
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by the presence of intracellular neurofibrillary tangles (NFTs) containing hyperphosphorylated tau, and the extracellular deposition of amyloid plaques (APs) with misfolded amyloid-β (Aβ) peptide. Glia maturation factor (GMF), a highly conserved pro-inflammatory protein, isolated and cloned in our laboratory, has been shown to activate glial cells leading to neuroinflammation and neurodegeneration in AD. We hypothesized that inflammatory reactions promoted by NLRP3-Caspase-1inflammasome pathway trigger dysfunction in autophagy and accumulation of Aβ which is amplified and regulated by GMF in AD. In this study, using immunohistochemical techniques we analyzed components of the NLRP3 inflammasome and autophagy- lysosomal markers in relation to Aβ, p-tau and GMF in human postmortem AD and age-matched non-AD brains. Tissue sections were prepared from the temporal cortex of human postmortem brains. Here, we demonstrate an increased expression of the inflammasome components NLRP3 and Caspase-1 and the products of inflammasome activation IL-1β and IL-18 along with GMF in the temporal cortex of AD brains. These inflammasome components and the pro-inflammatory cytokines co-localized with GMF in the vicinity and periphery of the APs and NFTs. Moreover, using double immunofluorescence staining, AD brain displayed an increase in the autophagy SQSTM1/p62 and LC3 positive vesicles and the lysosomal marker LAMP1 that also co-localized with GMF, Aβ and hyperphosphorylated p-tau. Our results indicate that in AD, the neuroinflammation promoted by the NLRP3 inflammasome may be amplified and regulated by GMF, which further impairs clearance of protein aggregates mediated by the auto-phagosomal pathway.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neuro-degenerative disorder characterized by the abnormal aggregation and accumulation of amyloid-β (Aβ), neurofibrillary tangles (NFTs), and microtubule associated protein tau. These plaques and tangles are generally associated with activated microglia and reactive astrocytes in AD brain [1]. The molecular mechanism underlying the accumulation and aggregation of Aβ and tau is not fully understood. However, abnormal post-translational modification, such as hyperphosphorylation, acylation, and impaired degradation of these proteins have been reported [2, 3]. Recent in vivo findings showed binding of Aβ peptide with tau to form a soluble stable complex and it is this association that promotes aggregation [4]. The association of Aβ peptide with tau causes sudden dissociation of tau from microtubules and collapse of axon and its transport leading to the loss of synapse and neuronal death [5]. Glia maturation factor (GMF) is a pro-inflammatory molecule that is present in glial cells and some neuronal cells of the central nervous system (CNS). Upregulation of GMF expression causes inflammation in neurodegenerative disorders [6]. It was for the first time discovered, purified, sequenced, and sub-cloned from bovine brain in our laboratory [7 –12]. Previous reports from our laboratory showed that GMF is mainly co-localized and expressed in the vicinity of Aβ and tau in the temporal cortex of human AD brain [13, 14]. Neuroinflammatory changes in AD are initiated by the robust activation of microglia and astrocytes and release of pro-inflammatory cytokines interleukin-1beta (IL-1β) and IL-18 [15]. The prolonged accumulation and aggregation of Aβ peptide stimulates the glial cells to secrete pro-inflammatory cytokines and other inflammatory mediators in AD [13]. The NLRP3infllammasome has been associated with several chronic inflammatory disorders and is activated by inflammatory mediators and aggregated proteins in AD [16, 17].
NLRP3 infllammasome is a multiprotein complex mainly expressed in myeloid cells and plays an essential role in the Aβ induced activation of caspase-1 and downstream secretion of two of its substrates, the pro-inflammatory cytokines IL-1β and IL-18 [18, 19] leading to neuronal cell death. However, these cytokines also have a beneficial role in promoting inflammation and eliminating harmful substances. Overproduction of IL-1β and IL-18 have been linked to inflammatory and autoimmune diseases [20, 21]. Previous studies have shown that the expression of NLRP3 and caspase-1 is extensively upregulated in the brain of AD patients [22, 23]. Aggregated Aβ releases IL-1β via activation of the NLRP3 inflammasome and caspase-1 in glial cells. NLRP3 and caspase-1 deficiency enhances Aβ clearance in AD brain [22]. Large number of glial cells are present around the Aβ plaques showing that microglia are playing a major role in clearance of amyloid deposits by phagocytic activities. However, with the progression of AD, glial cells get adapted to cytokines deposition, which disturbs the microglial clearance function [24, 25]. Production of IL-1β is mainly dependent on the activation of the infllammasome, a cytosolic protein complex that assembles in response to several factors, like reactive oxygen species (ROS) and misfolded proteins, triggering lysosomal rupture [26, 27]. Aβ peptide accumulates and is converted to the characteristic plaques in AD that activates the NLRP3 inflammasome.
Autophagy is a catabolic and degradative process that delivers cytoplasmic constituents into lysosome for degradation and eventual recycling of the resulting molecules [28]. Many cellular stress factors, such as organelle damage and pathogen infection, induces autophagy. The auto-phagosome, which is formed by elongation and closure of the isolated membrane; engulfs a portion of the cytoplasm and subsequently fuses with the lysosome leading to lysosomal degradation [29]. Autophagy is essential to maintain neuronal homeostasis and any defects in autophagy elicits excessive accumulation of Aβ proteins in brain [30]. Previous studies have shown the role of autophagy in different neurodegenerative disorders and the association between autophagy and AD [31]. The lysosome inside the cell is a major degradative organelle, responsible for degradation of long-lived cytoplasmic constituents and protein aggregates too large to be degraded by the proteasome [32 –34]. Several studies have shown that hyperphosphorylated tau can be cleared by the autophagy pathway [3 , 36] and aggregation of pathological p-tau and SQSTM1/p62 could result from defective autophagy lysosomal clearance [37, 38]. It is known that accumulation of autophagosome following defects in autophagosome-lysosome fusion leads to increase in LC3 expression and the lysosomal marker lysosomal stressed-associated protein (LAMP1) in the cytoplasm. Our study demonstrates that neuroinflammation in AD may be a result of activation of the NLRP3 inflammasome regulated by GMF, which is in conjunction with a defective autophagy-lysosomal pathway.
MATERIAL AND METHODS
Antibodies and reagents
Rabbit GMF polyclonal antibody and mouse GMF monoclonal antibodies were purchased from Proteintech (Chicago, IL, USA). Mouse anti-6E10 (Bio Legend San Diego, CA, USA), mouse anti-phospho-tau (EMD Millipore, Massachusetts, USA), mouse anti-NLRP3 (AdipoGen, San Diego, CA, USA), rabbit polyclonal anti-caspase-1(Santa Cruz, Dallas TX, USA), rabbit polyclonal anti-IL-1β (Abcam, Cambridge, MA, USA), mouse anti-IL-18 (Santa Cruz, Dallas TX, USA), rabbit anti-SQSTM1/p62 (Abcam, Cambridge, MA, USA), rabbit ant-LC3(Abcam, Cambridge, MA, USA), and rabbit anti-LAMP1(Abcam, Cambridge, MA, USA) and mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (H-1200; Vector Laboratories, Inc. Burlingame, CA, USA).
Human brain samples
Temporal lobes containing hippocampal region from human postmortem rapid brains of AD patient (n = 5) and age-matched Non-AD brains (n = 5) were obtained through the University of Iowa Deeded Body Program and fixed in 4% paraformaldehyde. They were cut into 40 μm thick coronal sections using a sledge freezing microtome and these sections were collected in phosphate buffer saline (PBS) and stored in cryo storage solution (glycerol 30 ml, ethylene glycol 30 ml, 40 ml 0.1 M PBS) until used for immunofluorescence staining. This study was approved by the University of Missouri Institutional Review Board (IRB #2008067; Exempt Application 224561), Columbia, MO, USA. This study was conducted under standard ethical procedures. All the appropriate personal protection safety procedures were followed to handle the human samples.
