reduced in Tat-exposed mice [107]. Tat also reduces the number of synapses between hippocampal neurons, indicated by a lower number of post-synaptic density-95 clusters [108]. Administration of Tat increases the number of microglial phagocytic inclusions that contains axon terminals, dendritic spines, and post-synaptic densities, indicating increased proteolyticdegradation [109].
Tat interacts with synaptosomes to induce oxidative stress, resulting in increased protein and lipid oxidation and decreased mitochondrial membrane potential [110]. Tat injection increases levels of malondialdehyde in the caudate-putamen of rat brains, indicating elevated oxidative stress [98]. Tat decreases levels of antioxidant enzymes, including glutathione peroxidase and superoxide dismutase, and administration of these antioxidant enzymes protects neurons against Tat-induced apoptosis[111, 112].
In an initial study by Cheng et al., Tat induced depolarization of human fetal neurons, indicating its direct excitatory effect [113]. Tat promotes toxicity by affecting the release of various neurotransmitters. Tat facilitates the exocytosis of excitatory neurotransmitters such as glutamate and NMDA and decreases the release of GABA, an inhibitory neurotransmitter, from the cortex and hippocampal neurons [114, 115]. Furthermore, Tat-mediated glutamate toxicity is exacerbated by phosphorylation of NMDA receptors. Tat also promotes the release of acetylcholine through the activation of metabotropic glutamate receptors and mobilization of intracellular calcium stores [116]. Tat potentiates the toxic effect of glutamate by impairing its reuptake by astrocytes [117].
Tat increases levels of intracellular calcium by interacting with NMDA receptors and promoting the release of inositol triphosphate-sensitive intracellular calcium stores [118–120]. Increased intracellular calcium overload promotes toxicity by inducing oxidative stress and producing pro-inflammatory cytokines such as TNF-α [121, 122].
Role of Gp120
Gp120 is expressed on the surface of HIV and facilitates its interaction with various surface receptors [123]. Free protein, shed from the virus, promotes neuronal toxicity by directly interacting with neurons and increasing the release of toxic mediators from surrounding immune cells. Gp120 induces neuronal apoptosis by increasing the expression of Fas ligand [124]. By interacting with neurons, Gp120 disrupts calcium homeostasis and causes significant intracellular calcium accumulation [125]. Furthermore, Gp120 acts as a partial agonist to CXC4 receptors expressed on Cajal-Retzius neurons and increases their excitability via calcium-dependent chloride channels [126]. Activation of calcium channels by Gp120 also triggers the expression of various pro-inflammatory cytokines, including TNF-α and IL-6, through activation of the wnt5a/CaMKII pathway [127]. Up-regulation of IL-1β by Gp120 from glial cells induces the expression of ferritin heavy chain and mediates the phosphorylation of NMDA receptors in neurons, resulting in the loss of spines and subsequent neuronal death [128, 129].Furthermore, Gp120 directly interacts with neurons and reduces neurite outgrowth [130].
Gp120-mediated production of cytokines, including IL-1β and TNF-α, from microglia and astrocytes, promotes glutamate-associated toxicity by increasing its release. Furthermore, glutamate release promotes calcium influx in astrocytes, and thereby decreases the expression of Na+-dependent glutamate/aspartate transporter, which plays an important role in glutamate uptake [131].
Gp120 decreases permeability of the BBB by various mechanisms [132, 133]. It decreases the expression of various TJPs, including occludin, ZO-1, and ZO-2, and increases the migration of monocytes into the CNS [134, 135]. Gp120 induces the production of reactive oxygen species (ROS) through NADPH to upregulate the expression of MMP2 and MMP9 in endothelial cells [136, 137]. Gp120 also interacts with neurons to decrease mitochondrial movement, resulting in mitochondrial dysfunction [125, 138]. Furthermore, Gp120 increases the induction of ROS by neurons and glial cells by various mechanisms. It decreases the expression of various antioxidant enzymes such as SOD2, glutathione peroxidase, and glutathione synthase in neurons and glial cells [139, 140]. It increases the production of ROS in microglia by increasing the expression of voltage-gated potassium channels, which is reversed by pretreatment with curcumin [141, 142]. It also increases ROS production in astrocytes by increasing the expression of CYP2E1 and activating NADPH oxidase enzymes [143].
