
Abstract
Human ATP-binding cassette (ABC) transporters mediate a critical function in the cell, namely the transport of molecules across lipid membranes. Associated to their ubiquitous tissue distribution, they are key players in cellular homeostasis but also potential causative or contributing factors for many pathologies, including Alzheimer’s disease (AD). In the central nervous system (CNS), numerous ABC transporters are present throughout the brain parenchyma and especially at the blood-brain barrier (BBB). AD is a neurodegenerative disorder mainly characterized by extracellular deposition of amyloid-β (Aβ) peptides and intracellular accumulation of hyperphosphorylated forms of tau protein. Besides being degraded via proteolytic and phagocytic processes mediated by brain parenchymal cells, a major mechanism for eliminating cerebral Aβ is through its transport across the BBB into the peripheral blood. In fact, many AD cases are associated with impaired Aβ clearance. Consistently, several studies have recently uncovered important roles for ABC transporters in AD pathophysiology. Hence, this review focuses on the relevance of ABC transporters in CNS homeostasis by highlighting AD as a strong example of the deleterious consequences that might result from the former’s altered expression and/or activity in the brain. The potentiality of human ABC transporters as novel pharmacological targets for both the diagnosis and therapeutics of AD is emphasized.
Keywords
INTRODUCTION
In the last decades, the physiological functions and tissue distribution of ATP-binding cassette (ABC) transporters have been extensively characterized, allowing for their association to several human diseases. ABC transporters use the energy released from adenosine triphosphate (ATP) hydrolysis to translocate molecules across lipid membranes of cells and/or intracellular organelles [1, 2]. Since the discovery of ABCB1, in 1976, as the first member of this transporter superfamily [3], many other related transporters have been identified; currently, almost 50 ABC transporters divided into seven subfamilies (ABCA–ABCG) are known to be expressed in the human genome [4]. Interestingly, ABC transporters have been shown to play important roles in Alzheimer’s disease (AD), a highly prevalent neurodegenerative disorder that starts with memory loss and progresses to severe cognitive decline, being the most common cause of age-related dementia. Among other alterations in the central nervous system (CNS), the two main AD pathological hallmarks include extensive accumulation of amyloid-β (Aβ), a peptide resulting from amyloid-β protein precursor (AβPP) processing, in the brain parenchyma and intraneuronal deposition of hyperphosphorylated tau protein [5, 6]. ABC transporters have been implicated in the control of cerebral Aβ levels given their ability to export Aβ across the blood-brain barrier (BBB) into the peripheral blood, which is complemented by their participation in other Aβ clearance mechanisms (e.g., proteolytic degradation and phagocytosis). Moreover, some molecules related to neurodegeneration (e.g., cholesterol) are substrates for ABC transporters and the regulation of their cerebral levels might influence AβPP metabolism [5, 7]. The first ABC transporter recognized for its role in AD was ABCB1, in 2001 [8]; since then, a total of nine ABC transporters have been associated with AD, of which ABCA5 was only identified in 2015 [9].
This review will focus on the roles of human ABC transporters in AD pathogenesis. Initial insights on the basic structure and functional mechanism of ABC transporters will be discussed, as well as on members expressed in the CNS and implicated in neurological disorders. The involvement and physiological relevance of ABCA1, ABCA2, ABCA5, ABCA7, ABCB1, ABCC1, ABCG1, ABCG2, and ABCG4 in AD will also be described. Lastly, the association of ABC transporters with pharmacological therapeutic efficacy and their application as therapeutic targets in AD will be considered.
ABC TRANSPORTERS
The ABC transporter superfamily, a major group of energy-dependent transmembrane proteins in both prokaryotes and eukaryotes, mediates the active transport of a wide spectrum of molecules, from simple sugars, fatty acids, nucleosides, and amino acids to complex polysaccharides, lipids, oligonucleotides, and proteins [2]. ABC transporters act by coupling the passage of substrates across lipid membranes to the hydrolysis of the phosphate group bond located between γ- and β-phosphates of an ATP molecule [1]. The high free energy released when ATP is converted to adenosine diphosphate (ADP) and orthophosphate (Pi) is applied to remove the carried molecules from, or to accumulate them in, intracellular compartments against concentration gradients [1, 10]. ABC transporters are located in the plasma membrane of prokaryotic and eukaryotic cells, as well as in the membrane of intracellular organelles in eukaryotes (e.g., lysosome, mitochondria, endoplasmic reticulum, and peroxisome) [1, 4]. According to the direction of the transport movement, ABC transporters act as importers or exporters. The ABC importers transfer extracellular substrates into the cytoplasm of prokaryotic cells, while the ABC exporters found in both prokaryotes and eukaryotes typically move cytoplasmic substrates out of cells and/or into cytosolic organelles [1, 10]. Some of the ABC transporters, however, carry out other functions, such as DNA repair, chromatin organization, and regulation of K+ channels [10].
Structural diversity
Most ABC transporters share a typical architecture comprising four functional units: 1) two nucleotide-binding domains (NBDs), which bind ATP molecules and induce their hydrolysis to provide the driving force required for the transport; and 2) two transmembrane domains (TMDs), which create a pathway for substrate translocation across lipid bilayers. At the sequence level, NBDs are recognized by seven highly conserved motifs with characteristic amino acid residues (A-loop, Walker A and B motifs, SALD motif, H-loop, Q-loop, and ABC signature motif), comprising the characteristics that distinguish ABC transporters from other ATP-binding proteins. While NBDs are conserved in sequence and structure between all ABC transporters, TMDs are structurally more diverse, containing distinct sets of membrane-spanning α-helices among different ABC transporters. Despite lacking significant sequence similarities, TMDs share a specific folding within certain groups of ABC transporters, based on which four classes can be recognized: 1) type I, 2) type II, and 3) type III ABC importers involved in influx activities; and 4) ABC exporters exhibiting efflux functions [1, 2, 10].
Prokaryotic ABC transporters are typically composed of four subunits expressed as individual proteins. In eukaryotes, however, the four domains composing ABC exporters are usually encoded as fusion proteins that can be organized as a single polypeptide comprising all subunits (full transporters) or as a dimer of two polypeptide chains, each comprising one TMD and one NBD (half transporters) [1, 11].
General mechanism of action
Under physiological conditions, ABC transporters adopt two conformations. During each catalytic cycle, TMDs alternate between inward- and outward-facing positions so that the substrate-binding site is initially accessible from one side of the lipid bilayer for substrate loading and, then, is oriented toward the opposite side for substrate delivery. The “ATP switch model” proposes that this conformational switching is controlled by the formation and dissociation of a NBD dimer that, in turn, is coordinated by ATP binding and hydrolysis. According to this model, the binding of a substrate to TMDs increases the affinity of NBDs for two ATP molecules. The chemical energy associated with NBD dimerization after ATP binding is converted into elastic conformational energy that is transferred to TMDs, allowing them to switch between the two main positions for substrate transport across the membrane. Subsequent ATP hydrolysis with release of Pi and ADP induces destabilization and dissociation of the NBD dimer, permitting TMDs to return to a basal configuration, ready for a new beginning of the transport cycle[2, 10, 12].