Immunofluorescence staining
Coronal sections of temporal cortex (n = 3–5) from different brains were washed with 0.1% PBS-TritonX-100for 20 min. For most antibodies used in this study, antigen retrieval was done using sodium citrate buffer with 0.1% Triton-X. Sections were blocked with blocking solution containing 10% donkey serum for 1 h at room temperature. Sections were incubated at 4°C with the respective primary antibodies as shown in the figure legends. The following primary antibodies were used: mouse anti-amyloid beta (6E10) (1 : 250), mouse anti-phospho-tau (1 : 250), mouse anti-GMF (1 : 200), mouse anti-NLRP3 (1 : 250), rabbit anti-capase-1 (1 : 200), rabbit anti-IL-1β (1 : 200), mouse anti-IL-18 (1 : 200), rabbit anti-LC3 (1 : 200), rabbit anti- SQSTM1/P62 (1 : 250), and rabbit anti-LAMP1(1 : 250). The sections were washed three times followed by incubation with the respective Alexa Fluor- 488 (green) and/or Alexa Fluor -568 (red) tagged secondary antibodies (1 : 500) for 1 h at room temperature. Thereafter sections were washed and mounted with a cover slip using Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (H-1200; Vector Laboratories, Inc., CA, USA). Images were acquired on a fluorescence Nikon (DIAPHOT) fluorescence microscope. Staining intensity and area measurement was undertaken using the MetaMorph image analysis software.
Morphological analysis
Staining intensity and measurement of area
We quantified the average intensity and the area of positive antibody labeling in the temporal cortex of human AD brain. An integrated morphology analysis was undertaken using MetaMorph image analysis software as described previously [39]. For each section, the level of non-specific staining was adjusted to a set level to ensure a standard background across different groups and the average intensity and the area of immunofluorescence staining were calculated. Measurements were conducted on 3–5 representative sections per group.
Statistical analysis
Statistical analysis was performed using the GraphPad Instat 3 software and using unpair t test. Results are expressed as mean±SEM. The p values less than 0.05 were considered statistically significant.
RESULTS
Co-localization of GMF with GFAP and IBA1 in the temporal cortex of human AD brain.
Human postmortem AD and age-matched Non-AD brain tissue from different cases were analyzed by immunofluorescence staining to check the cell type where GMF is expressed. GMF showed more expression in activated astrocytes (GFAP positive) and microglia (IBA1 positive). Our results showed (Fig. 1A) higher expression and co-localization of GMF with GFAP in the temporal cortex of AD brain compared with age matched Non-AD brain. Quantification based on average labelled intensity and positive area is shown in (Fig. 1B). Higher expression and co-localization of GMF was also seen in the microglia (IBA1-positive) (Fig. 1C) of AD brain compared with Non-AD brain. Quantification for the average intensity and total labeled positive area is as shown in Fig. 1D.
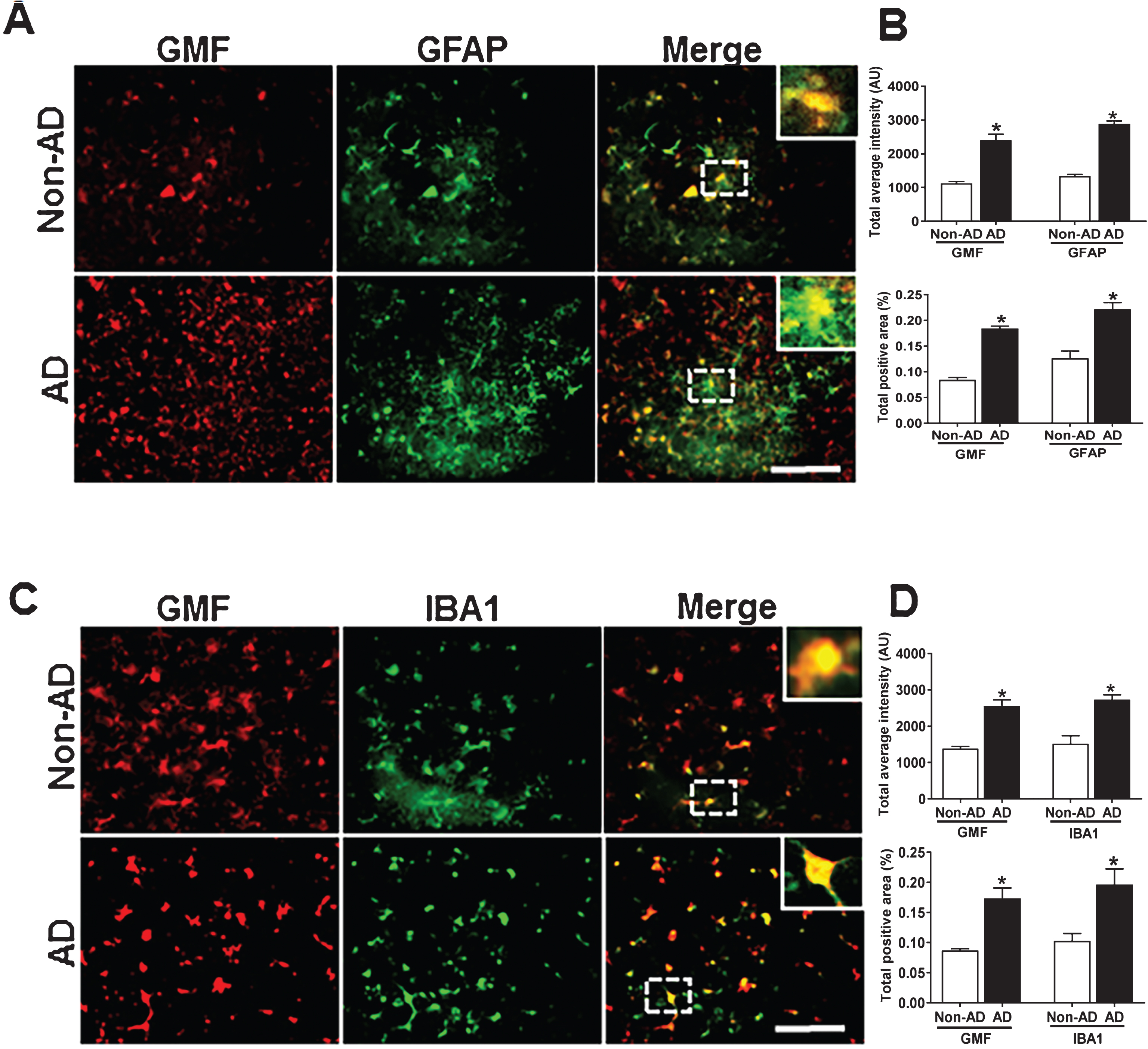
Co-localization of GMF with GFAP and IBA1 in the temporal cortex of AD and non-AD control brain. A) Sections were immunostained with ant-GMF and anti-GFAP respectively. Representative images showed higher expression and co-localization of GMF (red) with GFAP (green), which clearly indicates astrocytes are one of the site of expression of GMF in human AD brain. C) Sections were immunostained with anti-GMF and anti-IBA1 respectively. Representative image displays the higher expression of and co-localization of GMF (red) with IBA1 (green) which clearly indicates microglia are another site in addition to the astrocyte and some neuronal cell for the GMF expression in human AD brain. The values are expressed as mean±standard error of determination from each group (n = 3–5). *p < 0.05 versus non-AD was considered statistically significant. Boxed area shows the enlarged view of co-localization of GMF (red) with GFAP (green) and IBA1 (green) Scale bar = 50 μm and AU (Arbitrary Unit). B, D) Quantification of GMF, GFAP, and IBA1 based on average labeled intensity and labeled positive area in AD and non-AD brains.