Role of Nef
HIV Nef is an accessory viral protein found in post-mortem brains of HIV-infected patients [144, 145]. Nef is also expressed in astrocytes from post-mortem brains of SIV-infected macaques [146]. Mutations in the Nef open reading frame affect disease progression [147, 148]. The presence of Nef protein is favorable for HIV-infection of astrocytes [149]. Nef alone plays a role in the development of HAND by various mechanisms. Recombinant Nef decreases the viability of neurons and astrocytes and induces astrogliosis [150, 151]. Nef induces the production of various pro-inflammatory cytokines such as IL-6, IL-8, CCL5, TNF-α, and IFN-γ from astrocytes and microglia. Nef-mediated up-regulation of cytokines is promoted through the activation of MAPKs and calmodulin signaling pathways [152–155]. Nef serves as a chemotactic agent by promoting the infiltration of leucocytes and monocytes into the CNS [152, 156]. Nef also induces the production of quinolinic acid from macrophages, a neurotoxin that is detected in the brain and cerebrospinal fluid (CSF) of HIV-infected individuals [157, 158]. A study by Van Marle and colleagues showed that overexpression of brain-derived Nef in neurons promotes astrocytic cell death. Furthermore, supernatants from Nef-expressing astrocytic cultures induce the death of neurons mainly through the actions of IP-10 [159]. Nef expression alone can induce cognitive deficits in rat models, as rats that are injected with Nef-expressing astrocytes show impaired object and spatial recognition memory [160, 161].
Nef affects the transcriptional level of anaplastic lymphoma kinase, which activates MMPs that disrupt the integrity of the BBB [162]. Treatment of astrocytes with Nef increases the production of complement factor C3 and nitric oxide and induces lipid peroxidation [163]. Expression of Nef in astrocytes triggers autophagy and inhibits the fusion of autophagosomes to lysosomes [164]. Furthermore, by colocalizing with an important autophagic protein, Beclin 1, Nef affects autophagic maturation and protects HIV from degradation [165]. Nef is released from primary human fetal astrocytes in extracellular vesicles, and this release is increased by regulators that influence the autophagic pathway. Neurons treated with purified Nef-enriched extracellular vesicles exhibit symptoms of toxicity, as shown by a decrease in levels of the antioxidant enzyme glutathione and degeneration of neurites and axons [166].
Role of Vpr
Vpr is one of six auxiliary proteins produced by HIV and is detected in the serum and CSF of HIV-infected patients [167]. Vpr plays an important role in the pathogenesis of SIV in macaques (reviewed in [168]). It induces viral reactivation by promoting the degradation of histone deacetylase 1 and 3 and inducing the production of IL-6 [169, 170]. It promotes activation of HIV promoter by inducing the expression of hypoxia-inducible factor 1-α through increased production of ROS [171, 172]. Loss of Vpr decreases HIV antigen production by more than 1,000-fold in macrophages [173]. Vpr and its fragments permeabilize and result in the apoptosis of various CD4-positive and non-CD4-positive cell types [174, 175]. Vpr protein treatment inducesapoptosis of human neurons by activating caspase-8 [176]. Furthermore, intracellular Vpr expression induces apoptosis in human neurons, indicating that both extracellular and intracellular Vpr are toxic to cells [177]. The region of Vpr between 70–96 amino acids is essential for inducing apoptosis of neurons [178]. Vpr elevates intracellular calcium concentration in neurons [179, 180]. Infection of monocyte-derived macrophages with Vpr-deleted HIV decreases the production of pro-inflammatory cytokines, such as IL-1β, IL-8, and TNF-α, as compared with infection with wild-type HIV. Vpr alone induces the production of pro-inflammatory cytokines, such as IL-6, IL-8, MCP-1, and CCL5, from astrocytes and macrophages [169, 182]. Induction of cytokines involves the activation of p38 MAPK, JNK MAPK, TLR4/MyD88, and NF-κB signaling pathways[169, 183]. Infusion of Vpr-transfected astrocytes into the hippocampus impairs spatial and recognition memory in rats and induces morphological changes in neurons with reduced expression of synaptophysin [184]. Furthermore, supernatant from Vpr-deleted HIV results in decreased neurotoxicity compared with wild-type HIV [183]. Addition of extracellular Vpr to human astrocytes affects the glycolysis pathway and decreases production of ATP, resulting in an increased amount of oxidative stress. An elevation of oxidative stress is induced partly by decreased levels of the antioxidant enzyme glutathione [181]. Vpr affects the expression of miRNAs, particularly miR-34a, which alters the expression of various genes associated with neuronal dysfunction [179].
