
Abstract
Background:
The translocase of the outer membrane (TOM) is a vital mitochondrial transport system facilitating the importation of nuclear encoded proteins into the organelle. While mitochondrial dysfunction, including perturbation of oxidative phosphorylation (OXPHOS) complex, is evident in Alzheimer’s disease (AD), it remains unclear whether the observed OXPHOS deficits may be associated with TOM alterations.
Objectives:
To correlate TOM subunits with OXPHOS complex proteins in AD.
Methods:
Postmortem neocortex (BA40) from AD and age-matched controls were processed to obtain mitochondrial enriched homogenates for the measurement of Tom20, Tom22, Tom40, and Tom70 as well as components of OXPHOS complex I–V by immunoblotting.
Results:
Tom20 and Tom70 immunoreactivities were significantly reduced in AD, as were components of OXPHOS complex I and III. Both Tom20 and Tom70 positively correlated with complex III and V, while Tom20 also correlated withcomplex IV.
Conclusion:
Reductions in certain TOM subunits and their correlations with specific OXPHOS complex proteins suggest that an impaired mitochondrial transportation system may contribute to previously observed oxidative phosphorylation deficits in AD. Follow-up studies are needed to corroborate the present correlative study.
INTRODUCTION
Mitochondrial dysfunction plays an important role in the pathogenesis and progression of various neurodegenerative diseases, including Alzheimer’s disease (AD). While energy hypometabolism and mitochondrial oxidative damage are one of the earliest events in AD [1–3], deficits in key mitochondrial macromolecules such as complex I, III, and IV of the oxidative phosphorylation (OXPHOS) pathway have also been observed [4–8]. Over the past decade, a “mitochondrial cascade hypothesis” has been proposed for sporadic AD, suggesting that an elevation of mitochondrial oxidative damage triggers 40- to 42-amino acid amyloid-β (Aβ) production, tau phosphorylation, and apoptosis [9, 10]. Notably, the bulk of mitochondrial proteins, including components of OXPHOS complexes, are encoded by nuclear DNA, and these proteins are synthesized in the cytosol before transportation into the mitochondria. The translocase of the outer mitochondrial membrane (TOM) is a major transport machinery (working in tandem with translocase of the inner membrane, TIM) facilitating the import of mitochondrial proteins from the cytosol [11]. TOM comprises of several subunits, namely Tom40 which form the central translocation pore, Tom20, and Tom70 which are the two receptor subunits that recognize incoming proteins from the cytoplasm, Tom22, which links Tom20 to Tom40, as well as other small subunits which stabilize the protein complex [12]. Given that TOM is involved in transporting most nuclear encoded proteins into mitochondria, its integrity is crucial to the functionality of the organelle and to cell survival in general [11]. However, the status of TOM subunits in AD is largely unclear at present. The literature on Tom40 expression in AD have been inconsistent [13–15], whilst levels of other TOM subunits are unknown. In this study, we measured the major TOM subunits, Tom20, Tom22, Tom40, and Tom70, in AD post-mortem neocortex, then correlated their immunoreactivities with those of OXPHOS complex I-V component proteins, on the working hypothesis that the OXPHOS deficits in AD are a result of impaired TOM-mediated mitochondrialimport of OXPHOS component proteins.
MATERIALS AND METHODS
Neuropathological assessment of AD cases
AD subjects were recruited into longitudinal studies in dementia and informed consent was received from next-of-kin for the removal of brains at death. At post-mortem, brains were divided into hemispheres, with one hemisphere fixed for standard neuropathological assessments, while the other was dissected and stored in –75°C. In total, 12 AD and 18 aged non-demented aged controls were obtained from the Brains for Dementia Research (BDR) UK network sites, which include the Thomas Willis Oxford Brain Collections, the London Neurodegenerative Diseases Brain Bank, and Newcastle University. Neuropathological confirmation of AD diagnosis was based on Consortium to Establish a Registry for Alzheimer’s disease (CERAD) criteria [16]. Furthermore, Braak neuropathological staging [17] found AD patients to be at Braak stages 4–6, while controls had Braak stages 0–2 only. Institutional Review Board (IRB) approvals were obtained from both the UK (National Research Ethics Service 08/H10104) and Singapore (National University of Singapore IRB 12-062E) research sites.
Sample preparation and enrichment of mitochondrial fraction
All chemicals were reagent grade and purchased from Sigma Aldrich (St Louis, MO, USA) unless otherwise stated. Frozen tissue samples from the inferior parietal lobe neocortex (Brodmann Area 40) were thawed on ice, dissected free of meninges and white matter, then homogenized in standard fractionation buffer (250 mM sucrose, 20 mM HEPES pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 1 mM EGTA, 1 mM ETDA, 1 mM DTT, with Complete™ protease inhibitor and PhosSTOP™ phosphatase inhibitor tablets [Roche Life Science, Penzberg, Germany]) using a dounce homogenizer and 26 G needle. The crude homogenate was then centrifuged at 10,000 g for 10 min, 4°C, followed by resuspension of the resulting pellet in mitochondrial fractionation buffer (with 10% glycerol, 0.1% SDS, and 2% CHAPS added to the standard fractionation buffer) and vortexing, then further centrifuged at 20,000 g for 2 min, 4°C. Supernatants containing the mitochondrial enriched fraction were collected and stored in aliquots at –75°C for later use. Protein concentrations of all extracted samples were determined by DCTM Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA).