Accumulation of Aβ peptide, hyperphosphorylated tau, and its co-localization with GMF in the temporal cortex of human AD brain
Increase in Aβ concentration followed by continuous changes in aggregation states of Aβ triggers innate immune reaction to cause chronic neuroinflammation in the brain followed by neuronal degeneration [40]. Earlier studies from our laboratory have shown the co-localization of GMF with Aβ and p-tau in human AD brains [41]. Human AD postmortem brain and age- matched non-AD control brain tissue from different cases were analyzed by immunofluorescence staining to determine the expression and co-localization of Aβ and hyperphosphorylated tau with GMF using the respective antibodies. Sections were labeled with antibody to amyloid plaque (AP) (6E10) and GMF antibody and representative image showed higher expression and co-localization of APs and GMF in AD brain (Fig. 2A) compared with non-AD brain. Quantification based on average labeled intensity and total labeled positive area is shown in Fig. 2B. We also tested the expression and co-localization of GMF with hyperphosphorylated p-tau. The sections were labeled with anti-phospho-tau and anti-GMF, as shown in Fig. 2C. Non-AD sections showed less expression and co-localization compared to AD brain which showed extensive expression and co-localization in the temporal cortex. Quantification for the average intensity and total labeled positive area is as shown in Fig. 2D.

Co-localization of Aβ (6E10) and p-tau with GMF in the temporal cortex of AD and non-AD control brain. A) Sections were immunostained with anti-Aβ (6E10) and anti-GMF respectively. Representative images display the higher expression and co-localization of 6E10 (red) and GMF (green) in AD as compared with non-AD control brain. C) Sections were immunostained with p-tau and with GMF respectively. Representative image displays higher expression and co-localization of GMF (green) with p-tau (red) in AD compared to age-matched non-AD brain. The values are expressed as mean±standard error of determination from each group (n = 3–5). *p < 0.05 versus non-AD was considered statistically significant. Boxed area shows the enlarged view of co-localization of GMF (green) with 6E10 (red) and p-tau (red) Scale bar = 50 μm and AU (Arbitrary Unit). B, D) Quantification of 6E10, p-tau, and GMF based on average labeled intensity and labeled positive area in AD and non-AD brains.
Double immunofluorescence detection of the NLRP3 inflammasome components and co-localization with Aβ, p-tau, and GMF in the temporal cortex of human AD brain
Tauopathy observed in AD brains has recently [42] been shown to be propagated extracellularly between neuronal cells mediated by neuronal activity. Therefore, it is very important to understand the neuronal activation pathways involved in propagation of neuroinflammation. To advance this objective, we analyzed by immunofluorescence staining the expression and co-localization of NLRP3 inflammasome protein and its activating protein components with Aβ, p-tau and GMF, as shown in Fig. 3A. AD brain showed higher expression and co-localization of GMF with 6E10 compared with non-AD sections. Quantification using average labeled intensity and labeled positive area is shown in Fig. 3B. We also found increased expression and co-localization of inflammasome NLRP3 with hyperphosphorylated tau shown in Fig. 3C. Quantification was done to check the average labeled intensity and total labeled positive area as shown in Fig. 3D. We also observed increased expression of GMF in AD brain, which co-localized with the increased NLRP3 expression in AD, as shown in Fig. 3E. Quantification determining the total intensity and total positive area showed (Fig. 3F) that in the AD group there was extensive expression and co-localization of NLRP3 with GMF compared with non-AD brain.

Co-localization of NLRP3 with Aβ, p-tau, and GMF in temporal cortex of AD and non-AD brain. A) Sections were immunostained with anti-NLRP3 and 6E10, respectively. Representative immunofluorescence staining displayed the higher expression and co-localization of NLRP3 (green) and 6E10 (red) in AD as compared with non-AD. C) Sections were immunostained with NLRP3 and with anti-p-tau respectively to check the co-localization of NLRP3 with p-tau. Representative immunofluorescence staining displayed the higher expression and co-localization of NLRP3 (green) and p-tau (red) in AD compared with non-AD. E) Sections were immunostained with NLRP3 with GMF respectively to check the co-localization of NLRP3 with GMF. Representative immunofluorescence staining displayed the higher expression and co-localization of NLRP3 (green) and GMF (red) in AD compared with non-AD (F). The values are expressed as mean±standard error each group (n = 3–5). *p < 0.05 versus non-AD was considered statistically significant. Zoom of boxed area shows the enlarged view of co-localization of NLRP3 (green) with 6E10 (red), p-tau (red), and GMF (red). Scale bar = 50 μm and AU (Arbitrary Unit). B, D, F) Quantification of NLRP3, 6E10, p-tau, and GMF based on average labeled intensity and labeled positive area in AD and non-AD brains.