Role of Aβ
With the advent of cART, the lifespan of people infected with HIV-1 has significantly increased. According to Joint United Nations Programme on HIV and AIDS, there are 4.2 million people over the age of 50 who are living with HIV (http://www.unaids.org/sites/default/files/media_asset/12_Peopleaged50yearsandolder.pdf). According to the Centers for Disease Control and Prevention, in the United States alone, the number of people over the age of 50 who were living with HIV increased by 42% by the end of 2013, whereas the number of new cases of HIV in 2014 decreased by 17% (https://www.cdc.gov/hiv/group/age/olderamericans/). With the widespread reach of cART, the number of older patients with HIV is projected to increase in the future, which increases their risk of developing AD-likepathology. Post-mortem analysis of brain sections from HIV-infected people over 50 years of age showed an increase in the deposition of Aβ plaques [185–187]. Furthermore, elevated hippocampal deposition of hyperphosphorylated Tau has been noted in HIV-infected patients on cART [188]. All accumulating evidence suggests a role of Aβ in the development of HAND in the HIV-infected population.
CONTRIBUTION OF HIV VIRAL PROTEINS AND CART TO THE DEVELOPMENT OF AD-LIKE PATHOLOGY
Increasing evidence suggests a role of HIV viral proteins in Aβ production and the development of HIV-associated neurotoxicity.
Role of HIV-1 Tat
Silver staining analysis of the frontal cortex in post-mortem brains of HIV-infected patients with HAD shows an increased presence of neuritic plaques, which is a common feature in AD patients [187]. Furthermore, immunostaining of SIV-infected monkeys shows elevated deposition of Aβ precursor protein (AβPP) in neurons in close proximity to cells stained for Tat [189]. Tat increases Aβ secretion in neuron cultures [187]. Injection of lentiviral Tat construct into the hippocampus of mice results in elevated levels of Aβ production and increases the size and number of Aβ plaques [190]. Moreover, Tat further increases the burden of Aβ in AβPP/PS1 mice crossed with HIV-1 Tat transgenic mice [191]. Several mechanisms are implicated in the Tat-mediated increased burden of Aβ. Tat binds to lipoprotein receptor protein 1 (LRP1) on the surface of neurons, which inhibits the uptake of AβPP (a precursor molecule for the generation of Aβ) and results in an elevated burden of Aβ. LRP1 plays a critical role in the clearance of Aβ, and binding of Tat to LRP1 decreases its clearance [189, 192]. Tat also binds to heparan sulfate proteoglycan receptors on the microglia surface and decreases the transfer of Aβ to LRP1, thereby inhibiting phagocytosis [193]. Tat enhances Aβ burden by promoting the accumulation of AβPP into lipid rafts [190]. Tat and peptides derived from Tat decrease the activity of neprisylin, a major Aβ-degrading enzyme [187, 194].
Tat also promotes the production of pro-inflammatory cytokines, such as IFN-γ, which act together to decrease the microglial uptake of Aβ. By interacting with endolysosomes, Tat increases their pH and the activity of beta secretase (BACE1), thereby increasing the accumulation of Aβ [195]. Tat further increases the activity of BACE1 by promoting the release of glutamate [196]. Tat-mediated toxicity is potentiated by the production of Aβ [197, 198]. Caffeine has been shown to be a therapeutic intervention that overcomes Tat-mediated endosomaldysfunction [199].
Role of Gp120
The addition of Gp120 to hippocampal cell cultures and brain microvascular endothelial cell cultures increases the production of Aβ in cell culture supernatants [200, 201]. Zhang et al. (2011) showed that Gp120 treatment of rat brain slices increases the accumulation of AβPP in the corpuscallosum. Furthermore, they showed that the Gp120-mediated increase in AβPP accumulation was decreased by the addition of CXCR4 antagonist but not NMDA receptor antagonist, implying an important role of CXCR4 [202]. Generation of Gp120/AβPP/PS1 triple-knockout mice revealed an increase in the number and size of Aβ deposits in the hippocampus and cortex. These mice also showed elevated levels of lipofuscin, a characteristic protein associated with decreased lysosomal function, and enlarged Aβ-accumulated lysosomes. Furthermore, experiments with these mice also showed that Gp120 alters the trafficking of Aβ distribution in neurons and increases the activities of BACE1 and γ-secretase, which affects Aβ clearance [203]. Gp120-mediated deposition and transport of Aβ are attenuated by an α7 nicotinic acetylcholine receptor antagonist [201].