Brain Aβ40 and Aβ42 measurements
Frozen brain samples from BA40 were thawed on ice and dissected as above before homogenization in 5M guanidine Tris HCl buffer (pH 8.0) to measure total (soluble + insoluble) Aβ40 and Aβ42 by enzyme-linked immunosorbent assay (ELISA) kit (Invitrogen, Carlsbad, CA, USA) as previously described [18]. Aβ40 and Aβ42 levels (expressed in pg/mg brainprotein) were then used to derive Aβ42:Aβ40 values.
Immunoblotting
Immunoblotting of mitochondrial enriched brain homogenates were as previously described [18]. Briefly, all extracted mitochondrial fractions were added to Laemmli Sample Buffer (Bio-Rad Laboratories, Hercules, CA, USA) followed by heating to 95°C, 5 min. Samples were loaded for SDS-PAGE at 9μg of extracted protein per lane on 12% polyacrylamide gels, transferred onto polyvinyl difluoride (PVDF) membranes, and blocked with 5% skim milk in 10 mM phosphate buffered saline, pH7.4 with 0.1% Tween 20 (PBST) at room temperature for 1 h. Membranes were immunoblotted overnight at 4°C in bovine serum albumin with primary antibodies against Tom40, Tom20, Tom22, Tom70 (1:500 dilution; from Abcam, Cambridge, UK), porin (1:1000 dilution; from Cell Signaling Technology, Danvers, MA, USA), or epidermal growth factor receptor (EGFR, 1:1000 dilution; from Cell Signaling Technology, Danvers, MA, USA). Furthermore, an OXPHOS antibody cocktail containing antibodies against specific components of OXPHOS complex I-V (complex I: 18 kDa NADH dehydrogenase [ubiquinone] 1β subcomplex subunit 8, NDUFB8; complex II: 29 kDa succinate dehydrogenase [ubiquinone] iron-sulfur subunit B, SDHB; complex III: 48 kDa ubiquinol-cytochrome c reductase complex core protein 2, UQCRC2; complex IV: 22 kDa cytochrome c oxidase II, COXII; complex V: 54 kDa ATP synthase Na+/K+ transporting subunit α1, ATPSA) was used (1:500 dilution; from Abcam, Cambridge, UK). This was followed by washings in PBST and incubating with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (1:5000 dilution; from Jackson ImmunoResearch, West Grove, PA, USA) at 25°C for 1 h. Immunoreactive bands on membranes were detected by enhanced chemiluminescence with Luminata™ Crescendo Western HRP substrate (Merck Millipore, Billerica, MA, USA) and quantified by an image analyzer (UVItec, Cambridge, UK). Membranes for TOM and OXPHOS components were stripped and reblotted with MTC02, an antibody against a 60 kDa non-glycosylated mitochondrial structural protein, for normalization of mitochondrial content (1:1000 dilution; from Abcam, Cambridge, UK), with immunoreactivities expressed in arbitrary units.
Statistical analyses
All analyses were performed using the SPSS software (version 22, IBM Inc., Armonk, NY, USA). Mann-Whitney U tests were used to compare mean immunoreactivities of each TOM subunit and components of OXPHOS complex I-V between control and AD groups, after normalization by mitochondrial structural marker MTC02. Spearman’s correlations assessed associations between immunoreactivities, disease, and demographic factors. A p-value < 0.05 was considered statistically significant.
RESULTS
Demographic and disease variables of study participants
Demographic and disease variables of the study cohort are listed in Table 1. Control and AD groups were well-matched in age, post-mortem interval, gender distribution, and brain pH as an indicator of tissue quality, with values >6.1 considered acceptable [19]. For brain Aβ concentrations, Aβ42 (42 amino acid Aβ fragment), but not Aβ40 (40 amino acid Aβ fragment), was significantly increased in AD compared to age-matched controls. The ratio of Aβ42 to Aβ40 (Aβ42:Aβ40) was also increased in AD (Table 1).
Demographic and disease variables of study participants
Data are mean±SEM unless otherwise indicated. PMI, postmortem interval; Aβ40, amyloid-β peptide (1–40); Aβ42, amyloid-β peptide (1–42). ap-values are for Mann-Whitney U tests except for gender distribution which employed χ2 tests. *Significantly different between control and AD.
