
Abstract
Amyloid-β (Aβ) induces a burst of oxidative stress and plays a critical role in the pathogenesis of Alzheimer’s disease (AD). Our previous results have shown that histone deacetylase 3 (HDAC3) inhibition ameliorates spatial memory deficits and decreases the Aβ burden in the brains of 9-month-old APPswe/PS1dE9 (APP/PS1) mice. In this study, we investigated the role of HDAC3 inhibition in oxidative stress in vivo and in vitro models of AD. HDAC3 was detected mainly in the neurons, and HDAC3 inhibition significantly decreased reactive oxygen species generation and improved primary cortical neuron viability. In addition, HDAC3 inhibition attenuated spatial memory dysfunction in 6-month-old APP/PS1 mice, and decreased the apoptotic rate in the hippocampi as demonstrated by TUNEL staining. HDAC3 inhibition also reduced markers of lipid peroxidation, protein oxidation, and DNA/RNA oxidation in the hippocampi of APP/PS1 mice. Moreover, HDAC3 inhibition inactivated the c-Abl/MST1/YAP signaling pathway in the hippocampi of APP/PS1 mice. In conclusion, our data show that HDAC3 inhibition can attenuate spatial memory deficits and inhibit oxidative stress in APP/PS1 mice; these results indicate a potential strategy for AD treatment.
INTRODUCTION
Alzheimer’s disease (AD), which affects approximately 5.4 million people in the United States, is the most common cause of dementia among the elderly [1]. The neuropathological hallmarks of AD include extracellular senile plaques composed of amyloid-β (Aβ) and intracellular neurofibrillary tangles. Growing evidence has suggested that Aβ induces a burst of oxidative stress, which plays a critical role in the early stage of AD [2]. Aβ accumulation induces mitochondrial dysfunction and increases reactive oxygen species (ROS) generation, which then initiates lipid peroxidation, protein oxidation, and DNA/RNA oxidation. In addition, oxidative stress promotes the production of Aβ and exacerbates memory deficits, leading to a vicious cycle of Aβ and oxidative stress [3–6]. Thus, inhibiting Aβ-induced oxidative stress is a potential strategy for treating AD.
Histone deacetylase 3 (HDAC3), a member of Class I HDACs, is abundantly expressed in the brain and exerts neurotoxic effects on the central nerve system (CNS) [7–9]. It has been shown that HDAC3 negatively regulates cocaine-induced conditioned place preference acquisition and that the inhibition of HDAC3 enhances long-term memory in object recognition tasks [10]. However, whether HDAC3 contributes to the pathology of AD remains controversial. The levels of HDAC3 are not altered in the brains of AD patients compared with those in healthy controls [11]. RGFP966, a selective HDAC3 inhibitor, does not affect memory function in 6-month-old APPswe/PS1dE9 (APP/PS1) mice [12]. On the other hand, RGFP966 attenuates the impaired long-term potentiation (LTP) induced by the Aβ42 oligomer [13]. Recently, we have shown that HDAC3 levels are increased in the nuclei of the hippocampi in APP/PS1 mice, and the inhibition of HDAC3 decreases Aβ levels and attenuates spatial memory deficits in APP/PS1 mice; these results indicate that HDAC3 might be a negative regulator of spatial memory [14]. However, the underlying molecular mechanism by which HDAC3 inhibition improves spatial memory remains unknown.
The Hippo/Mammalian Ste20-like kinase 1/2 (MST1/2) signaling pathway is evolutionarily conserved in Drosophila and mammals and plays a critical role in regulating cellular responses to oxidative stress [15–18]. Oxidative stress induced by hydrogen peroxide activates MST1 in primary neurons, while the inhibition of MST1 rescues neuronal cell death [19]. In addition, MST1 activation leads to the phosphorylation of several components of the Hippo pathway, including large tumor suppressor kinase 1/2 (Lats1/2), which subsequently phosphorylates and inactivates YAP and TAZ to inhibit TEAD-mediated gene transcription [19, 20]. C-Abl, an upstream tyrosine kinase of MST1, has been shown to affect the pathogenesis of AD. The levels of c-Abl in the brains of AD mice are significantly higher than those in the brains of wild-type mice, and Aβ treatment of primary neuronal cells induces Tau hyperphosphorylation by activating c-Abl [21, 22]. STI571, a c-Abl inhibitor, attenuates cognitive impairment and decreases Aβ deposits and Tau hyperphosphorylation in AD mice [21, 23]. Furthermore, c-Abl contributes to oxidative stress-induced neuronal cell death by activating the MST1/forkhead box O3 (FOXO3) signaling pathway [16, 25]. In this study, we have shown that HDAC3 inhibition attenuates oxidative stress and cell apoptosis in primary cultured neurons and in the hippocampi of 6-month-old APP/PS1 mice, which might be associated with the c-Abl/MST/YAP pathway.