A recent study by Khan et al. showed that exosomes containing Nef mRNA, isolated from patients with HAD, exhibit increased production and secretion of Aβ when added to a SH-SY5Y neuroblastoma cell line [204].
Role of ART drugs in AD progression
Different ART drugs have distinct capabilities of BBB penetration as demonstrated by CNS penetration-effectiveness scores. Patients on cART with higher CNS penetration-effectiveness scores are expected to benefit from a higher bioavailability of these drugs in the brain, resulting in better control of HIV replication. However, several reports show that there is lack of substantial neurocognitiveimprovement with cART. Evidence also indicates a shifting pattern of neurocognitive impairment in HIV patients, from deficits in motor ability, speed of information processing, and verbal speed in the pre-cART era to deficits in memory and executive function in the post-cART era [47]. There are several potential explanations for this clinically meaningful finding. One is ART-induced neurotoxicity, which is now known to result in part by dysregulated AβPP processing and subsequent deposition of Aβ plaques in the brain. Based on neuropathological data [11, 205], genetic screening [206, 207], and CSF markers [208], researchers have raised the possibility that AD may be becoming more common in HIV patients on cART. Although the exact mechanisms through which cART contributes to Aβ deposition in the brain are unknown, neurotoxic effects of ART drugs such as mitochondrial dysfunction [81, 209], increased oxidative and ER stress [82, 211], and neuronal damage and synaptic loss [212, 213] are now linked to altered AβPP processing and Aβ deposition in the brains of HIV patients.
In the first study of its kind, Giunta and colleagues [214] examined the effect of various ART drugs on neuronal Aβ production and clearance by microglial phagocytosis. ART drugs, especially in combination, were observed to increase the synthesis of Aβ1 - 40,42 from SweAβPP N2a neurons (i.e., murine N2a cells transfected with the human ‘Swedish’ mutant form of AβPP) and decrease the clearance of Aβ1 - 42 peptides by preventing their phagocytosis by N9 microglial cells. The combination of lamivudine, indinavir, and abacavir was found to have the most significant amyloidogenic effects, with indinavir and abacavir also having additive damaging effects on Aβ microglial clearance. In another study, Lan et al. [215] examined the effect of several PIs on the clearance of Aβ42 in macrophages. They observed that ritonavir, saquinavir, and atazanavir slightly suppressed Aβ42 clearance, whereas lopinavir, nelfinavir, and ritonavir increased the number of undegraded Aβ peptides. Additionally, Lan et al. found that all aforementioned PIs except atazanavir reduced Aβ40 synthesis in neurons through the inhibition of BACE1 and γ-secretase. However, these PIs slightly suppressed purified BACE1 enzyme activity in in vitro studies, indicating that ART drugs may have an indirect action on BACE1 activity in neurons. The authors also studied the effect of nelfinavir and lopinavir/ritonavir on Aβ production in immunodeficient AβPP SCID mice (with a double-mutant form of AβPP695 (KM670/671NL + V717F) in homozygosity for the SCID allele of Prkdc). Although nelfinavir achieved a significant concentration in the mouse brain, no changes in Aβ accumulation were observed. In general, PIs do not readily cross the BBB because of high levels of protein binding. Therefore, the interpretation of these effects of PIs on amyloidosis in the brain should consider their low infiltration into the CNS.
In another study, Brown et al. [83] found that efavirenz, in combination with lamivudine and zidovudine, induces mitochondrial dysfunction as evidenced by reduced cellular ATP stores, diminished mitochondrial membrane potential, and enhanced release of ROS in SweAβPP N2a neurons. Also, efavirenz increases Aβ production via BACE1 activation in vitro and in vivo. Combination treatment also inhibits microglial phagocytosis of Aβ1-42. The authors propose that an efavirenz-induced high ROS microenvironment in the CNS of HIV patientspromotes BACE1-mediated AβPP processing and also inhibits phagocytic clearance, leading to the production of Aβ. In a very recent study [211], ritonavir and saquinavir were found to induce the expression of classical ER stress markers BiP, p-eIF2α, and XBP-1 in neuroglial cultures, leading to activation of the unfolded protein response. Induction of ER stress is associated with PERK-dependent BACE1 activation. PIs also increase Aβ in multiple cell types, which is dose-dependently blocked by BACE1 inhibitor. These findings were also validated in macaques and rodents that received chronic PI treatment.