Validation of mitochondrial enrichment in immunoblot samples
To confirm mitochondrial enrichment after the subcellular fractionation procedure, processed brain homogenates were immunoblotted for the mitochondrial marker, porin (also known as voltage-dependent anion channel VDAC), and for plasma membrane marker EGFR. Figure 1A shows a representative immunoblot, with EGFR immunoreactivity found in both mitochondrial and cell membrane fractions, whereas porin immunoreactivity was detectable only in the mitochondrial fractions. Subsequent immunoblots of the mitochondrial fractionsnormalized for total protein content (9μg per lane) showed unchanged immunoreactivities of porin as well as MTC02, both structural markers for mitochondria [20] (Fig. 1B, C), suggesting that any differences in TOM or OXPHOS complex immunoreactivities between control and AD were not due to unequal quantities of mitochondria.
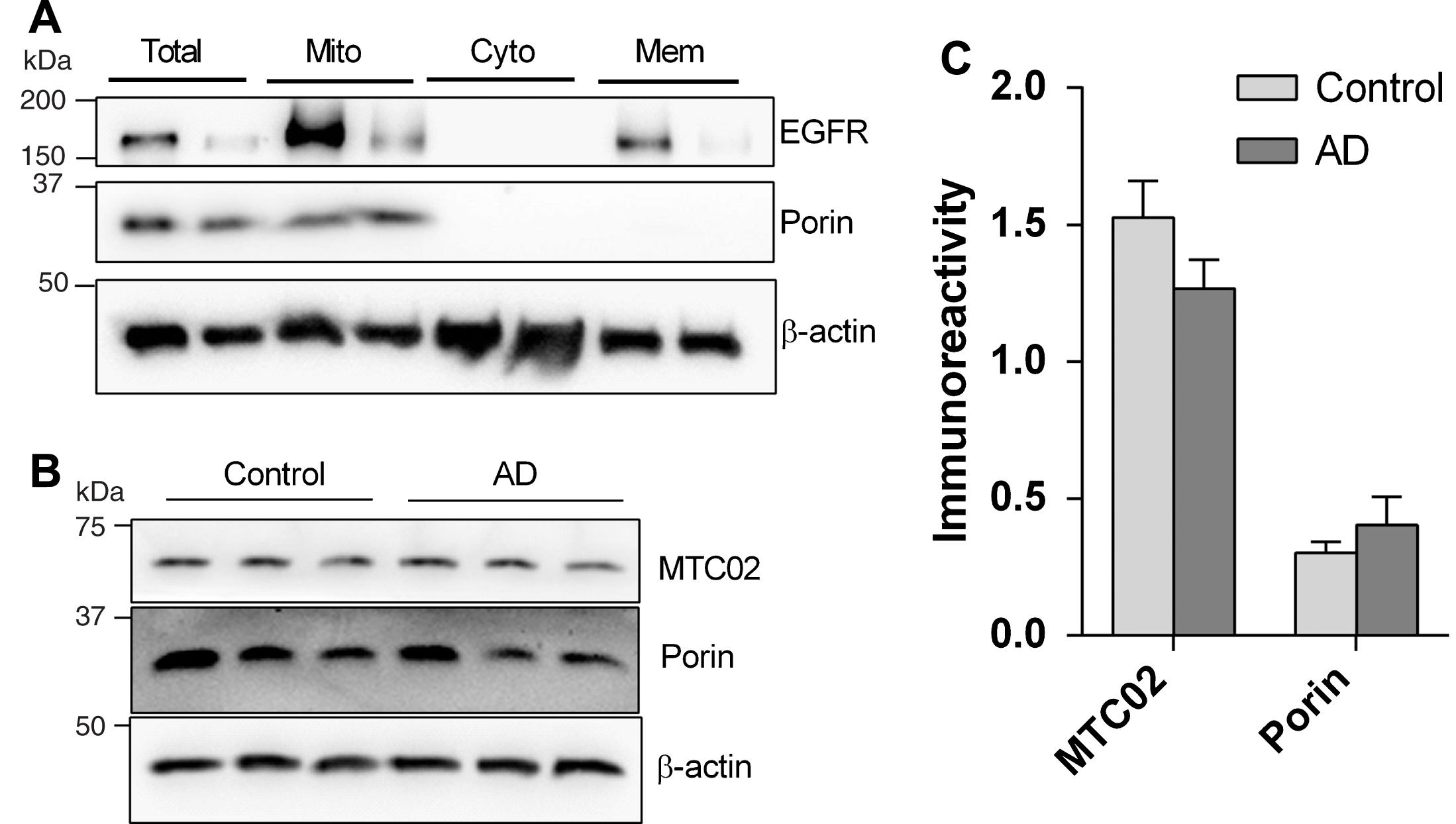
Immunoreactivities of mitochondrial markers in mitochondrial enriched fractions of human neocortex. A) Representative immunoblot of brain homogenates before (“total”) and after mitchondrial enrichment (“Mito”, “Cyto” and “Mem”). B) Representative immunoblot of mitochondrial enriched fraction showing C) no significant difference in the immunoreactivities (mean±S.E.M. arbitrary units) of mitochondrial markers porin and MTC02 between control (n = 18) and AD (n = 12). EGFR, epidermal growth factor receptor; Mito, mitochondrial fraction; Cyto, cytosolic fraction; Mem, membrane fraction.