Human ABC transporters
In eukaryotes, ABC transporters are categorized into eight subfamilies (ABCA–ABCH). Among these, ABCE and ABCF transporters are soluble molecules lacking TMDs that have no efflux function and the ABCH transporter subfamily is not present in mammals, fungi or plants [11].
Currently, 49 genes encoding for ABC transporters are known to integrate the human genome [4], which have been mapped to almost all chromosomes [13]. Based on sequence homology, domain organization and functional similarity, human ABC transporters are arranged in seven subfamilies (ABCA–ABCG). In the ABCA transporter subfamily, all 12 members are full transporters that can be subdivided into the ABCA1-like (ABCA1–ABCA4, ABCA7, ABCA12, and ABCA13) and the ABCA5-like (ABCA5, ABCA6, and ABCA8–ABCA10) subgroups. The ABCB transporter subfamily contains 11 members, including full transporters (ABCB1, ABCB4, ABCB5, and ABCB11) and half transporters (ABCB2, ABCB3, and ABCB6–ABCB10). In the ABCC transporter subfamily, only nine full transporters out of 12 members are associated with export activities (ABCC1–ABCC6 and ABCC10–ABCC12) [4, 13]; although another locus was described, ABCC13 seems to be a pseudogene incapable of codifying a functional protein [14]. Both ABCD and ABCG transporter subfamilies consist of half transporters, comprising four (ABCD1–ABCD4) and five (ABCG1, ABCG2, ABCG4, ABCG5, and ABCG8) members, respectively. In contrast, the members of ABCE and ABCF transporter subfamilies (ABCE1 and ABCF1–ABCF3, respectively) are not associated with transport activities [4, 13].
Although they are ubiquitously distributed, playing pivotal roles in many tissues/organs, human ABC transporters are highly expressed at: 1) physiological barriers (e.g., blood-placental barrier, blood-testis barrier, blood-spinal cord barrier, blood-cerebrospinal fluid (CSF) barrier, and BBB); 2) tissues involved in excretory functions/metabolism (e.g., liver and kidney); and 3) absorptive tissues (e.g., gut and lung) [15].
The following section will focus on human ABC transporters that are expressed in the CNS, due to increasing evidence supporting their association with several neurological pathologies, including AD.
ABC TRANSPORTERS IN THE CNS
The maintenance of homeostasis in the cerebral microenvironment is crucial for the proper functioning of the CNS. This physiological equilibrium is strongly regulated by CSF circulation and two permeability barriers, the first present in the brain capillary endothelium (BBB) and the second in the choroid plexus epithelium (blood-CSF barrier). Together, these are the main interfaces between the CNS and the systemic circulation, forming the first line of defense for the brain against xenobiotics (e.g., drugs and toxins) and metabolites, in addition to controlling the tissue distribution of nutrients and other endogenous molecules.
At the molecular level, different factors contribute to the highly specialized structure of the BBB and the blood-CSF barrier, including tight junctional attachments between adjacent brain capillary endothelial cells (BCECs) and contiguous choroid plexus epithelial cells (CPECs), limited vesicular trafficking as well as the presence of influx and efflux transport systems. Several ABC transporters complement the passive diffusion barrier created by intercellular tight junctions, not only by mediating the active removal of metabolites from the brain interstitial fluid (ISF) and CSF into the peripheral blood, but also by selectively restricting the passage of potentially toxic circulating compounds into the CNS. Thus, ABC transporters exert essential detoxifying and neuroprotective roles in the brain [15–17].
Similarly, the expression of ABC transporters in neurons and glial cells provides a “second” selective permeability barrier within the brain parenchyma to limit the entry of xenobiotics into the CNS as well as to regulate the intracellular concentration of waste products [18].
Expression of ABC transporters in the CNS
Current knowledge on the expression of ABC transporters (considering primarily those expressed in the human genome) in distinct cell types composing the BBB, blood-CSF barrier, and brain parenchyma in humans and other mammalian species is summarized in Table 1. Given that the primary function is the ATP-mediated transport of substrates across lipid membranes, ABC transporters that do not exhibit an efflux activity in eukaryotic cells were excluded from the analysis.
CNS expression of ABC transporters in humans and other mammals
Obs., The expression of ABC efflux transporters at the mRNA and/or protein levels is described. BCEC, brain capillary endothelial cell; CP, choroid plexus; CPEC, choroid plexus epithelial cell; VEC, ventricular ependymal cell.
Association of ABC transporters with neurological diseases
ABC transporters are critical players for cellular homeostasis maintenance. Loss-of-function mutations in their encoding genes have been causatively linked to genetic disorders that manifest in diverse tissues/organ systems, namely: Tangier disease (ABCA1; cardiovascular system); Stargardt disease (ABCA4; eye); progressive familial intrahepatic cholestasis (ABCB4 and ABCB11; liver); cystic fibrosis (ABCC7; lung and gut); persistent hyperinsulinemic hypoglycemia of infancy (ABCC8; pancreas); X-linked adrenoleukodystrophy (ABCD1; adrenal glands, CNS and testes); and sitosterolemia (ABCG5 and ABCG8; intestine and liver) [11, 13]. Considering that human ABC transporters have been consistently found at the BBB, blood-CSF barrier and/or brain parenchymal cells (Table 1), it is not surprising that they are also associated with CNS disorders.
ABCA and ABCG transporter subfamilies
ABCA transporters mediate the export of cholesterol and phospholipids across cell membranes through interaction with lipid-free or lipid-poor apolipoproteins in the brain, predominantly apolipoprotein (apo)-E and apoA-I, generating nascent high-density lipoproteins (HDL). ABCG transporters, especially ABCG1 and ABCG4, also stimulate the transport of sterols, phospholipids and retinoids to lipid acceptor proteins, preferentially apoE discs partially lipidated by ABCA1 and HDL [19]. In the CNS, cholesterol is essential for brain development and is mostly synthesized in situ, where it is concentrated in the myelin sheaths as well as in the plasma membrane of astroglial and neuronal cells [20]. ABCA transporters, in particular ABCA1, are major regulators of brain cholesterol homeostasis by promoting its redistribution between brain parenchymal cells [19]. Cholesterol synthesized mostly by astrocytes is released via ABCA1 and further captured by poorly lipidated apoE and apoA-I that deliver it mainly to neurons, which require large amounts of this sterol for key cellular events such as synaptogenesis [21–23]. Similarly, ABCG1 and ABCG4 can contribute to the recycling of cholesterol and related biosynthetic precursors from astrocytes to neuronal cells through their efflux to HDL particles [24]. Furthermore, ABCA1 facilitates the removal of cholesterol from BCECs [25], pericytes [26], and neurons [22, 27], supported by microglial ABCA7 [28] and by both neuronal ABCA5 [9] and ABCG1 [27], which might be important to prevent excessive intracellular accumulation and subsequent cholesterol-induced toxicity.