MATERIAL AND METHODS
Animals and treatment
Male APP/PS1 mice were obtained from the Model Animal Research Center of Nanjing University, and male age-matched wild-type (WT) C57BL/6 (B6) littermates were used as controls. A lentivirus (Lv-shHDAC3) was used to inhibit the expression of HDAC3 and was injected into the bilateral hippocampus (anterior-posterior position –2.0 mm, medial-lateral position±1.6 mm, 1.5 mm dorsoventral from the bregma) of 6-month-old APP/PS1 mice by using a stereotaxic apparatus as described previously [26]. One month later, Morris water maze (MWM) tests were performed to access cognitive function. All animal experiments were approved by the Animal Care Committee of Nanjing University, and all efforts were made to reduce the number of mice and their suffering.
Morris water maze test
As previously described [27], MWM tests were performed for 6 consecutive days. Briefly, during the acquisition trial, the mice were allowed to swim freely to find the submerged platform, and the times were recorded as the latency. During the probe trial, the mice were allowed to swim for 1 min, and the number of platform crossings, the time spent in the target quadrant, and the latency to find the target quadrant were recorded. The data were analyzed using the ANY-maze system (Stoelting, USA).
Primary cell culture
Primary cortical neurons were prepared from E15-17 C57/BL6J mice embryos as previously described [28]. The cells were maintained in neurobasal medium with B27 (Invitrogen, USA) and 25 nM glutamine at 37°C in a humidified 5% CO2 incubator. The cortical neuron purity was more than 90% according to microtubule-associated protein-2 (MAP-2) staining. At 3–4 days in vitro (DIV), Lv-shHDAC3 or Lv-con was added to the medium (MOI = 5). After infection for 72 h, synthetic Aβ42 (2 μM) (Millipore, USA) was added for another 24 h, and the following experiments were performed. Primary glial cells were isolated from <48 h-old newborn B6 mice as previously described [29]. When most of the cells were floating, the microglia cells were collected directly from the mixed glial cells in the medium at 10–12 DIV and seeded into 6-well plates (5×105 cells/mL) for 2–3 days. Astrocytes (the rest cells) were seeded into 6-well plates (5×105 cells/mL) and passaged for 2–3 generations. The primary glial cell purity was more than 95% according to Iba-1 and GFAP staining.
MTT assay
Cell viability was determined by using the conventional 3-[4,5-diethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay as described previously [30]. Briefly, after treatment with Aβ42 for 24 h, a 0.5 mg/ml MTT solution was added to primary cortical neurons for 4 h at 37°C. The dark blue formazan crystals were dissolved in 100 μl of DMSO, and the absorbance was measured at 570 nm with a microplate reader (Bio-Rad, USA). Cell survival rates were presented as the percentage of live cells compared to untreated cells.
Detection of ROS
ROS production by cells and in the hippocampus was measured using a ROS assay kit (GENMED, China). According to the manufacturer’s instructions, after removing the medium, 200 μl of Dihydroethidium (DHE) working solution was added to 24-well plates. The plates were incubated in the dark at 37°C for 20 min, and stock solution was added. Fresh brain tissues were immediately removed and cut into 15 μm-thick cryosections (Leica, Germany). Then, 200 μl of DHE working solution was added to the slices and incubated in a dark, humidified chamber at 37°C for 20 min. DHE fluorescence was measured using a microscope (Olympus, Japan).
Measurement of 3-nitrotyrosine (3-NT), 4-hydroxy-nonenal (4-HNE), and 8-hydroxy-2’-deoxyguanosine (8-OHdG)
To measure the levels of 4-HNE, 3-NT, and 8-OHdG in the hippocampi from APP/PS1 mice, ELISA kits were used according to the manufacturer’s instructions (CUSABIO, Mlbio, China). Briefly, samples of homogenized hippocampus tissue were added to a plate pre-coated with mouse 4-HNE, 8-OHdG or 3-NT antibody, and incubated for the indicated times at 37°C. After adding the TMB substrate solution and stop solution, absorbances were measured at a wavelength of 450 nm. The 4-HNE, 8-OHdG, and 3-NT concentrations were calculated according to the standard curve.