Caspase-1 co-localizes with Aβ, p-tau and GMF in temporal cortex of human AD brain
The NLRP3 inflammasome represents the molecular platform for the activation of caspase-1. AD brain sections showed higher expression of caspase-1 as compared to non-AD brain and its co-localization with Aβ and p-tau is shown in Fig. 4A, C, respectively. In addition to increased caspase-1 expression in AD, there was a strong co-localization of caspase-1 with GMF in temporal cortex in the region of APs and NFTs (Fig. 4E). Quantification of the immunofluorescence staining patterns shown in the above figures was calculated by determining the total intensity of staining and total positive area as shown inFig. 4B, D, F.
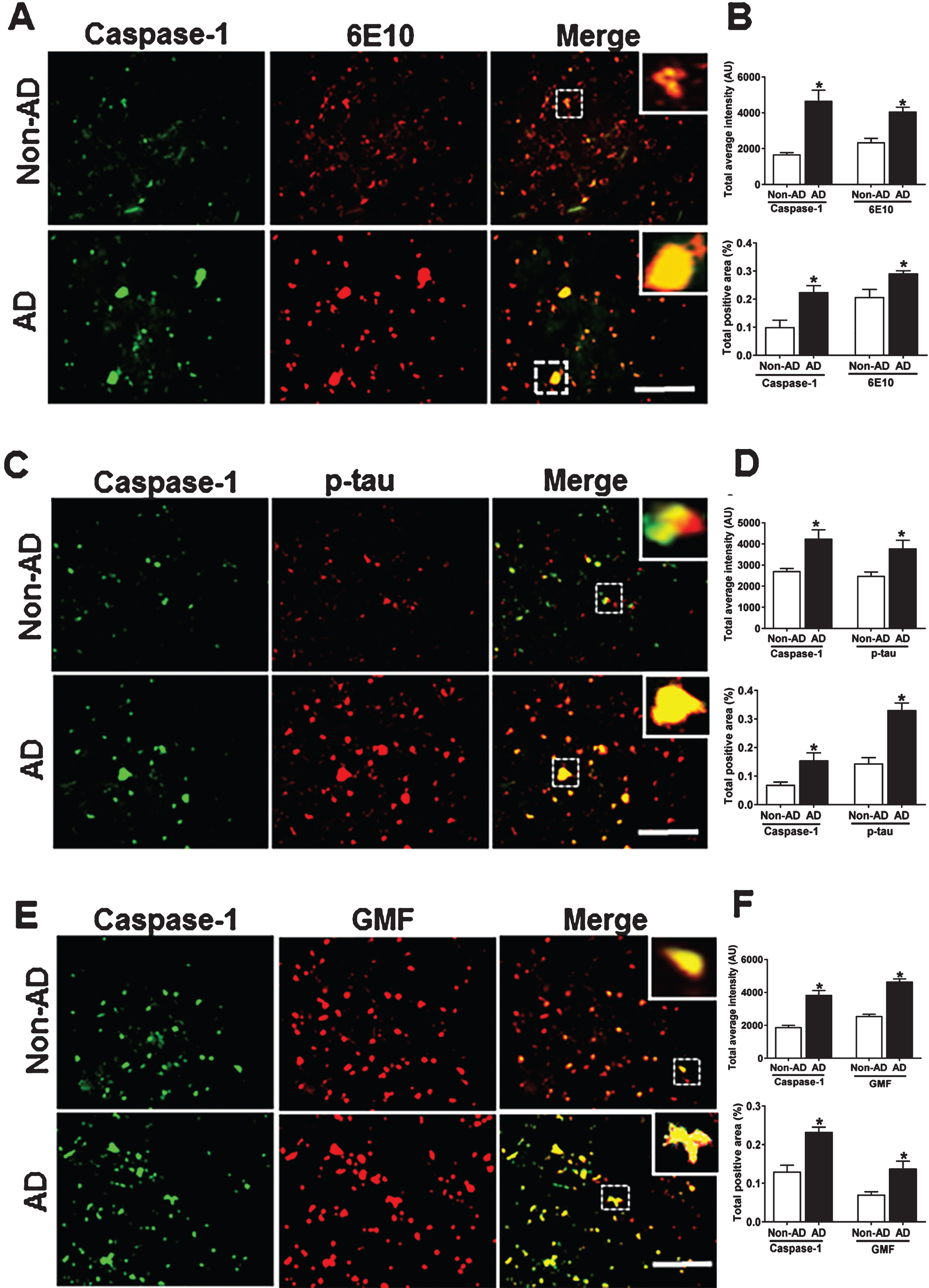
Co-localization of caspase-1 with Aβ, p-tau and GMF in the temporal cortex of AD and non-AD brain. A) Sections were immunostained with anti-caspase-1 and 6E10, respectively. Representative immunofluorescence staining displays the higher expression and co-localization of caspase-1 (green) and 6E10 (red) in AD as compared with non-AD. C) Sections were immunostained with anti-caspase-1 and anti-p-tau, respectively, to check the co-localization of caspase-1 with p-tau. Representative immunofluorescence staining displayed the higher expression and co-localization of caspase-1(green) and p-tau (red) in AD compared with non-AD. E) Sections were immunostained with anti-caspase-1 and anti- GMF respectively to check the co-localization of caspase-1with GMF. Representative immunofluorescence staining displayed the higher expression and co-localization of caspase-1 (green) and GMF (red) in AD compared with non-AD. The values are expressed as mean±standard error, from each group (n = 3–5). *p < 0.05 versus non-AD was considered statistically significant. Zoom of boxed area shows the enlarged view of co-localization of caspase-1 (green) with 6E10 (red), p-tau (red), and GMF (red). Scale bar = 50 μm and AU (Arbitrary Unit). B, D, F) Quantification of Caspase-1, 6E10, p-tau, and GMF based on average labeled intensity and labeled positive area in AD and non-AD brains.
IL-1β co-localizes with Aβ p-tau and GMF in the temporal cortex of human AD brain
Inflammasome–mediated activation of caspase-1 leads to the processing of the pro-inflammatory cytokine IL-1β. We next performed the co-localization of IL-1β and GMF in the vicinity of APs and NFTs in the temporal cortex of AD brain. Representative microscopic images showed (Fig. 5A, C, E) higher expression and co-localization of IL-1β with GMF and this also co-localized with APs (6E10) and NFTs (p-tau) in AD. Non-AD sections did not show much expression or co-localization of the proteins. Also, intensity of staining showing the co-localization of IL-1β with AP; probably represents extracellularly released IL-1β proteolytic fragment with the extracellular AP. Quantification determining total labeled intensity and total positive area is shown in (Fig. 5B, D, F).
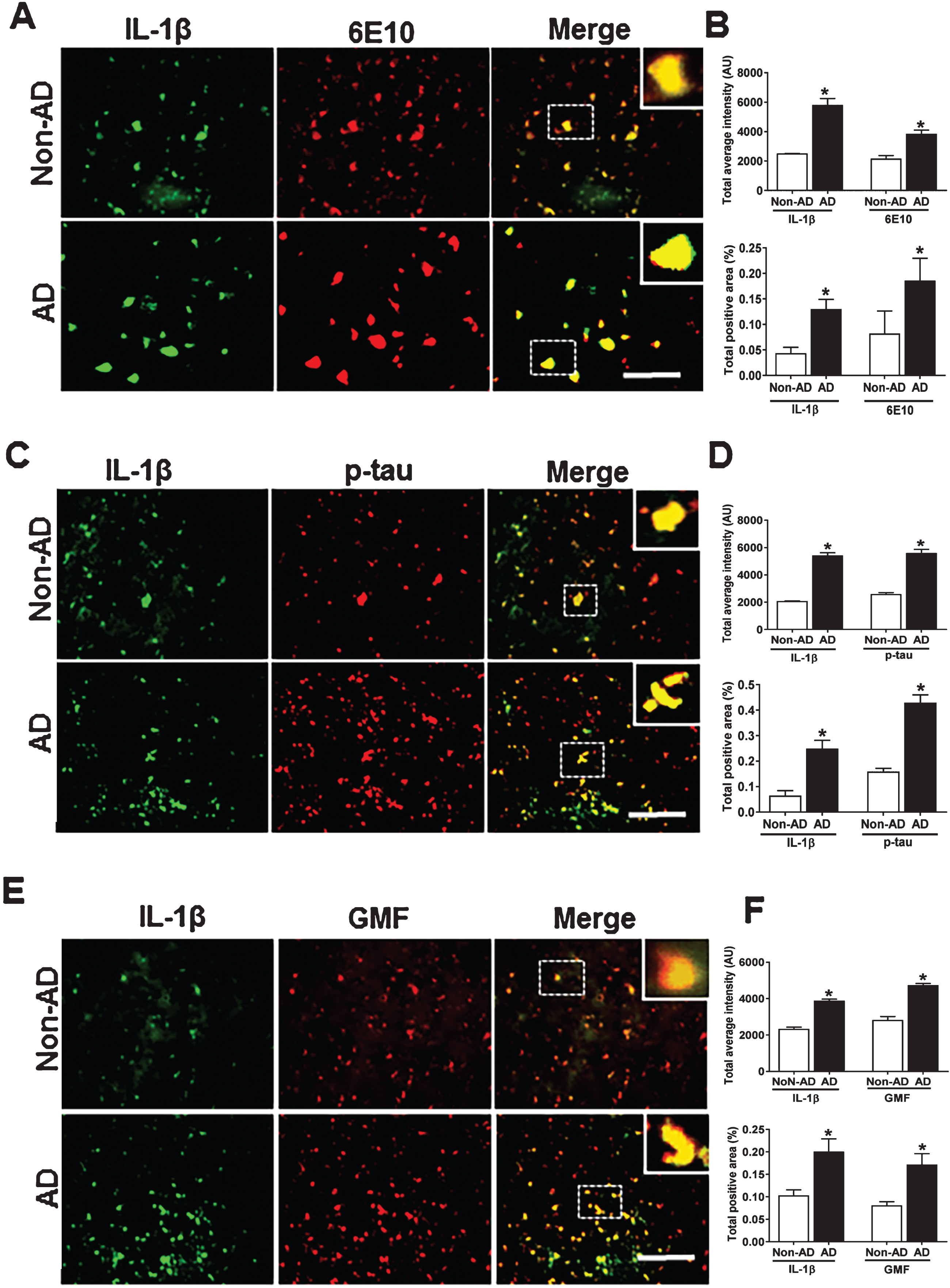
Co-localization of IL-1β with Aβ, p-tau, and GMF in temporal cortex of AD and non-AD brain. A) Sections were immunostained with anti-IL-1β and 6E10, respectively. Representative immunofluorescence staining displays the higher expression and co-localization of IL-1β (green) and 6E10 (red) in AD as compared with non-AD. C) Sections were immunostained with anti-IL-1β and with anti-p-tau, respectively, to check the co-localization of IL-1β with p-tau. Representative immunofluorescence staining displays the higher expression and co-localization of IL-1β (green) and p-tau (red) in AD compared with Non-AD. E) Sections were immunostained with anti-IL-1β and anti-GMF, respectively, to check the co-localization of IL-1β with GMF. Representative immunofluorescence staining displays the higher expression and co-localization of IL-1β (green) and GMF (red) in AD compared with non-AD. Data are expressed as mean±standard error, from each group (n = 3–5). *p < 0.05 versus non-AD was considered statistically significant. Zoom of boxed area shows the enlarged view of co-localization of IL-1β (green) with 6E10 (red), p-tau (red) and GMF (red). Scale bar = 50 μm and AU (Arbitrary Unit). B, D, F) Quantification of IL-1β, 6E10, p-tau, and GMF based on average labeled intensity and labeled positive area in AD and non-AD brains.