Deficits in TOM subunits and OXPHOS complex immunoreactivities in AD
Of the TOM subunits measured, Tom20 and Tom70 were significantly reduced in AD, with a similar, albeit non-significant, trend for Tom40 (p = 0.37), while Tom22 remained unchanged (Fig. 2A, B). To determine if Tom20 and Tom70 immunoreactivities may be associated with cortical Aβ burden, we correlated the TOM subunits with Aβ40, Aβ42, and Aβ42:Aβ40 values, and found that both Tom20 and Tom70 correlated negatively with Aβ42:Aβ40 values (Fig. 2C, D), while individual Aβ fragments did not (data not shown). Moreover, Fig. 3A, Bshows significant reductions of OXPHOS complex I and III immunoreactivities. Complex IV (p = 0.13) and V (p = 0.20) showed non-significant trends toward decreases in AD, while complex II remained unchanged (p = 0.26) between controls and AD, showing mitochondrial OXPHOS deficits in agreement with previous observations [4, 5]. Although some of the TOM and OXPHOS components were significantly correlated with age and brain pH (data not shown), multivariate analyses were not performed as age and brain pH were not significantly different between control and AD (Table 1).
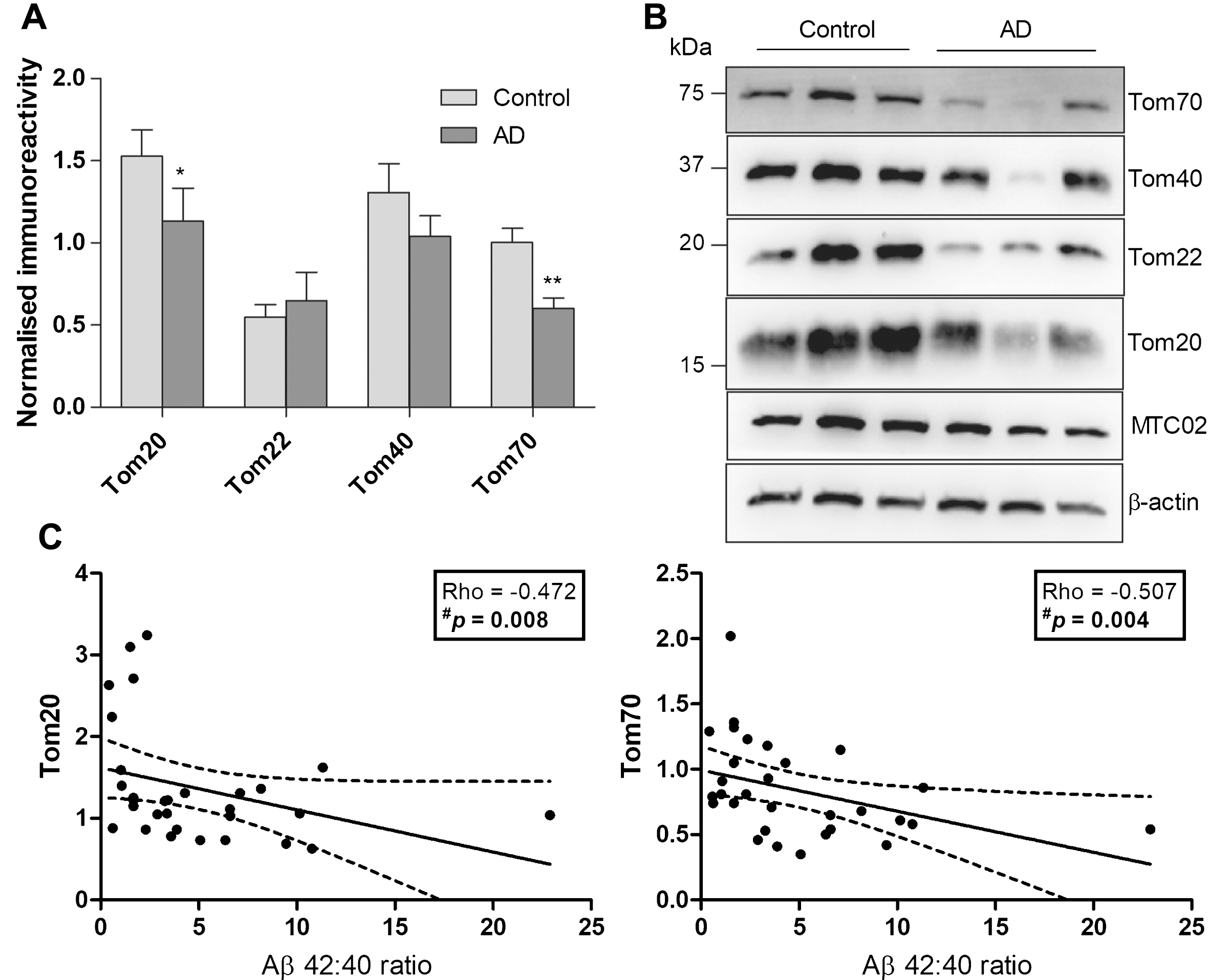
Immunoreactivities of TOM subunits and correlations with Aβ in a cohort of aged controls and AD patients. A) Bar charts of TOM subunit immunoreactivities (mean±S.E.M. arbitrary units) normalized to MTC02 in control (n = 18) and AD (n = 12), with B) showing a representative immunoblot. C) Scatter plots of Tom20 and Tom70 with Aβ42:Aβ40. Solid lines indicate linear regressed best-fit curves while dashed lines indicate their respective 95% confidence intervals. *p < 0.05 and **p < 0.01, significantly different from control (Mann-Whitney U tests). #Significant Spearman correlation.