Given that the maintenance of constant cholesterol levels is mandatory for the proper functioning of the CNS, research data indicates that dysfunction of ABCA and ABCG transporters is associated with neurodegenerative disorders. A significant contribution of these transporters to AD pathophysiology has been proposed based on functional and genetic association studies, with evidence suggesting either a putative preventive function (e.g., ABCA1, ABCA5, ABCA7, and ABCG4) or a predisposing role (e.g., ABCA2), and this is correlated with cellular cholesterol trafficking in some cases [5, 7]. Additional linkage of ABCA transporters to neurodegeneration can be found, for example, in Niemann-Pick type C disease and multiple system atrophy. Impaired ABCA1 expression and reduced apoA-I-mediated efflux of intracellular cholesterol occur in Niemann-Pick type C disease, a severe lipidosis characterized by lysosomal cholesterol storage [29]. In earlier stages of multiple system atrophy, ABCA8 upregulation induces sphingomyelin synthesis, resulting in dysregulated lipid homeostasis that likely contributes to abnormal α-synuclein production/aggregation and myelin dysfunction, two characteristic features of this α-synucleinopathy [30].
ABCD transporter subfamily
Among ABCD transporters expressed in the CNS, ABCD1 is particularly important in neurons and glial cells, where it facilitates the translocation of fatty acids across the peroxisomal membrane for enzymatic degradation within peroxisomes, a function that might be complemented by ABCD2 [31]. Several genetic variations in ABCD1 were identified as the underlying cause of the neurodegenerative demyelinating disease X-linked adrenoleukodystrophy, leading to accumulation of very-long-chain fatty acids in myelin and consequent destabilization of myelin sheaths [31, 32].
ABCB1, ABCG2, and ABCC transporter subfamily
ABCB1 is the main gatekeeper on the luminal side of the BBB, acting as an efflux pump that prevents xenobiotics (e.g., drugs) from entering the brain and also removes endogenous molecules with the potential to damage brain cells (e.g., Aβ peptides). Interestingly, ABCG2 has a substrate multi-specificity that overlaps with ABCB1, suggesting that they act synergistically at the BBB to confer neuroprotection and pharmacoresistance [15]. Likewise, ABCC efflux transporters also contribute to multidrug resistance, in addition to mediating the export of organic anions and drug conjugates. Of these, ABCC1 is a landmark of the blood-CSF barrier and, allied to its presence in the luminal side of the BBB, is a critical player for the protection of brain cells against harmful xenobiotics [33]. Moreover, astroglial ABCC1 is required for brain tissue defense during oxidative stress by supplying the antioxidants glutathione and glutathione disulfide to other cells in the CNS [34].
By preventing the access of therapeutic agents to brain parenchymal cell targets through the main interfaces between the peripheral blood and the CNS, ABCB1, ABCG2, and ABCC efflux transporters are implicated in the progression of pathological states in neurological disorders. For instance, in brain tumors, diseased cells are able to increase the expression of ABC transporters involved in the multidrug resistance phenotype (e.g., ABCB1, ABCC1, ABCC3–ABCC5, and ABCG2), forming the so-called brain-tumor cell barrier [33, 35]. The achievement of suboptimal intracellular drug concentrations due to upregulation of ABC drug efflux transporters is also one of the contributory mechanisms to therapeutic failure in human immunodeficiency virus encephalitis (e.g., ABCB1, ABCC1, ABCC2, ABCC4, ABCC5, ABCC11, and ABCG2) [33, 36] and epilepsy (e.g., ABCB1, ABCC1, ABCC2, ABCC5, and ABCG2) [33, 37]. On the other hand, since neurotoxic Aβ peptides are endogenous substrates for ABCB1, ABCC1, and ABCG2, and given the role of ABCC1 in the glutathione-dependent antioxidant system, research data points to a protective function of these transporters in brain detoxification and against oxidative stress in AD [5, 7].
As described above, increasing evidence suggests a correlation between the dysregulation of human ABC transporters and the pathogenesis of neurological diseases. Among these, AD clearly stands out, since multiple ABC transporters have been identified as key players in this disorder. Thus, current knowledge regarding the association of ABC transporters with AD development will be discussed below.
ABC TRANSPORTERS AND THEIR FUNCTION IN AD PATHOGENESIS
AD is a progressive neurodegenerative disorder typically characterized by three major alterations in the CNS: 1) extracellular accumulation of neurotoxic fibrillary Aβ peptides in the brain parenchyma (amyloid plaques), which interferes with synaptic communication; 2) deposition of hyperphosphorylated forms of microtubule-associated tau protein inside neurons (neurofibrillary tangles), which blocks the transport of molecules required for cell survival; and 3) extensive neuronal loss and atrophy of the hippocampus and cerebral cortex [5, 6].
Generation of Aβ peptides from AβPP processing is one of the critical factors for AD development. Newly synthesized AβPP suffers proteolysis via two main pathways, a process that can be modulated by AβPP phosphorylation or by phosphorylation-dependent events [38–44]. In the non-amyloidogenic pathway, most AβPP in the plasma membrane is progressively cleaved by α- and γ-secretase inside the Aβ sequence, inhibiting Aβ production. In the amyloidogenic pathway, after endocytosis of a small amount of cell surface-attached AβPP, β- andγ-secretase sequentially cleave AβPP in the endosomal membrane to generate different Aβ species that are secreted into the extracellular medium [45], ranging from 37 to 43 amino acids in length [46, 47]. Among these, the most common Aβ species are Aβ40 and Aβ42: Aβ42 is more hydrophobic, less soluble, and more neurotoxic than the other species, providing the core for Aβ oligomerization; in contrast, Aβ40 promotes the viability of neurons by protecting them against the oxidative damage induced by Aβ42 [6].
Sporadic (late-onset) AD accounts for more than 95% of all cases and the ɛ4 allele of APOE gene is the greatest genetic risk factor, wherein two copies of this allele increase the risk of AD up to 15-fold relative to the most common ɛ3 allele. Three genetic variants of APOE encode apoE2, apoE3, and apoE4, of which apoE2 and apoE3 bind to Aβ and stimulate its removal from the brain whereas apoE4 is less efficient at promoting Aβ clearance. Non-sporadic (early-onset) AD is intrinsically associated with autosomal dominant mutations that typically occur in AβPP, PSEN1, and PSEN2, increasing the overall production of Aβ or the ratio of Aβ42 relative to Aβ40 [6, 48].
Two main mechanisms leading to Aβ accumulation in AD are: 1) excessive Aβ production in the brain and peripheral tissues, as a result of mutations in AβPP or in genes encoding for AβPP processing enzymes; and 2) impaired clearance of cerebral Aβ, due to alterations in Aβ solubility/aggregation properties and/or in Aβ elimination processes. Alternatively, an increase in Aβ influx from the systemic circulation into the CNS mediated by receptor for advanced glycation end products (RAGE) at the BBB might also contribute to Aβ deposition. Among the mechanisms involved in the elimination of cerebral Aβ are: 1) Aβ degradation via protease (e.g., neprilysin and insulin-degrading enzyme, IDE) activity; 2) Aβ phagocytosis by astrocytes and microglia; 3) Aβ clearance through the brain ISF/CSF bulk flow into the bloodstream; 4) Aβ removal by perivascular lymphatic drainage; 5) Aβ transcytosis across the BBB via low-density lipoprotein (LDL)-related protein 1 (LRP1); and 6) Aβ efflux to the peripheral blood driven by ABC transporters [5, 49].