Apoptosis analysis
To investigate the apoptotic rate of hippocampal cells, an In Situ Cell Death Detection Kit AP (Roche, Germany) was used as previously described [27]. Briefly, brain cryosections were incubated with 0.1% Triton X-100 on ice for 2 min. After being rinsed in 15% acetic acid, the TUNEL reaction solution was added to the sections. Then, the slices were incubated in a dark, humidified chamber, and the chromogenic substrate BCIP/NBT was added. Images of the TUNEL-positive cells were taken using a microscope (Olympus, Japan).
Immunofluorescence assay
To determine the protective effects of HDAC3 inhibition, neurons were fixed in pre-cooled 4% paraformaldehyde (PFA) after Aβ42 treatment. The neurons were blocked with 2% BSA for 1.5 h and incubated with MAP-2 antibody (1 : 500, Bioworld, USA) at 4°C overnight. After washing off the primary antibody three times, the secondary antibody was added and incubated in the dark for 2 h at room temperature; then, DAPI reagent (1 : 1000, Bioworld, USA) was used to stain the nuclei. Images were taken with an inverted fluorescence microscope (Olympus, Japan).
The mice were anesthetized, and perfused with pre-cooled 0.9% saline and 4% PFA. After being cut with a cryosection apparatus, the brain sections were blocked with 2% BSA for 1.5 h and incubated overnight at 4°C with the following primary antibodies: anti-HDAC3 (1 : 200, Abcam), anti-GFAP (1 : 200, Cell Signaling, USA), anti-Iba1 (1 : 200, Abcam), anti-NeuN (1 : 100, Millipore, USA) or anti-DCX (1 : 500, Abcam, USA). The sections were then incubated with the secondary antibody for 2 h at room temperature, and DAPI reagent (1 : 1000, Bioworld, USA) was used to stain the nuclei. Images were taken with a fluorescence microscope or confocal laser-scanning microscope (Olympus, Japan). All qualitative immunostaining analyses were conducted by using Image J software (National Institutes of Health, USA), and the analyses were performed in a randomized and blinded manner.
Western blotting
Whole protein was isolated with RIPA lysis buffer (Thermo, USA), and the concentrations were measured using a BCA protein assay kit (Thermo, USA). Equal quantities of protein were loaded and run on SDS-PAGE gels and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, USA). The membranes were blocked with 5% non-fat milk for 1 h at room temperature, followed by an overnight incubation at 4°C with the following primary antibodies: anti-HDAC3 (1 : 800, Cell Signaling, USA), anti-Bax (1 : 1000, Cell Signaling, USA), anti-Bcl-2 (1 : 1000, Bioworld, USA), anti-NeuN (1 : 1000,Millpore, USA), anti-DCX (1 : 1000, Abcam, USA), anti-c-Abl (1 : 1000, Cell Signaling, USA), anti-p-MST1 (1 : 500, Cell Signaling, USA), anti-MST1 (1 : 1000, Cell Signaling, USA), anti-p-YAP (1 : 500, Cell Signaling, USA), anti-YAP (1 : 1000, Cell Signaling, USA), or anti-GAPDH (1 : 5000, Bioworld, USA). After washing with TBST, the membranes were incubated with the secondary antibodies for 2 h at room temperature. The protein signals were visualized by using an ECL kit (Thermo, USA), and the band intensities were quantified using Image J software.
Statistical analysis
All data were expressed as the mean±standard error of the mean (SEM) and were analyzed using SPSS 16.0 software (SPSS, USA). Differences in the escape latency for the MWM tests were analyzed using two-way analysis of variance (ANOVA) with repeated measures. Independent student’s t test was performed to analyze the differences between groups. A value of p < 0.05 was considered statistically significant.
RESULTS
HDAC3 inhibition protects against Aβ42-induced neuronal death
HDAC3 plays an important role in the function of the CNS [10, 31]; thus, we first measured the levels of HDAC3 in neurons, microglia and astrocytes. As shown in Fig. 1A, B, the levels of HDAC3 were highest in the primary cortical neurons, whereas they were much lower in the microglia and astrocytes. Similar results were observed using immunostaining (Fig. 1C). Next, we used a lentivirus to inhibit the expression of HDAC3, which was confirmed by western blotting (Fig. 2A, B). As shown in Fig. 2C, Aβ42 treatment significantly decreased neuronal viability (p < 0.01), whereas HDAC3 inhibition partially reversed these effects (p < 0.05). In addition, the loss of neurons and neurite structures was ameliorated by HDAC3 inhibition as demonstrated by MAP-2 staining (Fig. 2D, E, p < 0.05).