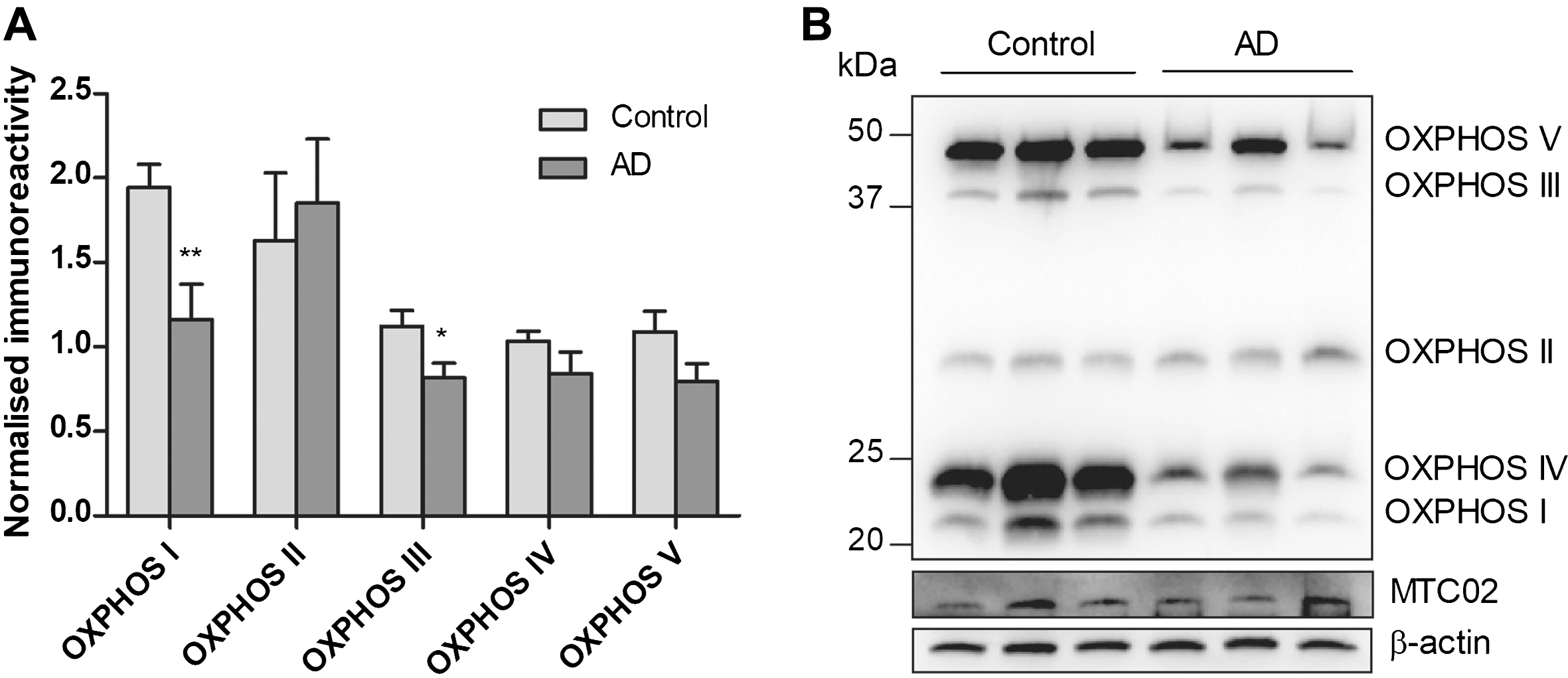
Immunoreactivities of OXPHOS complex I - V in a cohort of aged controls and AD patients. A) Bar charts of OXPHOS complex component immunoreactivities (mean±S.E.M. arbitrary units) normalized to MTC02 in control (n = 18) and AD (n = 10–11), with B) showing a representative immunoblot. *p < 0.05 and **p < 0.01, significantly different from control (Mann-Whitney U tests).
Correlations between TOM complex subunits and OXPHOS complex immunoreactivities in AD
Within the AD group, Fig. 4 shows general trends of positive correlations between Tom20/Tom70 subunits and components of OXPHOS complex I, III, IV and V, with complex III and V reaching statistical significance for both TOM subunits, while complex IV also correlated with Tom20. Furthermore, while Tom22 and Tom40 immunoreactivities were not significantly altered in AD, significant correlations were observed between Tom22 and OXPHOS complex II (n = 11, Rho = –0.68, p = 0.021); and between Tom40 and complexes IV (n = 11, Rho = 0.62, p = 0.043) and V (n = 10, Rho = 0.72, p = 0.019).