Through the past years, ABC transporters were shown to be involved in AD pathophysiology, mainly by preventing the accumulation of amyloid plaques in the CNS, either directly via active transport of Aβ across the BBB or indirectly through modulation of Aβ production and/or degradation in the brain. Additionally, several single nucleotide polymorphisms (SNPs) in ABC transporter-encoding genes have also been associated to AD predisposition. The following discussion will focus on the linkage of ABC transporters to AD based on genomic and functional studies; these include, by order of relevance, ABCA7, ABCA1, ABCB1, ABCG2, ABCA2, ABCC1, ABCG1, ABCG4, and ABCA5.
ABCA7
Although the APOE ɛ4-carrier status represents the most significant genetic risk factor for sporadic AD, large-scale genome-wide association studies have recently identified novel candidate genes that implicate cholesterol metabolism, the immune system, and synaptic dysfunction in AD etiology [50]. In line with this, a strong genetic association between ABCA7 and AD has been proposed based on the observation that multiple SNPs increase AD risk. Among these, the ABCA7 SNP rs3764650 is a common susceptibility variant for sporadic AD [51, 52], wherein intron13 carrying the major T allele acts as a promoter capable of enhancing ABCA7 expression [53]. Carriers of the minor (risk) G allele of this SNP present a decrease in ABCA7 expression, which potentiates the risk for the onset of AD [54] and has been correlated with amyloid plaque burden [55]. Similarly, a strong linkage of other ABCA7 SNPs (e.g., rs3752240 and rs4147912) to Aβ deposition in the brain has also been reported [56]. Moreover, the ABCA7 SNP rs115550680 has a risk effect size comparable to that of the APOE ɛ4-determining SNP (rs429358) [57] and several ABCA7 loss-of-function variants (e.g., c.441612T>G, p.R1489X, and E1679X) are significantly enriched in AD cases [58, 59], confirming that ABCA7 is a major genetic risk locus for sporadic AD. Of note, interactions between two ABCA7 SNPs (rs3764650 and rs3752246) and the APOE ɛ4 allele were shown to influence cognitive function in AD. In the absence of the AD-risk G allele of these ABCA7 SNPs, each APOE ɛ4 allele is linked to learning and memory decline; conversely, in carriers of the ABCA7 minor allele, each additional APOE ɛ4 allele is associated with better cognitive indicators. Thus, it is tempting to speculate that the presence of the minor G allele of these ABCA7 SNPs might promote memory preservation in individuals that, despite carrying the APOE ɛ4 risk allele, do not develop AD [50].
Regarding its functional role in AD development, research indicates that ABCA7 modulates AβPP processing (Fig. 1). In accordance with this, ABCA7 expression inhibits Aβ production in cells co-expressing AβPP [53, 60], whereas its suppression has the opposite effect [61, 62]. Different mechanisms have been proposed to explain this protective action of ABCA7 in AD. A study described that the reduction in Aβ levels induced by ABCA7 is caused by perinuclear retention of AβPP, which alters its transit through the secretory pathway where amyloidogenic proteolysis occurs [60]. Other authors reported an accelerated endocytosis of cell surface-attached AβPP in Abca7-knockout cells, which is mechanistically consistent with increased Aβ production in the secretory pathway [61]. Furthermore, it was recently demonstrated that the minor G allele of ABCA7 SNP rs3752246 is associated with higher levels of secreted Aβ due to elimination of a post-translational modification site on ABCA7, an alteration that is suspected to favorβ-secretase activity [53]. In line with the hypothesis that ABCA7 regulates this enzyme, Abca7 suppression in vitro facilitates AβPP amyloidogenic processing by increasing β-secretase levels and activity, which also exacerbates amyloid plaque burden in AD mouse models [61, 62]. It was proposed that ABCA7 prevents Aβ generation and amyloid pathology by modulating the levels of sterol regulatory element-binding protein 2 (SREBP2), which acts as a transcription factor for β-secretase [62].
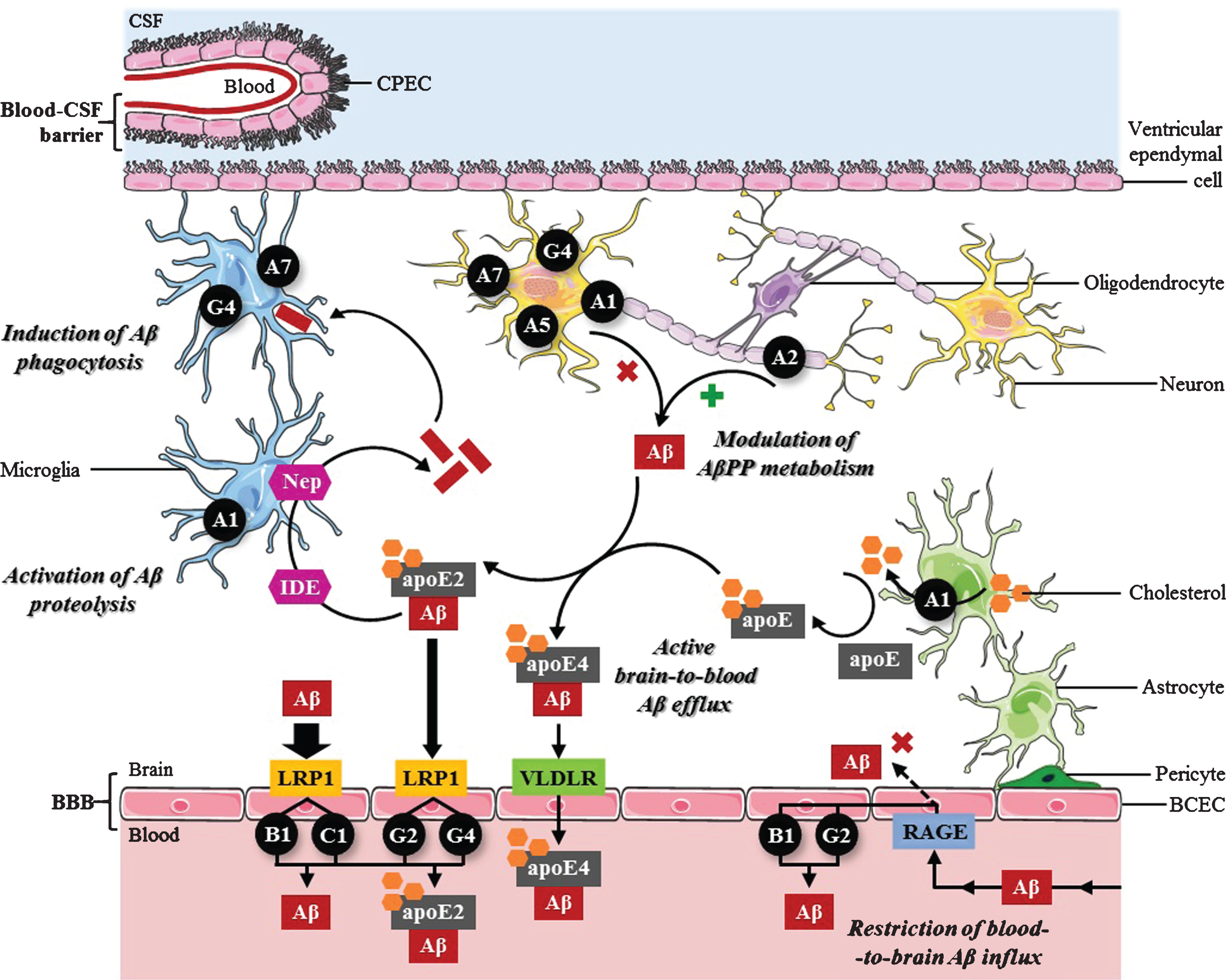
Proposed roles of ABC transporters in the control of cerebral Aβ levels. Modulation of AβPP metabolism: ABCA1, ABCA5, ABCA7, and ABCG4 are able to suppress AβPP proteolysis to generate Aβ in the CNS, whereas ABCA2 enhances Aβ production. Active brain-to-blood Aβ efflux across the BBB in collaboration with LRP1: LRP1 initially mediates Aβ endocytosis from the brain ISF into BCECs, followed by Aβ transcytosis from the abluminal to the luminal side of the BBB. Subsequently, ABCB1, ABCC1, ABCG2 and ABCG4 export Aβ from the apical membrane of BCECs to the systemic circulation. By controlling apoE levels and lipidation status, ABCA1 might also affect Aβ clearance through the BBB: 1) free Aβ is more rapidly endocytosed by LRP1 than apoE-bound Aβ; 2) lipidated apoE4/Aβ complexes are more slowly internalized via VLDLR compared to lipidated apoE2/Aβ complexes via LRP1. Activation of Aβ proteolysis: ABCA1-mediated apoE2 lipidation facilitates the proteolytic degradation of soluble Aβ within the brain by microglial neprilysin (Nep) or extracellular IDE. Induction of Aβ phagocytosis: ABCA7 and ABCG4 stimulate the elimination of cerebral Aβ via microglial phagocytic activity. Restriction of RAGE-mediated blood-to-brain Aβ influx across the BBB: ABCB1 and ABCG2 selectively restrict the apical-to-basolateral permeability of BCECs to circulating Aβ that is transported by RAGE through the BBB into the CNS by pumping it back to the peripheral blood.