Disruption of the BBB by ongoing HIV infection and ART drugs can also decrease the brain-to-blood clearance of Aβ, leading to its accumulation [216]. A balance between two transporters, (LRP1, which transports Aβ from brain to blood) and the receptor for advanced glycation end products (RAGE, which transports Aβ into the brain), have been found to regulate levels of Aβ in the brain [217]. LRP shares a 63% homology with the catalytic region of HIV-1 protease [218]. In one study, PIs were reported to decrease LRP levels in HepG2 cells [219]. LRP mediates the endocytosis and degradation of AβPP. Therefore, decreasing LRP levels by PIs could increase Aβ accumulation in the brain. Further conclusive studies are needed to delineate the effects of ART drugs on LRP1 and RAGE and their role in Aβ accumulation in thebrain.
Accumulating evidence shows that Aβ proteases, such as neprilysin (NEP), insulin-degrading enzyme (IDE), and endothelin-converting enzymes (ECE) 1 and 2, play an important role in regulating the level of Aβ peptides in the brain. IDE degrades Aβ in neuronal and microglial cell cultures [220–223]. Similarly, NEP and ECE degrade Aβ in vivo [224, 225]. In a study by Hamel et al. [226], nelfinavir was found to inhibit IDE in vitro. Hypofunction of IDE by PIs might inhibit Aβ degradation, promoting its accumulation in the brain. However, more studies are needed to gather conclusive evidence for the role of ART-mediated proteolytic activity in the development of amyloidogenesis.
OTHER NEURODEGENERATIVE DISEASES LINKED TO HIV INFECTION
Parkinson’s disease
PD is primarily characterized by the loss of pigmented dopaminergic neurons from the midbrain substantia nigra pars compacta (SNpc) region due to the formation of α-synuclein inclusion bodies (also called Lewy bodies). However, there are occurrences of PD without Lewy bodies called secondary Parkinsonism that are thought to involve viral infections. A large subset of HIV-infected patients present akinetic Parkinsonian syndromes at the early stage of disease onset, but PD-like syndromes are mostly reversed by HAART depending on the age and genetic predisposition of the patient [227, 228]. Patients who survive beyond 50 years of age most often exhibit accelerated degradation of neural networks compared with age-matched controls, mostly in the basal ganglia and hippocampal regions, due to the synergistic effects of immune-senescence and sustained viral load, despite being under control [229]. Multiple studies, in patients and animal models, show that dopaminergic neurons are the most vulnerable to HIV-infection. Interestingly, increased levels of α-synuclein are found in the substantia nigra of HIV-infected patients compared with that of normal age-matched controls [229], which is one major cause of the depletion of dopaminergic neurons in α-synuclein gene triplication-associated PD. In a separate study, Riederer and colleagues showed in a primate model that HIV infection can decrease dopamine levels by at least 44% within only 2 months of infection, which is consistent with observations of HIV-infected patients [230]. This reduction in the dopamine level could be attributed to a proportional decrease in the level of dopamine transporter, which is further consistent with the lower neuropsychological performance of HIV-infected patients [231, 232]. In thiscontext, it is important to mention that not only HIV, but also several other viruses, cause neurological complications [233]. For example, infection of rats with Japanese encephalitis B virus, the most common encephalitis-causing agent in Asia, induces marked gliosis in the SNpc similar to that observed in PD, and behavioral studies show that bradykinesia is recovered by treatment with L-DOPA and MAO inhibitor, suggesting that the virus induces typical symptoms of PD in infected animals [234]. Furthermore, other evidence that viruses can cause PD-linked pathology comes from a risk assessment of people living in “Parkinsonian clusters” who share common lifestyles and environments and who exhibit at least two times more disease susceptibility than people living outside the cluster[235, 236].