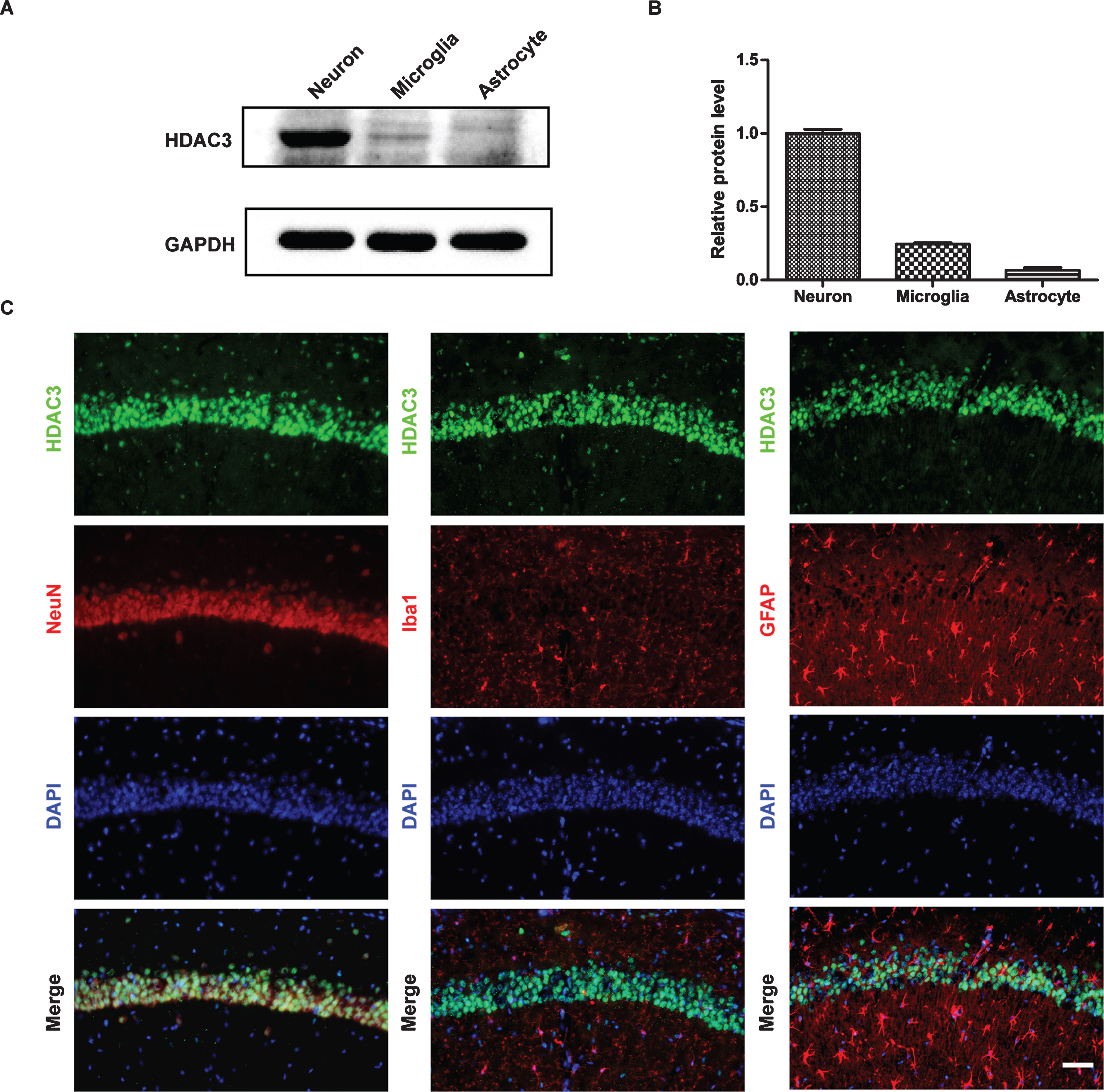
HDAC3 was detected mainly in primary cortical neurons. A) Western blotting was used to examine the levels of HDAC3 in primary neurons, microglia and astrocytes. (n = 4–6 per group). B) Quantification of the intensities normalized to GAPDH as a loading control. C) Colocalization of HDAC3 and NeuN/Iba1/GFAP in the hippocampi of wild-type B6 mice (n = 4–6 mice per group). Scale bar = 50 μm.