In addition, another mechanism may underlie the modulatory effect of ABCA7 on the susceptibility to AD. Some authors postulated that ABCA7 acts as a regulator of phagocyte function (Fig. 1) [28, 63], which is consistent with the high levels of ABCA7 detected in microglia and when monocytes differentiate into macrophages [28]. These reports showed that Abca7 deficiency substantially reduces the capacity of bone marrow-derived macrophages [28, 63] and brain-derived microglia [28] to phagocytose Aβ oligomers both in vitro and in vivo. Surprisingly, an increase in ABCA7 expression is found in the brain of AD patients [28, 54], and it is associated with more advanced cognitive decline [64]. Although this alteration appears to reflect that ABCA7 contributes to AD risk, this is unlikely considering the protective roles of ABCA7 against AD development that were suggested by functional studies (i.e., inhibition of Aβ production and induction of phagocytic clearance of cerebral Aβ). Further, the presence of the AD-protective major T allele of ABCA7 SNP rs3764650 correlates with increased ABCA7 expression [53, 54] whereas, due to a loss-of-function disease mechanism, the AD-risk minor G allele is associated with accumulation of amyloid plaques [55], strongly suggesting that higher levels of ABCA7 would reduce the risk of developing AD. Hence, the most likely explanation for these findings in humans is that a compensatory increase in ABCA7 expression occurs in response to AD, which, however, seems to be insufficient to prevent AD progression, potentially due to SNP-driven alteration of normal protein function and/or accumulation of non-functional protein [53, 54]. Supporting this hypothesis, the AD-risk minor G allele of ABCA7 SNP rs3752246 was shown to cause the expression of a modified form of ABCA7 protein in which the lack of myristoylation alters its normal function, contributing to enhanced β-secretase activity and consequent Aβ production [53]. Also, several ABCA7 genetic variants detected in AD cases were predicted to be deleterious to the coding protein, leading to ABCA7 loss-of-function [59].
ABCA1
Following ABCA7, the second ABC transporter-encoding gene with the strongest linkage to sporadic AD is ABCA1, which resides in close vicinity to chromosome 9q where several loci have been predicted to influence AD [65]. Diverse genetic association studies identified SNPs in ABCA1 linked to AD risk, which were correlated with a protective effect against AD development (e.g., rs2230806, rs2230808, and rs9282543) [65–67] and/or were associated to AD predisposition (e.g., rs2230806, rs2230808, rs4149313, rs46292819, and rs1800977) [65, 69]. The fact that some of these ABCA1 SNPs were described to have opposing effects in sporadic AD cases may be explained by gender differences in the incidence and prevalence of this disease, with women being significantly more affected (almost two-thirds) than men [70]. Interestingly, a linkage has been proposed between this disproportional gender-specific AD risk and both the APOE ɛ4 allele and the ABCA1 219K allele (ABCA1 SNP rs2230806). The deleterious effects of APOE ɛ4-carrier status are substantially more pronounced in women than in men; women carrying one APOE ɛ4 allele were shown to have a two-fold increase in the susceptibility to AD compared to heterozygous men [71]. Moreover, the presence of the ABCA1 219K allele in women was also associated to a 1.75-fold increased risk of developing AD compared to female non-carriers, which contrasts with the protective effect observed in men [65]. As in the presented case, it is possible that gender-specific differences could explain the linkage of other ABCA1 SNPs to AD predisposition or, alternatively, protection against the disease. Furthermore, the presence of the APOE ɛ4 allele may also modulate the association of ABCA1 SNPs to AD risk. For instance, carriers of the APOE ɛ4 allele who are homozygous for the ABCA1 –14T allele (ABCA1 SNP rs1800977) were found to be more susceptible to AD than those who have other allelic variants of this ABCA1 SNP [68]. Although the physiological mechanisms that underlie the ABCA1 SNP-driven effects on AD risk have not been completely clarified, these data indicate that ABCA1 is a good candidate gene involved in AD.
Based on functional studies, a potential role for ABCA1 in AD pathophysiology is related to its function as a major cholesterol exporter in the CNS. Cholesterol distribution in membrane lipid rafts regulates AβPP localization and its availability to proteolytic enzymes, since the raft cluster environment is required for β- [72] and γ-secretase [73] activity, while α-cleavage occurs outside these cholesterol-rich microdomains [72]. Several groups described that ABCA1 induction in AβPP-expressing cells leads to reduced cellular cholesterol content, resulting from ABCA1-mediated efflux to apoA-I and apoE, and to inhibition of Aβ production. While some authors showed an increase in α-secretase proteolytic activity [22], others reported a decrease inβ- and γ-cleavage of AβPP [74], revealing that ABCA1 might downregulate Aβ generation (Fig. 1) via different mechanisms.