Amyotrophic Lateral Sclerosis (ALS)
Although classical ALS-like symptoms largely differ from HIV-associated ALS-related neurological symptoms, the viral etiology of ALS could be a significant concern due to the selective vulnerability of motor neurons to certain viruses, including HIV. The expression of human endogenous retroviral (HERV)-linked sequences in motor neurons in the CNS dramatically increases in ALS patients [237], suggesting an undetermined link between viral infection and motor neurodegenerative disease. Transactive response DNA binding protein of 43 Kd (TDP-43), which was earlier discovered as a regulator of HIV infection in humans, also contributes to a major ALS disease subtype called ALS-TDP-43. Interestingly, recent findings by Nath and colleagues show that expression of HERV-K or its envelope protein is controlled by TDP-43, leading to neurodegeneration and muscular atrophy [238]. After the first discovery of human retrovirus T-lymphotrophic virus-1 in postmortem brain tissue of two ALS patients in Guam in the 1970 s [239], exhaustive survey and medical evidence collected to date shows that only a small subset of retrovirus-infected patients develop symptoms like those in ALS. However, at the same time, it is critical to note that neuropathological symptoms in patients infected with different types of retroviruses exhibit distinct patterns of disease phenotypes similar to classical ALS phenotypes. Particularly, HIV-infected patients with a median age of ∼40 years exhibit neuropathological symptoms very similar to those of classical ALS patients with a median age of ∼55 years for sporadic disease onset, but their recovery is quite common following ART, unlike the invariable deterioration in sporadic ALS. HIV-associated ALS can also develop at any stage of HIV disease progression, with the majority of patients affected by both upper and lower motor neurodegenerative disorder. However, in most cases, following ART, even a considerably improved immunosuppression condition and almost undetectable viral load may cause mild neuropathological symptoms that are mostly ignored at the beginning. Because the expression of viral infectious proteins depends on the expression of reverse transcriptases in the human body, the level of reverse transcriptases in body fluids primarily serves as a confirmatory indicator of viral load in the body. Based on earlier studies, patients with HIV or other retrovirus-related neurological complications show serum reverse transcriptase activity levels 53% higher than those of non-HIV control patients.
RELATIONSHIP BETWEEN BRAIN STRUCTURE ALTERATIONS AND AGING IN HIV INFECTION
Although brain aging is associated with mild volumetric alterations, comparative analysis of total brain volume between aging HIV-positive and HIV-negative individuals may not be useful for advanced diagnosis, as cognitive or neuropsychological deficits are mainly related to microstructural alterations in specific brain regions. Instead, because of fluid-filled spaces in the brain, imaging the diffusion or movement of molecular water could increase our understanding of brain degeneration. Unlike gray matter (GM), diffusion of water molecules is more directional (i.e., anisotropic) in white matter (WM) due to its compact microstructure consisting of axonal cell membranes, myelin sheaths, and neurofilaments. MND or dementia is most frequently associated with WM abnormalities in elderly people. Despite successful cART/HAART treatment in HIV-positive patients, they continue to have persistent untraceable viral load in the CNS, resulting in WM abnormalities and neuro-inflammation, and thus leading to mild to severe cognitive impairment and brain atrophy at older ages [240, 241]. Multiple studies show a significant interaction between age and HIV infection, as HIV-infected patients frequently exhibit a greater extent of structural deformation in cognitive regions of the brain with aging, including abnormalities in whole-brain WM hyperintensities and fronto-subcortical WM integrity, compared with age-matched HIV-negative individuals [242–245]. Diffusion tensor imaging (DTI) revealed the marked degeneration of WM microstructures, expressed as large differences between fractional anisotropy and mean diffusivity, that is highest in the corpus callosum and projection fibers of the corona radiata in HIV-positive elderly patients compared with age-matched controls, which is consistent with earlier studies suggesting that WM-associated microstructural alterations lead to severe dementia and motor dysfunction [246, 247]. Interestingly, recent DTI studies reveal that loss of WM integrity correlates with duration of HIV infection. Chronic HIV-infected patients present both the loss of WM integrity as well as disruption of the BBB. On the other hand, HIV-negative and early HIV-infected (≤1 year of viral exposure) patients exhibit only disruption of the BBB and no significant alterations in WM integrity [248]. Moreover, aging HIV-infected patients exhibit greater WM hyper-intensities and lower fractional anisotropy in the anterior corona radiata due to hepatitis C virus coinfection, the more likely development of AIDS, and higher CD4-positive cell counts as a marker of hyper-activation of inflammatory responses [245, 249].