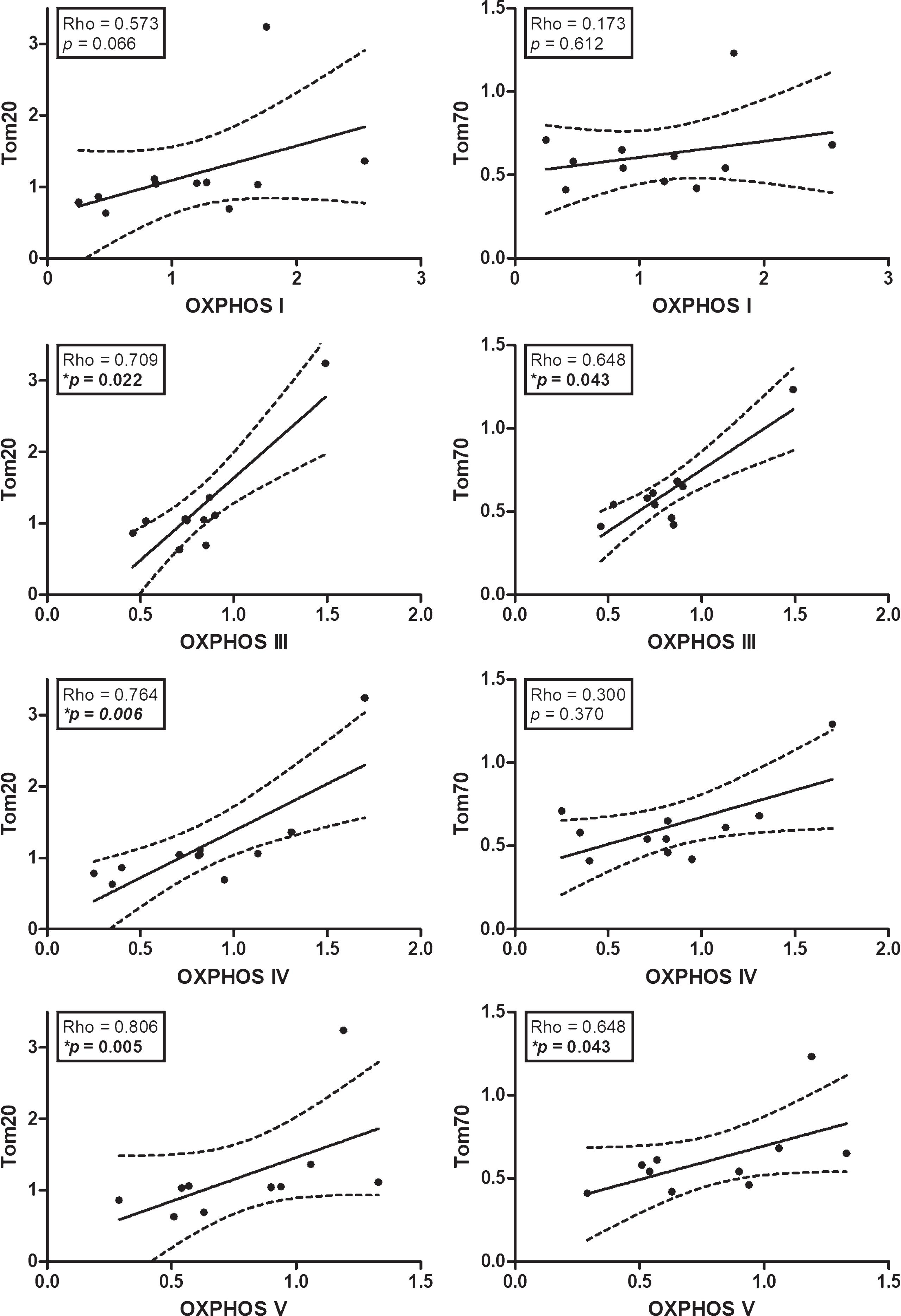
Correlations between TOM subunits and OXPHOS complexes in AD. Scatter plots of normalized immunoreactivities of Tom20 and Tom70 with components of OXPHOS complexes I, III, IV, and V. Solid lines indicate linear regressed best-fit curves while dashed lines indicate their respective 95% confidence intervals. *Significant Spearman correlations.