The association of ABCA1 with AD was also established given its role in the control of apoE concentration and lipidation status [23, 75]. In line with this, Abca1 deficiency considerably reduces the levels of soluble apoE and its lipidation in AD mouse models, causing increased amyloid deposition in the brain parenchyma and microvasculature [76, 77], whereas its overexpression produces the opposite effects [78]. Moreover, induced Abca1 and Apoe expression via treatment with liver X receptor (LXR) ligands significantly ameliorates amyloid pathology in vivo. Surprisingly, it was described that ABCA1-mediated apoE lipidation markedly enhances Aβ degradation by inducing the proteolytic activity of microglial neprilysin and extracellular IDE. Such effects were shown to be isoform-specific, with lipidated apoE2 and apoE4 constituting the most and least efficient isoforms, respectively, in promoting the proteolytic clearance of soluble Aβ (Fig. 1) [79].
Despite this protective role within the brain parenchyma, several reports suggested that the interaction of apoE with Aβ might disrupt its elimination from the brain ISF into the peripheral blood in an isoform- and lipidation-dependent pattern (Fig. 1). While free Aβ is efficiently transported across the BBB via LRP1, its association with poorly-lipidated apoE slows the clearance process, and this inhibitory effect is accentuated by apoE lipidation [81]. Furthermore, the binding of apoE to Aβ affects its clearance through the BBB by redirecting the rapid efflux of unbound Aβ40 and Aβ42 via LRP1 to the less effective very-low-density lipoprotein receptor (VLDLR), wherein apoE4 markedly increases brain retention of Aβ-apoE4 complexes relatively to apoE3 and apoE2, especially in its lipidated state [82]. These results are consistent with the fact that APOE ɛ4 allele stands for the strongest genetic risk factor of sporadic AD [83]. By directly binding to Aβ, apoE is able to change its conformation and affect its capacity to undergo fibrillation. Among the three isoforms, lipidated apoE4 is more prone to induce the conversion of soluble Aβ peptides into amyloid fibrils and is associated with a higher increase in amyloid burden than apoE3 and apoE2, as demonstrated in AD mouse models [84, 85] and supported by human data[85, 86].
Hence, although not being directly involved in Aβ transport across the BBB [87], ABCA1 is a major determinant of amyloid pathology. Overall, genetic association and functional studies strongly suggest that the physiological role of ABCA1 may predispose to AD (i.e., apoE4-associated inhibition of Aβ clearance across the BBB and induction of Aβ fibrillation) or prevent its development (i.e., suppression of Aβ production and apoE2-associated proteolytic degradation of cerebral Aβ), mainly depending on whether or not patients carry the APOE ɛ4 risk allele. In essence, interactions of this risk allele with ABCA1 allelic variants modulate ABCA1 actions and can impact gender differences in AD prevalence, as demonstrated by the significant associations between the APOE ɛ4 allele [71], or the ABCA1 219K allele [65], and the higher susceptibility to AD found in women but not in men. Interestingly, hippocampal ABCA1 mRNA and protein expression is increased in the brain of AD patients, which is positively correlated with the amount of neurofibrillary tangles and amyloid plaques as well as with cognitive impairment [88]. Although this study did not evaluate gender-specific differences in ABCA1 expression in AD cases, one can speculate that the deleterious effects associated with ABCA1 dysregulation most likely affect female patients, whereas the opposite association of increased ABCA1 expression with protective effects against AD is expected to occur in males.
ABCB1
Although the ABCB1-encoding gene is highly polymorphic, genetic association studies have only described a modest linkage between two common ABCB1 SNPs (rs1045642 and rs2032582) and the susceptibility to sporadic AD [89, 90]. Of these, the ABCB1 SNP rs2032582 can modify AD risk in a gender-specific and APOE ɛ4 allele-dependent manner. The frequency of the variant A/T alleles was reported to be higher among women that do not develop AD and this enrichment was even more pronounced in non-carriers of the APOE ɛ4 allele [89], indicating that the ABCB1 allelic variants and the APOE ɛ4 risk allele drive opposite effects regarding AD predisposition. Supporting this hypothesis, carriers of the variant genotype of this ABCB1 SNP tend to present increased levels of ABCB1, whereas those carrying the AD-risk APOE ɛ4 allele show a significant decrease in ABCB1 expression [91].
According to functional studies, evidence has accumulated to support that ABCB1 has an essential preventive function at the BBB against AD development by limiting Aβ deposition in the brain. It is well established that, by directly interacting with Aβ40 and Aβ42 [8], ABCB1 actively mediates Aβ transport across the apical membrane of BCECs (Fig. 1), either to restrict the entry of blood-borne Aβ into the CNS [92–95] or to promote Aβ elimination from the brain ISF into the systemic circulation [96, 97]. Consistent with these findings, it was demonstrated that the functional consequence of ABCB1 SNP rs2032582 is an increase in the transport activity of the variant form of ABCB1 protein [98]; thus, one can speculate that an enhanced capacity of the modified protein to transport Aβ through the BBB may reflect the molecular mechanism whereby the allelic variants of this ABCB1 SNP are associated with an AD-protective effect. Of note, the levels of ABCB1 are decreased [99], and its activity at the BBB is significantly compromised [100, 101] in the brain of AD patients. Moreover, ABCB1 expression in BCECs is inversely correlated with the density of amyloid plaques and neurofibrillary tangles in AD cases [102]. Hence, these results strongly support that loss of the protective physiological function of ABCB1 at the BBB is one of the molecular mechanisms that triggers Aβ accumulation in the brain of AD patients. In addition to a progressive decline of ABCB1 levels that occurs in brain microvessels during normal aging [103], it was also found that Aβ peptides can mediate ABCB1 downregulation at the BBB, either by interacting with RAGE and inducing the nuclear factor kappa B (NF-κB) signaling pathway, which consequently leads to a reduction in ABCB1 expression [104], or by driving ubiquitination, internalization and proteasome-dependent degradation of ABCB1 [105].
ABCG2
A recent genetic association study described a modest linkage of the ABCG2 SNP rs2231142 with a higher susceptibility to develop sporadic AD. Interestingly, the moderate risk effect associated with the C/C genotype of this ABCG2 SNP was shown to be more pronounced in combination with the APOE ɛ4 allele [106].
In accordance with the well accepted collaborative role that it plays with ABCB1 to protect the CNS against neurotoxic molecules, several functional studies have established that ABCG2 also interacts directly with Aβ [107] and promotes the cellular efflux of Aβ40 and Aβ42 across the luminal side of the BBB (Fig. 1). In this way, ABCG2-mediated Aβ transport prevents amyloid deposition in the brain parenchyma, not only by clearing cerebral Aβ to the peripheral blood [108] but also by acting as a gatekeeper at the BBB that efficiently restricts the passage of circulating Aβ into the CNS [92, 107].