However, the normalization of DTI diffusivity parameters within specific regions of the corpus callosum and centrum semiovale is possible in HIV-positive patients who receive long-term cART or merely initiate treatment [250]. This same study suggests that the initial modulation in DTI parameters could be due to alterations in inflammatory responses rather than cognitive performance, as neuropsychological testing reveals similar scoring before and after receiving cART regimens. However, to better understand the underlying pathogenesis, DTI scanning of the corpus callosum should be performed. Interestingly, the Hawaii Aging with HIV Cohort Study revealed that WM hyper-intensities are associated with reduced frontal GM volume in aging HIV-infected patients, suggesting that the frontal lobes may be more susceptible to small vessel ischemic vascular disease [251]. With CD4-positive cell count acting as an index for immune suppression or activation in HIV-infected patients, a CHARTER group study showed that extent of CD4-positive T-cell recovery is associated with increased abnormal WM and subcortical GM volumes, suggesting the involvement of neuro-inflammation in CNS pathogenesis [252]. As supported by other studies, an increase in subcortical GM volume could be attributed to a proportional increase in tissue water content and/or infiltration of inflammatory cells [253, 254]. These alterations in brain structure might be the outcome of diverse inflammatory responses from recovering immune systems across the brain. On rare occasions, severe immune reconstitution inflammatory syndrome can influence WM abnormalities in the CNS in settings of secondary opportunistic infections [255, 256]. Moreover, even with successful cART treatment of HIV-infected patients, neuropathological evidence of inflammatory responses has been observed in the basal ganglia [257].
Substance abuse is a serious threat to brain functions related to cognitive performance, which significantly reduces quality of life [258–263]. Substance abusers, particularly psychostimulant users, are most susceptible to infectious diseases like HIV acquired by intravenous drug abuse and unsafe sexual practices [264, 265]. Recent findings show that frontal WM and the corpus callosum are the most vulnerable brain regions to psychostimulant abuse as measured by DTI diffusivity parameters, leading to drastic cognitive impairment [266].
Another common type of substance abuse is alcoholism. A growing body of evidence indicates that long-term alcohol consumption causes significant metabolic dysfunction and cognitive impairment in middle-aged to older people. Chemical shift imaging analysis of brain metabolites reveals that levels of NAA, a marker of living neurons, decrease in alcoholics or short-term abstinent alcoholics, whereas NAA reaches normal levels in long-term abstinent alcoholics [267]. Notably, individuals with comorbid alcoholism and HIV infection show a drastic decrease in NAA levels in the parietal-occipital cortex, whereas neither HIV infection nor alcoholism alone exerts the same magnitude of effect, suggesting that alcoholics with comorbid HIV infection have a higher risk of neuronal compromise [268, 269].
DNA DAMAGE AND HIV-1 INFECTION: A CAUSAL CONNECTION?
HIV-infected patients develop multiple types of cancerous neoplasms depending on co-infection by secondary viral agents in the context of AIDS. However, non-AIDS HIV-patients also have a higher incidence of cancers, indicating that HIV has the potential to induce cancerous lesions before the development of AIDS. Multiple studies provide mechanistic insights into how viral agents interfere with host cell cycle signaling pathways and induce genomic instabilities that facilitate incorporation of viral gene elements into the host genome primarily through Vpr and integrase. Vpr is capable of inducing DNA strand breaks via both direct and indirect mechanisms. Vpr itself can bind to chromosomal DNA, inducing double-strand breaks (DSBs) by recruiting yet unknown nuclear factors with endonuclease activity, possibly through its carboxy-terminal domain [270]. Previously, it was assumed that Vpr interacts with the SLX4 protein complex to recruit structure-specific endonucleases for inducing the DNA damage response (DDR) in infected cells. However, a recent study shows that HIV-1, HIV-2, and other Vpr orthologs have inherent abilities to activate the DDR and cell cycle arrest without interacting with the SLX4 complex [271]. On the other hand, Vpr can interfere with the cell cycle by arresting infected cells at G2-phase by activating ataxia-telangiectasia-mutated and Rad3-related kinase (ATR)-Chk1-Wee1 DNA damage signaling [272–274]. Vpr modulates this signaling pathway by activating ataxia-telangiectasia-mutated (ATM) to induce phosphorylation of Chk2 and histone H2A.X. Homologous recombination (HR)-mediated DNA repair machinery is involved in the integration of viral genomic elements into the host genome, which is marked by the foci formation of HR-related proteins breast cancer susceptibility protein 1 (BRCA1) and Rad51 [272, 275]. Taken together, these results show that the ATM-mediated DDR pathway and HR play crucial roles in introducing a higher viral copy number into the host genome. This is further supported by evidence that treatment with caffeine (an inhibitor of both ATR and ATM) or the ATM inhibitor KU55933 significantly decreases viral copy number in infected cells [275, 276].