DISCUSSION
While mitochondrial dysfunction is evident in AD [1–3], the underlying pathogenic mechanisms remain unclear. Using well characterized postmortem brain tissues, this study is, to our knowledge, the first to comprehensively examine immunoreactivities of the major TOM subunits in AD neocortex. We report here that at least two subunits, namely Tom20 and Tom70, are significantly reduced in AD, along with reductions in the components of OXPHOS complex I and III (Figs. 2 and 3). Deficits in Tom20 and Tom70 may lead to impairment in the recognition and binding of cytosolic pre-proteins, and possibly affect subsequent TOM-mediated translocation of proteins (including components of OXPHOS complexes [21]) across the mitochondrial membrane (see Fig. 5). Indeed, the wide-ranging positive correlations between Tom20/Tom70 subunits and OXPHOS complex proteins (Fig. 4) suggest that these TOM subunit deficits may contribute to the mitochondrial OXPHOS deficits of AD. This finding has potential therapeutic implications, as it suggests that the restoration of TOM function to be a worthwhile therapeutic goal in AD which can help reverse mitochondrial dysfunction and prevent subsequent oxidative stress-relatedneuronal damage. While the antecedent events leading to TOM subunit deficits are not known, our data suggest an association with Aβ burden, specifically higher Aβ42 to Aβ40 ratios (Fig. 2C). The association between Tom20 alterations and Aβ has some support from in vitro studies which reported down regulation of mitochondrial Tom20 concomitant to increased oxidative stress and mitochondria morphological changes in cells chronically treated with Aβ [22]. It is at present unclear why only Aβ42:Aβ40, but not the individual Aβ alloforms, correlated with reduced Tom20. While Aβ40 and Aβ42 are the two most abundant Aβ fragments found in brain under physiological conditions, Aβ42 is known to be more neurotoxic, prone to aggregate, and induces more oxidative damage compared to Aβ40 [23–25]. Interestingly, higher Aβ42 to Aβ40 ratios have been associated with more aggressive disease [26]. Therefore, the present data suggest that TOM deficits may be related to Aβ neuro- or mitochondrial-toxicity potential as indicated by the relative abundance of Aβ42 over Aβ40. Interestingly, the mitochondrial dysfunction and associated-oxidative stress downstream of TOM may in turn exacerbate secretion of Aβ [10], leading to a vicious cycle [2]. However,further studies are required to elucidate the mechanistic links between Aβ and TOM/mitochondrial function, including delineating specific effects of Aβ40 versus Aβ42.
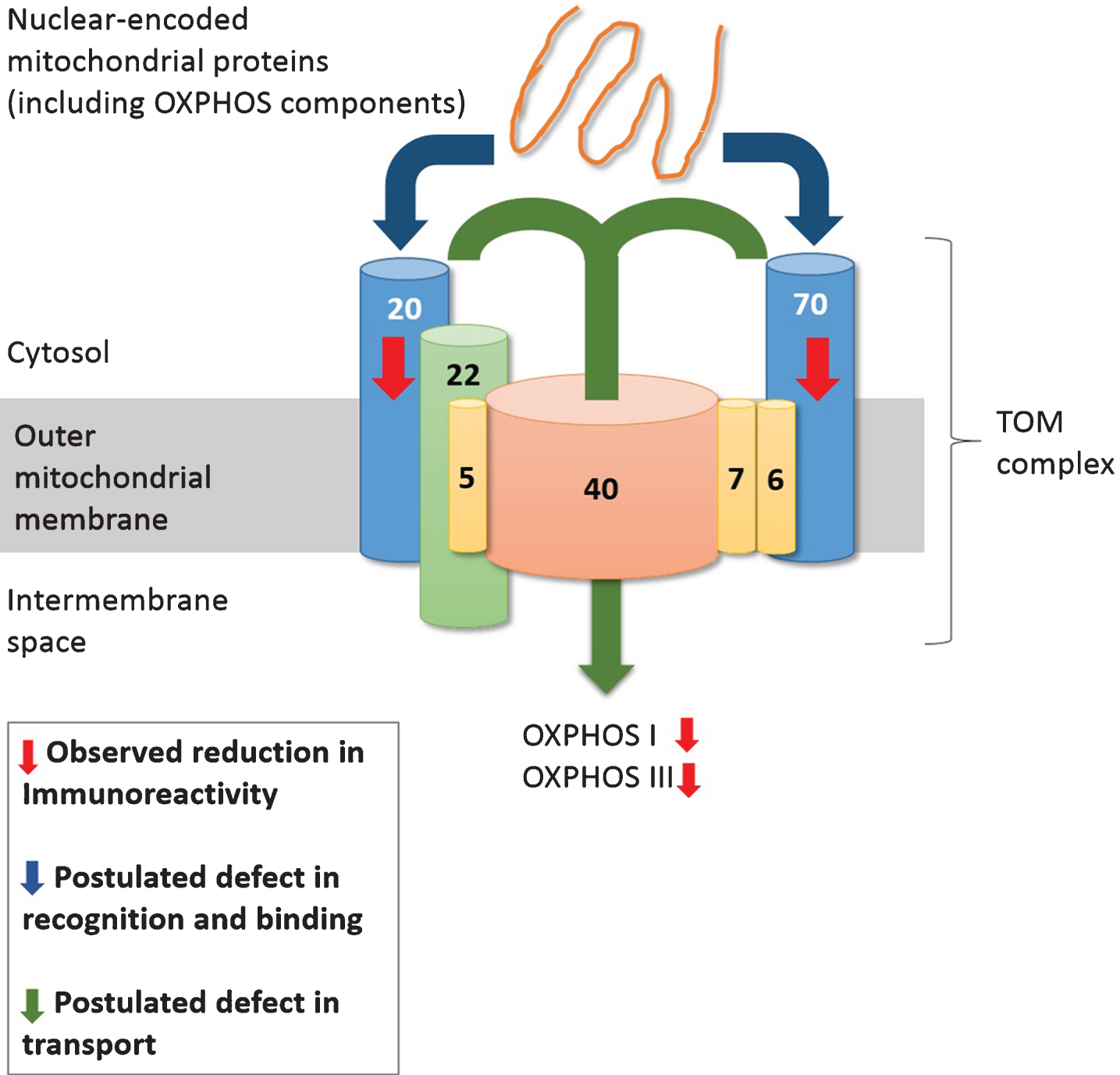
Model of TOM-mediated deficiency in protein transport leading to OXPHOS deficits in AD mitochondria. In BA40 of AD neocortex, TOM subunits Tom20 and Tom70 are reduced, thus compromising TOM-mediated recognition of proteins (including OXPHOS components) and their subsequent transport across the mitochondrial membrane through the Tom40 pore, leading to deficits in OXPHOS complex I and III, potentially resulting in mitochondrial dysfunction.