In addition, a protective role for ABCG2 against oxidative stress and neuroinflammation during AD progression was proposed. It appears that ABCG2 prevents reactive-oxygen species (ROS) generation in vitro and consequent activation of the ROS-responsive transcription factor NF-κB, leading to decreased expression of inflammatory genes. It was also demonstrated that ABCG2 overexpression reduces Aβ production, which might be associated with the inhibition of a positive modulatory effect of ROS on the activity of AβPP processing enzymes [109]. Further emphasizing the importance of ABCG2 in redox homeostasis and inflammation, another study concluded that Abcg2 loss correlates with reduced glutathione levels as well as with increased cellular lipid peroxidation, DNA oxidation and inflammatory gene expression in the brain of AD mouse models [110].
Surprisingly, ABCG2 mRNA and protein expression were found to be strongly upregulated in the brain of AD patients [107], and it was correlated with cerebral amyloid deposits in AD mouse models [107, 109]. In agreement with these observations, the risk effect of the C/C genotype of ABCG2 SNP rs2231142 appears to be associated with an increased transcription activity [106]. However, considering that the above functional studies strongly suggest a protective physiological function of ABCG2 against Aβ accumulation and oxidative stress associated with AD, an inverse correlation between cerebral ABCG2 levels and the risk of developing AD would be expected. As previously suggested [5], the increase in ABCG2 expression may be a compensatory mechanism at the BBB to counteract the age-related progressive decline of ABCB1-mediated Aβ clearance that occurs in the brain of AD patients [99] by preventing the access of peripheral Aβ to the CNS [107]. It has also been hypothesized that ABCG2 upregulation in AD cases reflects an adaptive mechanism during oxidative stress that inhibits the NF-κB signaling pathway and associated pro-inflammatory responses [109].
ABCA2
The association between ABCA2 and AD was proposed by a limited number of reports. Two studies identified a genetic variant in ABCA2— the SNP rs908832— that is correlated with increased risk of both sporadic [111] and non-sporadic [112] AD. The T allele of this ABCA2 SNP, which is overrepresented in sporadic AD cases, is associated with high cholesterol levels in the CSF [111], suggesting that, similarly to APOE and ABCA1, ABCA2 may also be a candidate susceptibility gene that reinforces the linkage of lipid homeostasis to AD pathogenesis.
At the functional level, several findings indicate that ABCA2 regulates AβPP metabolism (Fig. 1) through distinct mechanisms. It was described that ABCA2 increases AβPP mRNA and protein expression as well as the levels of secreted Aβ in cell culture models [113, 114], namely by inducingβ-secretase cleavage at the β’-site/Glu11 of the Aβ sequence in full-length AβPP [114]. Another report revealed that Abca2 knockdown leads to a reduction in Aβ generation both in vitro and in vivo, as a result of decreased γ-secretase processing of AβPP. In fact, Abca2 depletion was shown to alter the levels, glycosylation pattern, and subcellular localization of Nicastrin, affecting the formation and cleavage activity of the γ-secretase complex [115]. In line with this, ABCA2 co-localizes in vitro with AβPP and Aβ in endolysosomes [113], which further supports its role in stimulating AβPP amyloidogenic proteolysis. Recently, a new molecular mechanism linking ABCA2 to AD via modulation of AβPP levels was proposed; the authors postulated that ABCA2 expression positively regulates sphingosine levels, resulting in an inhibitory effect on protein kinase C activity that, in turn, induces endogenous AβPP transcription [116].
In essence, the physiological function of ABCA2 is predicted to contribute to AD progression, as confirmed by human data showing high levels of ABCA2 in the brain of AD patients, especially in temporal and frontal lobes, two regions particularly affected by amyloid pathology [113].
ABCC1
To date, few reports have proposed a functional association of ABCC1 to AD pathophysiology, but there is some evidence of a protective physiological role. A recent study described that ABCC1 efflux activity is of exceptional importance for the removal of cerebral Aβ through the BBB (Fig. 1). The authors showed that Abcc1 deficiency elevates Aβ40 and Aβ42 levels in the brains of AβPP/PS1 transgenic mice, resulting in a marked increase of amyloid plaque burden. These effects were not caused by altered expression of AβPP processing enzymes or Aβ degradation proteins, but were related to a reduction in ABCC1-mediated Aβ transport across BCECs, as demonstrated in an in vitro model. Further supporting the role of ABCC1 in Aβ clearance, the reversal of such effects via treatment with an ABCC1 agonist significantly decreased cerebral Aβ deposition in vivo [117]. Similarly, another study associated an increase in ABCC1 export activity at the BBB with a reduction of amyloid load in AD mouse models [118].
A protective function for ABCC1 against oxidative stress was also proposed, whereby ABCC1 mediates the efflux of 4-hydroxy-2-transnonenal (HNE) [119], a product of lipid peroxidation that is increased in the brain of AD patients [120]. In this way, ABCC1 was suggested to act in collaboration with glutathione S-transferase (GST), which initiates a detoxification process via inactivation of intracellular HNE by forming glutathione/HNE conjugates. Since GST function is reduced in the brain of AD patients, it was proposed that unconjugated HNE covalently binds to GST and ABCC1, disrupting their antioxidant activity in CNS cells [119]. Recently, another in vitro report revealed that monomeric Aβ increases the release of astrocyte-derived glutathione by inducing Abcc1 expression. This protective antioxidant effect was found to decline with Aβ oligomerization, which is consistent with a decrease of Abcc1 levels as amyloid pathology progresses in AD mousemodels [121].
ABCG1
Regarding the involvement of ABCG1 in AD, a limited number of contradictory functional studies exists. On the one hand, it was shown that ABCG1 and AβPP co-expression in vitro increases the levels of secreted Aβ by enhancing the availability of AβPP as a substrate for the amyloidogenic processing pathway, indicating that ABCG1-mediated regulation of AβPP trafficking potentially favors AD development [122]. However, other authors demonstrated that ABCG1 significantly reduces Aβ production in AβPP-expressing cells [27, 123], whereas its suppression has the opposite effect. Furthermore, the presence of ABCG1 not only inhibits γ-secretase function but also disturbs its localization in the lipid raft microdomains where its activity takes place, suggesting that ABCG1 prevents AD pathogenesis [123]. These findings and the fact that ABCG1 expression increases cholesterol efflux to apoE discs reveal that ABCG1-induced redistribution of brain cholesterol might contribute to such alterations in AβPP proteolysis [27]. Contrariwise, Abcg1 overexpression in AβPP transgenic mice was shown not to alter the levels of Aβ, AβPP, or apoE nor to influence amyloid plaque load in vivo, which argues against the previous in vitro reports [124]. Therefore, additional studies are needed to elucidate the exact mechanisms through which ABCG1 is involved in AD.
ABCG4
Although few functional studies evaluated the involvement of ABCG4 in AD, available data indicate that it prevents disease progression by somehow replicating the protective activities of other ABC transporters (Fig. 1). For instance, ABCG4 shares a role with ABCG1 in decreasing the levels of secreted Aβ in cell culture models while suppressing γ-secretase activity and altering its distribution in lipid rafts [123]. Furthermore, other authors revealed that ABCG4 is able to export Aβ in vitro and that its expression is significantly increased in brain microvessels of Abcb1/Abcg2-deficient mice, suggesting a possible collaborative function with ABCB1 and ABCG2 regarding the removal of cerebral Aβ across the BBB [108]. It was also demonstrated that ABCG4 mRNA and protein expression is markedly upregulated in microglial cells and correlated with amyloid plaques in the brain of AD patients. It was postulated that ABCG4 activity might contribute to Aβ degradation via phagocytosis by bone marrow-derived microglia [125], which were previously shown to migrate into the brain in AD mouse models to eliminate amyloid deposits [126], indicating that ABCG4 likely complements the role of ABCA7.