Integrase (Int), a 32Kd HIV protein, promotes the integration of reverse-transcribed double-strand DNA into the host genome [277]. During the transfer of viral DNA elements, the catalytic activity of Int, depending on the presence of the D,D(35)E motif in the central domain, generates two-nucleotide gaps in a single strand, which are presumably repaired by host repair machinery [277–279]. The involvement of different repair pathways has been proposed to repair single-strand gaps for efficient viral transduction. However, Daniel and colleagues suggest that the efficient repair of gaps by the post-integration repair mechanism, which geometrically differs from conventional DSB repair, prevents Int-dependent apoptosis through the association of DSB sensor protein Nijmegen breakage syndrome 1 protein (NBS1) and ATM kinase [280]. Surprisingly, this kind of repair recruits ATR independently of NBS1 and ATM, suggesting a distinct DSB repair pathway separate from conventional DSB repair as a potential therapeutic target [281]. However, HIV DNA can be integrated in the presence of DSB-inducing agents, including Vpr, in an Int-independent manner in Int catalysis-defective virus. Also, this virus has been shown to be resistant against raltegravir, an inhibitor of Int catalytic activity. These findings provide crucial insights into possible underlying mechanisms of uninterrupted viral replication in monocyte-derived macrophages, a persistent reservoir of viral particles [282].
Sakurai et al. showed that both HR and non-homologous end joining proteins are involved in the process of viral DNA integration into the host genome for efficient transduction [283]. Also, several other studies report controversial roles of DSB sensor proteins, such as poly(ADP-ribose) polymerase 1,ATR, ATM, and DNA-dependent protein kinase, in retroviral host integration [284–287], warranting further investigation into the mechanistic aspects of viral DNA integration to develop potential therapeutic strategies (Fig. 2).

Schematic presentation of HIV-associated CNS dysfunction via genome damage. The major CNS cell populations, neurons, astrocytes and microglia, are differentially affected by HIV factors, but may involve genome damage signaling. HIV-secreted Vpr protein can cause DNA strand breaks by directly binding to the chromosome along with other endonuclease partners. Vpr can also lead to constitutive DDR (p53 and ATM) activation through SLX4, which leads to NF-kB-activation, leading to neuroinflammation and which could contribute to neuronal dysfunction and apoptosis. Another HIV protein Integrase also causes neural loss by apoptosis via its interaction with NBS1 and ATM. Furthermore, Vpr interferes with cell cycle in replicating pool of brain cells including neural stem cells and cycling glia arresting them at G2-phase of cell cycle, finally leading to cell death by ATR/Chk1 activation in the infected brain cells.
CONCLUDING REMARKS
Diverse neurological complications are routinely associated with HIV-infection. As comprehensively described in this review, several mechanisms could cumulatively contribute to the neurotoxicity of HIV, which is also affected by factors such as aging and nature of HIV treatment regime. These complications include dementia, sensory dysfunction, motor function deficits, and overlap with other neurodegenerative diseases like PD, ALS, and AD. The treatment of HIV-associated neuropathy remains difficult, due to a multitude of challenges including a complex diagnosis that largely depends on clinical symptoms without the availability of a definitive biomarker. The success of treating neurocognitive impairment in aged HIV-patients relies on the early detection of the CNS dysfunctions, which most often remains undetectable due to their mild nature in the beginning. The rate of progression of motor and cognitive dysfunctions like dementia largely depends on the patient’s age and genetic make-up. The viral neurotoxins can also induce promote proteinopathies including TDP-43, α-synuclein and tau in an aggressive presentation. Thus, the cognitive test standards to clearlydistinguish between age-related and viral toxin-induced neuronal dysfunction is critical to diagnose and prevent cART or HAART-associated neurological complications in the elderly patients. Further research is required to elucidate the signature profile of HIV-associated neurological manifestation in different populations and to understand the molecular insight in to the specific as well as generic crosstalk of HIV and CNS cells, which would allow for development of mechanism-based treatmentstrategies.