IL-18 Co-localizes with Aβ, p-tau, and GMF in temporal cortex of human AD brain
As with IL-1β release, inflammasome–mediated activation of caspase-1 also leads to processing of its downstream pro-inflammatory cytokine IL-18. We next explored the co-localization of IL-18 and GMF in the vicinity of APs and NFTs in temporal cortex of AD brain. Representative microscopic images showed (Fig. 6A, C, E) higher expression and co-localization of IL-18 with GMF and this also co-localized with APs (6E10) and NFTs (p-tau) in AD. Non-AD sections did not show much expression or co-localization of the proteins. Quantification determining total labeled intensity and total positive area is shown in Fig. 6B, D, F.
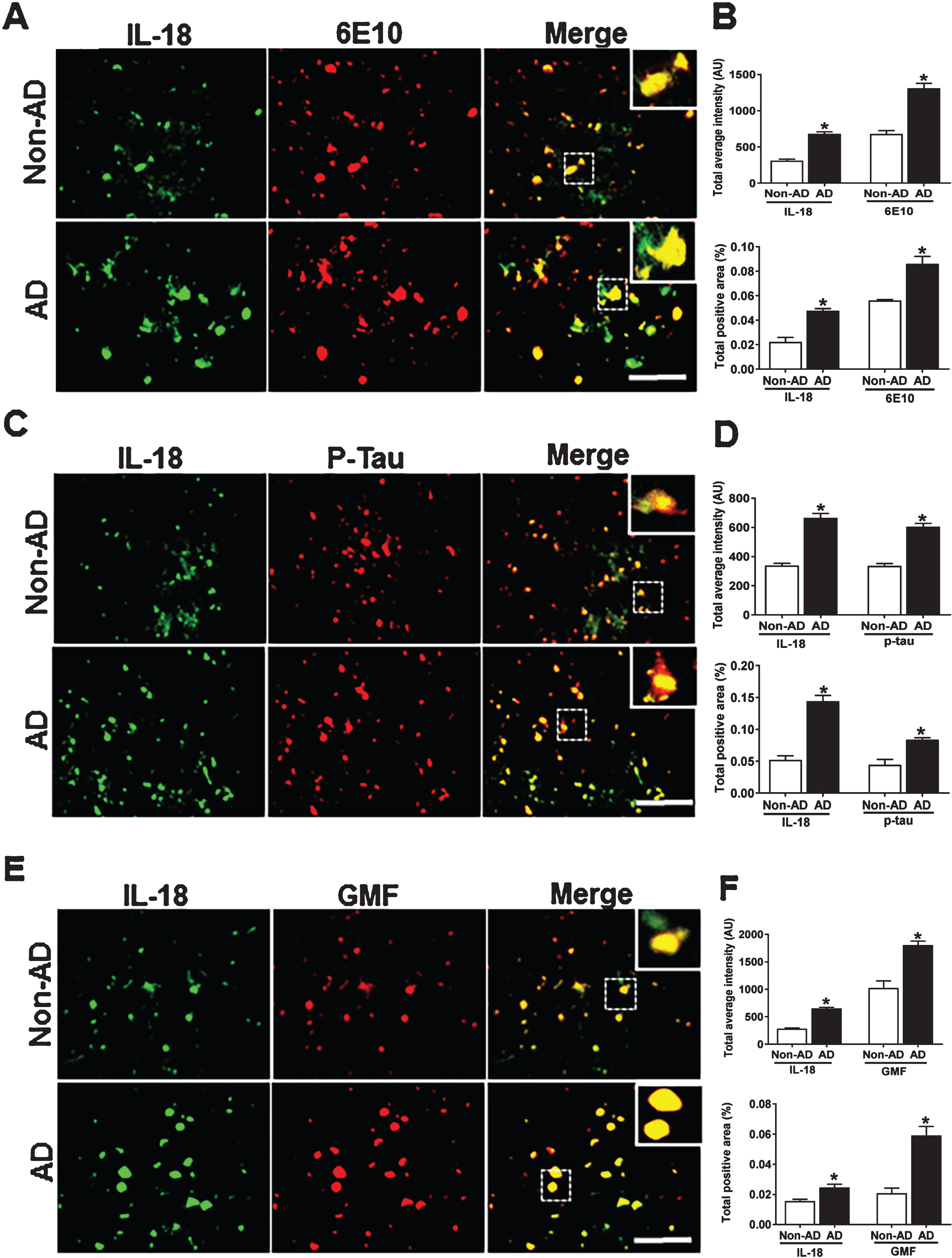
Co-localization of IL-18 with Aβ, p-tau, and GMF in temporal cortex of AD and non-AD brain. A) Sections were immunostained with anti-IL-18 and 6E10 respectively. Representative immunofluorescence staining displays the higher expression and co-localization of IL-18 (green) and 6E10 (red) in AD as compared with non-AD. C) Sections were immunostained with anti-IL-18and with anti-p-tau, respectively, to check the co-localization of IL-18 with p-tau. Representative immunofluorescence staining displays the higher expression and co-localization of IL-18 (green) and p-tau (red) in AD compared with non-AD. E) Sections were immunostained with anti-IL-18 and anti-GMF, respectively, to check the co-localization of IL-18 with GMF. Representative immunofluorescence staining displays the higher expression and co-localization of IL-18 (green) and GMF (red) in AD compared with non-AD. Data were analyzed as mean±standard error, from each group (n = 3–5). *p < 0.05 versus non-AD was considered statistically significant. Zoom of boxed area shows the enlarged view of co-localization of IL-18 (green) with 6E10 (red), p-tau (red), and GMF (red). Scale bar = 50 μm and AU (Arbitrary Unit). B, D, F) Quantification of IL-18, 6E10, p-tau, and GMF based on average labeled intensity and labeled positive area in AD and non-AD brains.
Accumulation of LC3-positive structure in temporal cortex and co-localization with Aβ, p-tau, and GMF in the temporal cortex of human AD brain
Previous studies using electron microscopy [43] have shown abundant presence of autophagic vacuoles (AVs) in AD brains. Using immunolabeling methods AVs in the brain have been shown to be the storehouse of intracellular Aβ since transport of AVs and their maturation to lysosomes is defective in AD [44]. In autophagy, aggregated proteins and other cytoplasmic debris are cleared by autophagosomes; the double membrane vesicles expressing the auto-phagosome marker LC3. To evaluate the auto-phagosome formation and its co-localization with Aβ, p-tau, and GMF, we used immunofluorescence staining. Coronal sections of temporal cortex of human AD and non-AD brains were stained with anti-LC3, anti-Aβ, anti-p-tau and ant-GMF. Representative images in Fig. 7A, C, E showed that there was very limited diffuse staining in non-AD but in AD there was increased cytoplasmic expression of LC3 and more intense staining. LC3 positive cells also co-localized with Aβ and p-tau stained cells in AD. Quantification data showed more of total labeled intensity and total positive area as shown in Fig. 7B, D, F.