ABCA5
A recent study identified a significant upregulation of ABCA5 expression in the hippocampus of AD patients. ABCA5 decreases Aβ40 and Aβ42 generation (Fig. 1) without changing AβPP mRNA or protein expression in vitro, leading the authors to propose that ABCA5 might alter the activity of AβPP processing enzymes [9]. Given the functional similarity between ABCA5 and its homologue ABCA1 as neuronal cholesterol transporters, one can speculate that ABCA5 probably has a similar protective function (i.e., suppression of Aβ production from AβPP amyloidogenic proteolysis); however, this hypothesis requires additional validation.
Overall, considering that many of the aforementioned ABC transporters show overlapping functions in AD pathophysiology (e.g., brain-to-blood Aβ efflux via ABCB1, ABCC1, ABCG2, and ABCG4; inhibition of Aβ production by ABCA1, ABCA5, ABCA7, ABCG4, and, possibly, ABCG1; and induction of Aβ phagocytosis by ABCA7 and ABCG4), studies searching for the concomitant presence of SNPs in two or more genes encoding for functionally related ABC transporters in AD cases are warranted. Further, it would also be important to investigate the levels of mRNA and protein expression of these sets of ABC transporters simultaneously in a particular brain region. These strategies could allow the researchers to understand if a compensatory mechanism may exist, in which an increase in Aβ toxicity induced by downregulation of a specific ABC transporter would be counterbalanced by upregulation of another functionally related ABC transporter (e.g., ABCB1 downregulation and ABCG2 upregulation in the brain of AD patients).
OTHER Aβ TRANSPORT PATHWAYS IN THE BBB
Besides ABC transporters, Aβ translocation across the BBB is also facilitated by two receptors operating in opposing directions: 1) LRP1, which transports Aβ from the brain ISF to the peripheral blood [127–129]; and 2) RAGE, which promotes the entry of circulating Aβ into the CNS [129, 130] (Fig. 1). Belonging to the LDL receptor family, LRP1 is able to recognize monomeric, oligomeric, and aggregated forms of cerebral Aβ [128, 129]. LRP1 mediates Aβ endocytosis on the abluminal side of the BBB followed by transcytosis, contributing to Aβ clearance. Direct LRP1/Aβ binding at the BBB preferentially eliminates soluble Aβ40, while the removal of the more hydrophobic Aβ42 via LRP1 possibly requires apoE3 intervention [127, 128]. In contrast, RAGE is an immunoglobulin superfamily receptor that acts as an influx transporter for soluble forms of peripheral Aβ40 and Aβ42. Upon direct binding on the luminal side of the BBB, RAGE transports Aβ from the systemic circulation to the brain ISF [129, 130]. Additionally, RAGE mediates Aβ-induced neurotoxicity on neurons, BCECs and microglia. As a consequence of Aβ/RAGE interaction at the BBB, Aβ binds to RAGE expressed on neurons’ surface and induces oxidative damage, which is accompanied by neurovascular stress [130, 131]. Binding of soluble Aβ to RAGE-expressing microglia also stimulates sustained pro-inflammatory cytokine secretion that further contributes to neuronal death [131].
In the brain of AD patients, an age-related decrease in LRP1 expression and function is found in BCECs, which accelerates Aβ accumulation in the cerebral tissue [127–129]. On the contrary, the Aβ-enriched environment created during AD progression upregulates RAGE expression at the BBB [129, 130] and in neurons [131], allowing for the propagation of several neurotoxic effects. Hence, this imbalance between LRP1-mediated Aβ clearance and RAGE-dependent Aβ influx at the BBB favors AD pathogenesis by increasing not only amyloid plaque burden but also the vulnerability of brain cells to Aβ [128–130].
ABC TRANSPORTER-BASED THERAPEUTIC STRATEGIES
ABC transporters are involved in the multidrug resistance phenotype common in many CNS diseases. ABCB1 regulates the entry of several antidepressant drugs in the brain and ABCB1 polymorphisms might influence treatment outcomes in depression [132]. Accordingly, ABCB1 genotyping can be used to optimize therapeutic strategies, as in the case of patients carrying ABCB1 SNPs who might benefit from higher antidepressant doses to achieve a positive response [133]. Interestingly, depression and dementia often overlap in the elderly. The successful treatment of depressed patients appears to correlate with a reduced risk of AD [134]. In fact, besides the antidepressive effects of Hypericum perforatum (St. John’s wort), this plant has been associated with enhanced ABC transporter activity and a concomitant decrease in amyloid burden [118].
Recently, pioneer work has been developed to identify novel inducers of ABC transporters able to improve Aβ clearance from the CNS. As discussed in the previous section, impaired brain-to-blood efflux of Aβ via ABC transporters at the BBB is a critical factor in AD development. The pharmacological activation of pregnane X receptor (PXR) increases the expression and transport function of ABCB1 and ABCG2 at the BBB in vitro [135], which is related to reduced cerebral Aβ levels in AβPP transgenic mice [97]. Similarly, the treatment of an AD mouse model with an ABCC1 agonist drug showed promising results, wherein the resulting increased ABCC1 transport activity lead to a marked reduction in the number and size of amyloid plaques [117]. Furthermore, the modulation of ABC transporters that regulate brain cholesterol distribution and, thus, AβPP processing and/or Aβ degradation might also be a new therapy option for AD. The pharmacological induction of liver X receptor (LXR) and retinoic X receptor (RXR) increases ABCA1 expression and apolipoprotein-mediated cholesterol efflux in cellular models [22], which is associated with decreased Aβ deposition [136] and improved cognition [137] in vivo. In essence, the pharmacological modulation of human ABC transporters holds great promise for the development of novel AD treatment and diagnosis strategies.
CONCLUSION
Although additional research is necessary to clarify the roles of ABC transporters in AD, increasing evidence suggests that several of them are key players in protecting the CNS against Aβ neurotoxicity, for which their ability to export cerebral Aβ to the peripheral blood across the BBB is of paramount importance. Based on this clearance mechanism, novel diagnostic tools and therapeutic approaches may emerge in the upcoming future to detect early and ameliorate amyloid pathology in the brain of AD patients through modulation of the expression and/or activity of specific ABC transporters.
Footnotes
ACKNOWLEDGMENTS
This work was financed by PTDC/DTP-PIC/5587/2014 and supported by Instituto de Biomedicina (iBiMED)— UID/BIM/04501/2013, the Fundação para a Ciência e Tecnologia (FCT) of the Ministério da Educação e Ciência, COMPETE program, the QREN, and the EU (Fundo Europeu de Desenvolvimento Regional). The authors acknowledge the support from the European Union Framework Programme for Research and Innovation HORIZON 2020, under the TEAMING Grant agreement No 739572 – The Discoveries CTR.