Due to the post-mortem nature of this study, several limitations need to be addressed with follow-up investigations. Firstly, while the correlative data suggest a biologically plausible mechanism linking reduced TOM subunits to OXPHOS deficits (see Fig. 5), further studies are needed to elucidate the physical interactions between TOM subunits and OXPHOS complexes, as well as the impact of TOM functional impairment on mitochondrial function. Furthermore, while selection of BA40 for this study was based on tissue availability and pathological relevance (see [27]), it is unclear whether these observations are representative of other brain areas affected by AD, and follow up studies looking at other cortical regions (for e.g., other neocortical areas or the hippocampal formation) are needed. Moreover, the study samples consisted exclusively of late-stage AD (Braak stages V/VI), resulting in the inability to determine the time-course of the observed TOM and OXPHOS alterations, although others have suggested that such pathological processes would likely have occurred early in the course of disease [3]. Follow-up studies using early or prodromal stages of AD will help clarify this issue. Notably, the OXPHOS immunoblotting antibody cocktail used in this study measured the immunoreactivities of only one component of each OXPHOS complex, and hence may not be generalizable to the entire mitochondrial OXPHOS system. More studies are needed to assess other mitochondrial proteins in order to further investigate the effects of TOM complex impairment on mitochondrial dysfunction in AD. The potential complexity of such studies is exemplified by the fact that not all OXPHOS components currently measured are nuclear DNA-encoded. Thus, while the mitochondrial DNA (mtDNA)-encoded COXII (component of complex IV) only showed non-significant decreases when normalized for mitochondrial content (by MTC02, see Fig. 3A), it is likely that perturbations in mtDNA, including oxidative damage and deletions [28, 29] will affect COXII expression levels. On the other hand, although TOM does not directly interact with COXII, other nuclear-encoded proteins such as COXIV [30] and Oxa1 [31], which play crucial roles in the assembly and enzymatic activity of complex IV, may be affected by altered TOM-mediated transport into the mitochondria [32]. Therefore, the expression and activities of the mitochondrial OXPHOS system are likely a product of complex interactions between TOM-dependent and independent processes.
In conclusion, the present study on AD post-mortem neocortex reports deficits in Tom20/Tom70 subunits which correlated with both higher Aβ42 to Aβ40 ratios and deficits in OXPHOS complex proteins. We speculate that the toxicity of Aβ downregulated certain TOM subunits which in turn leads to reduced mitochondrial OXPHOS complexes dueto impairment in TOM-mediated transport of OXPHOS components across the mitochondrial membrane. Our data thus point to TOM as a potential therapeutic target to restore protein transportation and mitochondrial deficits in AD. However, there is a need for further studies which help elucidate the underlying mechanisms linking Aβ burden, TOM-dependent as well as TOM-independent processes to mitochondrial dysfunction.
Footnotes
ACKNOWLEDGMENTS
This study was funded by the National Medical Research Council of Singapore (NMRC/CSA-SI/0007/2016) and the Yong Loo Lin School of Medicine, National University of Singapore (R-184-000-259-112). Tissues for this study were collected through tissue repositories supported by the UK Medical Research Council and by Brains for Dementia Research, (![]() ), a joint venture between the Alzheimer’s Society and Alzheimer’s Research UK. CGB would like to thank the National Institute for Health Research (NIHR) Mental Health Biomedical Research Centre and Dementia Unit at South London and Maudsley NHS Foundation Trust; and the Institute of Psychiatry, King’s College London.
), a joint venture between the Alzheimer’s Society and Alzheimer’s Research UK. CGB would like to thank the National Institute for Health Research (NIHR) Mental Health Biomedical Research Centre and Dementia Unit at South London and Maudsley NHS Foundation Trust; and the Institute of Psychiatry, King’s College London.