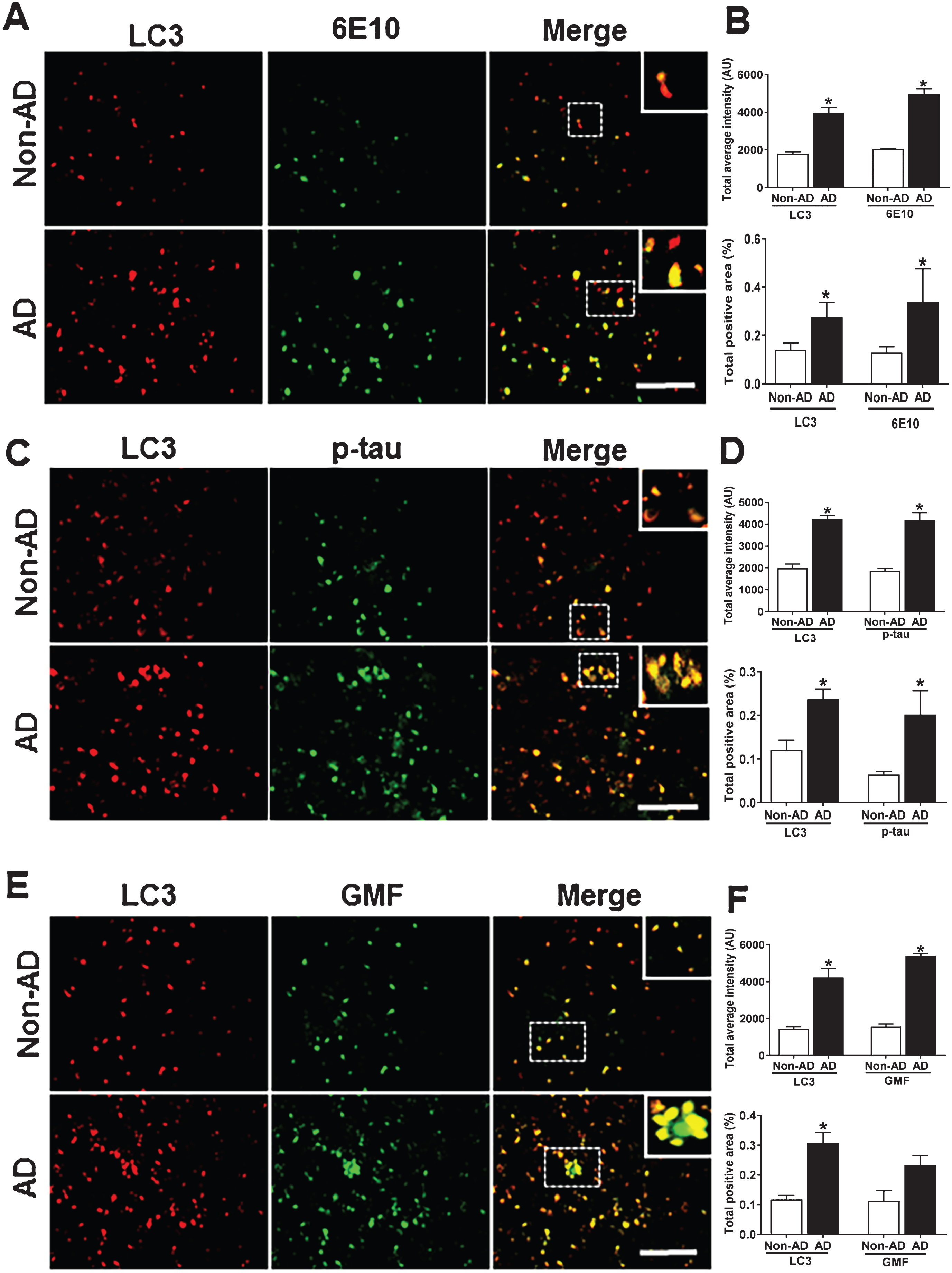
Accumulation of autophagy marker (LC3) and co-localization with Aβ, p-tau, and GMF in the temporal cortex of AD and non-AD brain. A) Sections immunostained with anti-LC3 and E10, respectively, displayed higher accumulation and co-localization of LC3 and 6E10 in AD compared with non-AD. C) Sections immunostained with anti-LC3 and anti-p-tau, respectively, displayed higher accumulation and co-localization of LC3 with p-tau in AD compared with non-AD. E) Sections immunostained with anti-LC3 and anti-GMF, respectively, displayed higher accumulation and co-localization of LC3 with GMF in AD compared with non-AD. Data were analyzed as mean±standard error, from each group (n = 3–5). *p < 0.05 versus non-AD was considered statistically significant. Zoom of boxed area shows the enlarged view of co-localization of LC3 (red) with 6E10 (green), p-tau (green), and GMF (green) Scale bar = 50 μm and AU (Arbitrary Unit). B, D, F) Quantification of LC3, 6E10, p-tau, and GMF based on average labeled intensity and labeled positive area in AD and non-AD brains.
Accumulation of SQSTM1/p62 in temporal cortex and co-localization with Aβ, p-tau, and GMF in the temporal cortex of human AD brain
The polyubiquitin-binding protein SQSTM1/p62 possesses a short LC3 interaction region that helps it to directly bind with LC3 for its degradation. Any defects in autophagy leads to accumulation of SQSTM1/p62 in AD. SQSTM1/p62 is necessary to target protein aggregates for degradation via autophagy [45, 46]. To evaluate whether the protein clearance was defective in AD brain, we used immunofluorescence staining to analyze the co-localization of SQSTM1/p62 with AP, p-tau, and GMF. Coronal sections of temporal cortex of human AD and non-AD brains were stained with anti- SQSTM1/p62 and anti-Aβ, anti-p-tau or GMF. Representative images in (Fig. 8A, C, E) showed that there was very limited diffuse staining in non-AD but in AD there was increased cytoplasmic expression of SQSTM1/p62 and more intense staining. SQSTM1/p62 positive cells also co-localized with 6E10 and p-tau stained cells in AD. Quantification data showed more of total labeled intensity and total positive area as shown in Fig. 8B, D, F. From the earlier sections and this current finding, the present study clearly demonstrates that the inflammation associated with AD is linked to lysosomal dysfunction and impaired autophagy.
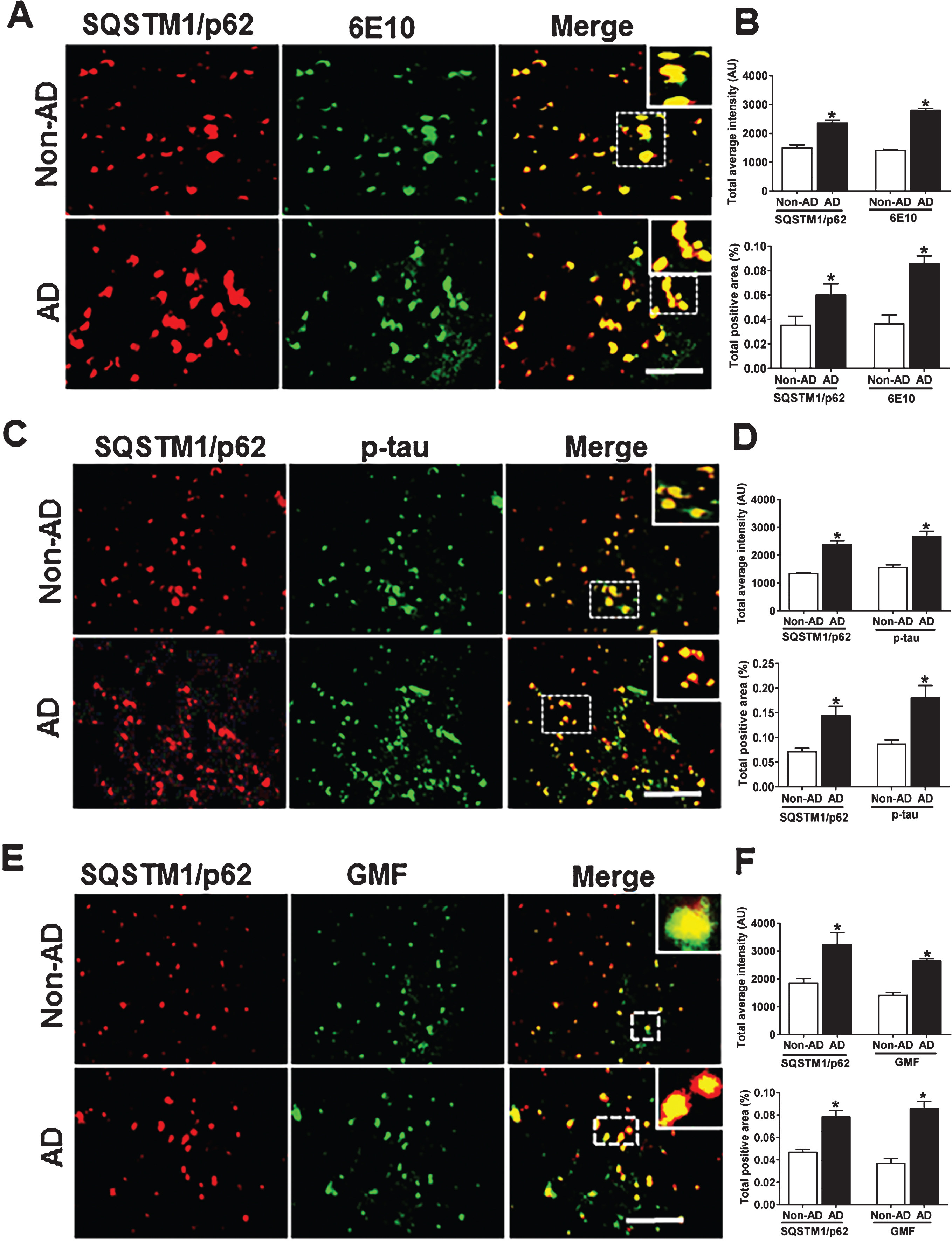
The autophagy marker SQSTM1/p62 accumulates and co-localizes with Aβ marker 6E10, p-tau, and GMF in the temporal cortex of human AD compared to non-AD brain. A) Sections were double immunofluorescence stained with anti-SQSTM1/p62 (red) and 6E10 (green), respectively and displayed higher accumulation and co-localization of SQSTM1/p62 and 6E10 in AD compared with non-AD. C) Sections were immunostained with anti-SQSTM1/p62 and anti-p-tau, respectively, and displayed higher accumulation and co-localization of SQSTM1/p62 with p-tau in AD compared with non-AD. E) Sections were immunostained with anti-SQSTM1/p62 and anti-GMF, respectively, and displayed higher accumulation and co-localization of SQSTM1/p62 with GMF in AD compared with non-AD. Data were analyzed as mean±standard error, from each group (n = 3–5). *p < 0.05 versus non-AD was considered statistically significant. Zoom of boxed area shows the enlarged view of co-localization of SQSTM1/p62 (red) with 6E10 (green), p-tau (green), and GMF (green). Scale bar = 50 μm and AU (Arbitrary Unit). B, D, F) Quantification of SQSTM1/p62, 6E10, p-tau, and GMF based on average labeled intensity and labeled positive area in AD and non-AD brains.
Accumulation of lysosomal marker LAMP1 and its co-localization with Aβ, p-tau and GMF shows probable impairment of lysosomal integrity in the temporal cortex of human AD brain
Accumulation of p-tau as seen in AD could be a direct result of defects in autophagy-lysosomal clearance pathway. The inhibition of autophagy flux can be equated to accumulation of the lysosomal stress-associated protein (LAMP1). We performed immunofluorescence staining of LAMP1 with 6E10, p-tau, and GMF in temporal cortex of human AD brain. Representative images in Fig. 9A showed higher expression and co-localization of LAMP1 with 6E10 in AD compared with non-AD brain. Quantification analysis showed increased total intensity and total positive area (Fig. 9B). Higher expression and co-localization of LAMP1 was also seen with p-tau in AD compared with non-AD brain (Fig. 9C, D). Co-localization of AP and p-tau with LAMP1indicates that these protein aggregates must have been targeted for autophagic clearance. We next performed LAMP1 co-localization with GMF. Co-localization of LAMP1 with GMF additionally shows that GMF must be a regulator of autophagy. Representative image (Fig. 9E) showed extensive co-localization of LAMP1 with GMF in AD brain compared with non-AD brain. Quantification analysis showed total labeled intensity and intensity of positive area (Fig. 9F).
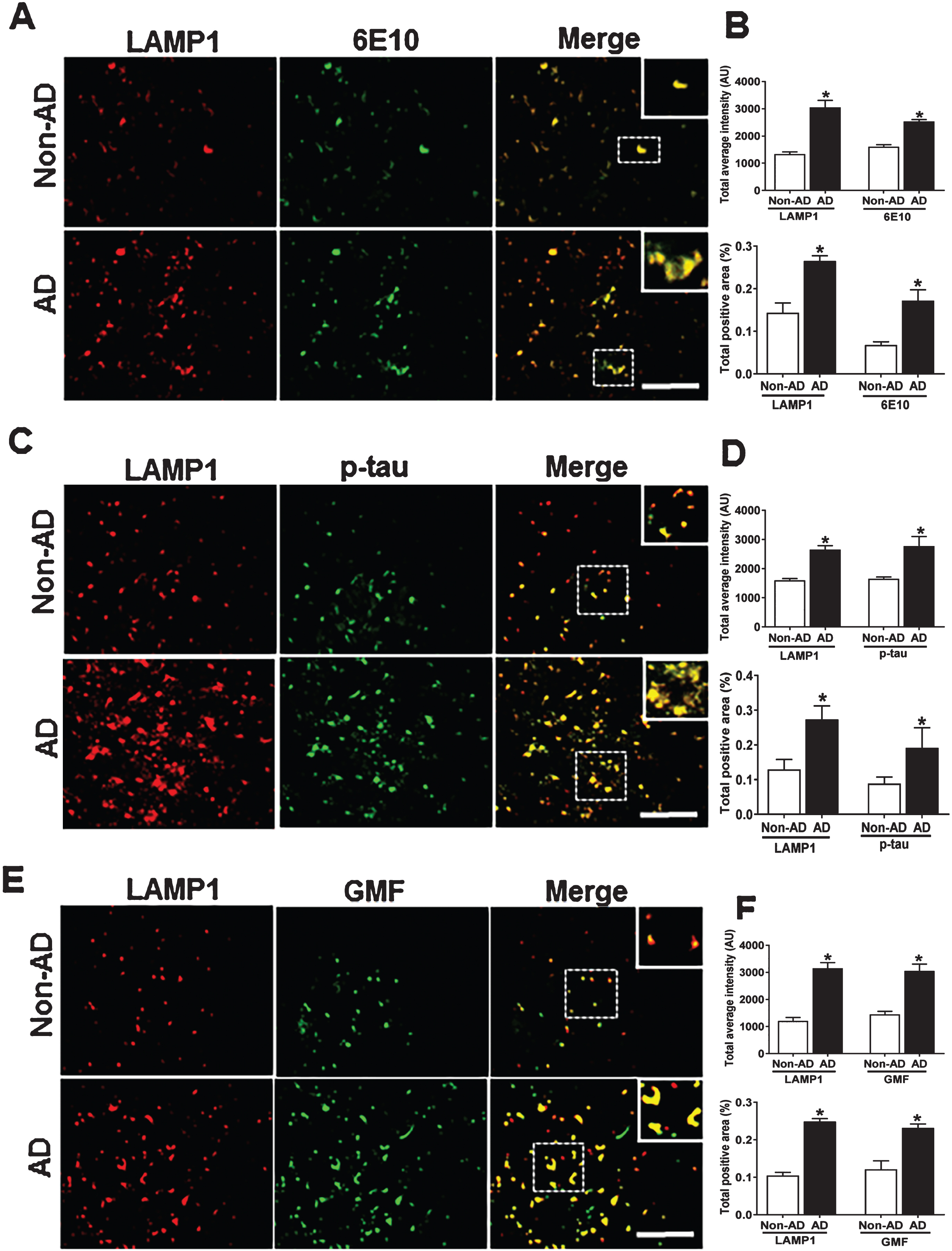
Accumulation of lysosomal marker (LAMP1) and co-localization with Aβ marker 6E10, p-tau, and GMF. Double immunofluorescence staining on postmortem brain temporal cortex of AD and non-AD brain. A) Sections immunostained with anti-LAMP1 and 6E10, respectively, displayed higher accumulation and co-localization of LAMP1 and 6E10 in AD compared with non-AD. C) Sections immunostained with anti-LAMP1 and anti-p-tau respectively, displayed higher accumulation and co-localization of LAMP1 with p-tau in AD compared with non-AD. E) Sections immunostained with anti-LAMP1 and anti-GMF respectively, displayed higher accumulation and co-localization of LAMP1 with GMF in AD compared with non-AD Data were analyzed as mean±standard error, from each group (n = 3–5). *p < 0.05 versus non-AD was considered statistically significant. Zoom of boxed area shows the enlarged view of co-localization of LAMP1 (red) with 6E10 (green), p-tau (green), and GMF (green). Scale bar = 50 μm and AU (Arbitrary Unit). B, D, F) Quantification of LAMP1, 6E10, p-tau, and GMF based on average labeled intensity and labeled positive area in AD and non-AD brains.
DISCUSSION
Using immunofluorescence staining of human postmortem brain tissue, we show that in AD, there is an abnormal accumulation of APs containing amyloid protein and NFTs with hyperphosphorylated p-tau both of which co-localized with pro-inflammatory pathway mediators; the NLRP3 inflammasome components and GMF. Also, in AD hyperphosphorylated tau and APs co-localized with the autophagosomal markers SQSTM1/p62 and LC3-containing structures and lysosomal marker LAMP1. Our current study demonstrates that in AD, increased pro-inflammatory GMF is found associated with the NLRP3 inflammasome protein and caspase-1, IL-1β, and IL-18. Similarly, increased GMF is also associated with markers of autophagy and the lysosomes. In this scenario of increased levels of GMF and inflammasome components in AD, it would appear that an increase in GMF would lead to increased ROS which could act as a trigger to ROS-dependent activation of the NLRP3 inflammasome resulting in increased oxidative stress mediated lysosomal damage and abnormalities in autophagy. We propose that the inflammasome pathway triggering autophagy and lysosomal dysfunction and aggregation of Aβ are amplified and regulated by GMF in human postmortem AD brain. Previous studies from our laboratory have reported the involvement of GMF in the pathogenesis of neurodegenerative disease [6, 47]. However, the precise relationship and mechanism of GMF in aggregation of Aβ and phosphorylation of tau in AD progression is not yet known and is presently under study. For this purpose, we used coronal sections of human postmortem brain tissue to demonstrate the expression and co-localization of GMF at the sites of Aβ aggregation and hyperphosphorylated tau through immunofluorescence staining. Our results demonstrate massive expression and co-localization of GMF with Aβ marker (6E10) and p-tau in AD brain compared with aged matched non-AD, which reflects the role of GMF in pathogenesis of AD.
Several factors including ATP, bacterial toxin, and various crystals [48 –50] are known to activate NLRP3 inflammasome. The present study using immunofluorescence staining of human AD brain section seems to confirm the in vitro finding that Aβ activates the NLRP3 inflammasome. We report that aggregated Aβ, which form insoluble fibrils in the brain of patient with AD probably associates with and activates the NLRP3 inflammasome. It is believed that NLRP3 inflammasome may function as a general sensor for the recognition of peptide or protein aggregates, involved in inducing inflammatory response and pathogenesis of AD. We found higher expression and co-localization of NLRP3 near Aβ and p-tau in temporal cortex. To check the potential role of NLRP3 inflammasome in the maturation and release of pro-inflammatory cytokines, we performed co-localization studies between NLRP3 inflammasome and GMF and the released products of inflammasome activation, IL-1β and IL-18, using immunofluorescence staining. Our results indicate that GMF and the inflammasome component NLRP3 along with the pro-inflammatory cytokines (IL-β, IL-18) are closely associated in the vicinity of Aβ and tau and probably upregulate aggregation of Aβ and phosphorylation of tau protein. We believe that upregulation of NLRP3 and GMF may, to begin with, lead to the initiation of autophagy in order to limit the lethality of inflammasome activation. However, with prolonged accumulation of aggregated APs and hyperphosphorylated tau, increased inflammasome activation under the regulation of GMF could damageautophagy.
Autophagy is a very important physiological and essential process for protein homeostasis and cell death. The initiation of autophagy promotes protein degradation and increase in the amino acid pool which supports energy production and allows for necessary protein synthesis. In the absence of basal autophagy in the CNS, neurodegeneration results, suggesting an important role of autophagy in continuous removal of protein aggregates responsible for the neurodegeneration. Previous studies have shown that dysfunction in autophagy is associated with several neurological disorders, including AD, PD, and Huntington disease [51, 52]. In the present study on postmortem brain of human AD patients, we show the possible relation between autophagy and the upregulation of GMF and NLRP3 in response to Aβ aggregation. We found that AD patient brain showed abnormal accumulation of autophagy markers SQSTM1/p62 and LC3, and both these markers co-localized with Aβ marker (6E10) and hyperphosphorylated tau. LAMP1 is an important part of lysosomal membrane, playing a very important role in facilitating autophagy-vacuole and lysosomal fusion. The lysosome is an important organelle for degradation of metabolic products through activation of autophagy, bringing about degradation of engulfed proteins [53, 54]. In a recent study [55], using macrophages from the lysosomal storage disorder Gaucher’s disease showed that disruption in lysosomal storage and impaired autophagy lead to inflammasome activation and that the elevated level of the autophagic adaptor SQSTM1/p62 prevented delivery of the inflammsomes to the autophagososmes. In the present immunohistochemical studies on human postmortem brain of AD patients, we found that besides elevated SQSTM1/p62, higher expression of LAMP1 in AD brain compared with age matched non-AD control, suggesting a defective lysosomal clearance and impaired autophagy which is also in line with previous reports in AD brain [56 –59]. Induction of autophagy could be beneficial for the treatment of neurodegenerative disorders, and increased autophagy has been found to ameliorate pathology in various diseases by promoting clearance of aggregated and hyperphosphorylated proteins inside the cytoplasm [35, 60]. However, this is the first study that demonstrates a possible role for GMF in the autophagy-lysosomal clearance pathway of protein aggregates Aβ and p-tau associated with AD.
Conclusions
Our findings provide support for a dysfunction in the autophagosomal-lysosomal pathway triggered and regulated by the NLRP3 inflammasome and GMF in human AD brain that results in aggregated Aβ and tauopathies. The autophagosomal-lysosomal pathway for clearance of Aβ and p-tau is impaired in AD. Our study highlights the fact that any sort of therapeutic strategy, which inhibits the upregulation of GMF and NLRP3-Caspase-1 pathway and targets the autophagosomal-lysosomal pathway, may be the potential way forward to combat neurodegeneration.