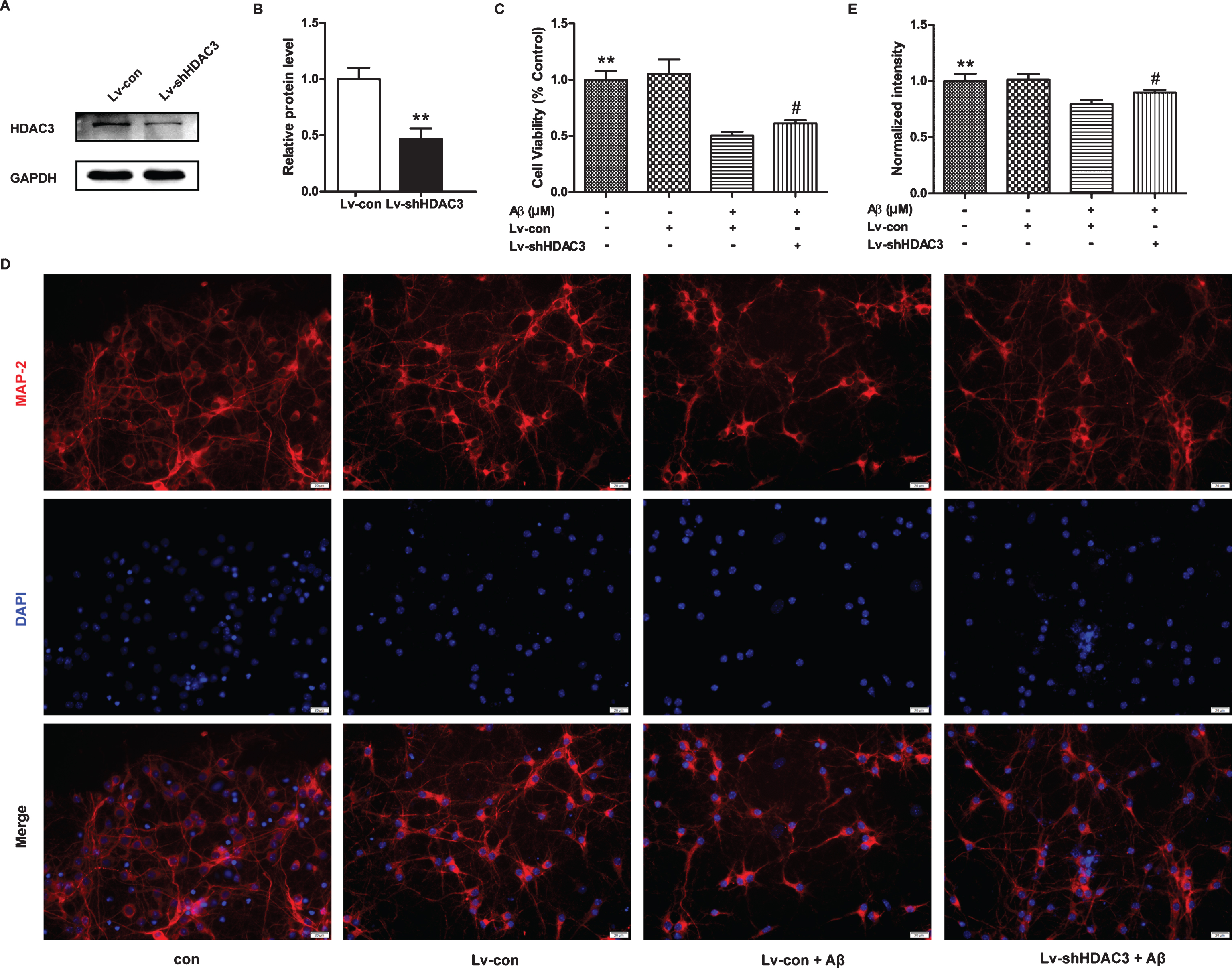
HDAC3 inhibition protected neurons from Aβ42-induced cell death. A) HDAC3 expression was determined by western blotting after lenti-shHDAC3 infection for 72 h (n = 4–6 per group). B) Quantification of the intensities normalized to GAPDH as a loading control. **p < 0.01. C) Cell viability in neurons infected with lenti-shHDAC3 was examined by MTT assays after Aβ42 treatment for 24 h (n = 6 per group), **p < 0.01 versus control, #p < 0.05 versus Lv-shHDAC3. D) HDAC3 inhibition prevented the loss of neurite structures in the neurons infected with lenti-shHDAC3 followed by Aβ42 treatment as determined by immunostaining for MAP-2 (n = 4–6 per group). Scale bar = 20 μm. E) Quantitative analysis of MAP-2 staining normalized to control neurons. **p < 0.01 versus control, #p < 0.05 versus Lv-shHDAC3.
HDAC3 inhibition reduces ROS production in Aβ42-treated neurons
It has been shown that Aβ42 induces mitochondrial dysfunction, which subsequently leads to the overproduction of ROS [32]. As shown in Fig. 3, the amount of ROS was significantly increased in neurons treated with Aβ42 for 24 h (p < 0.01), whereas the amount of ROS was attenuated by HDAC3 inhibition (p < 0.01).
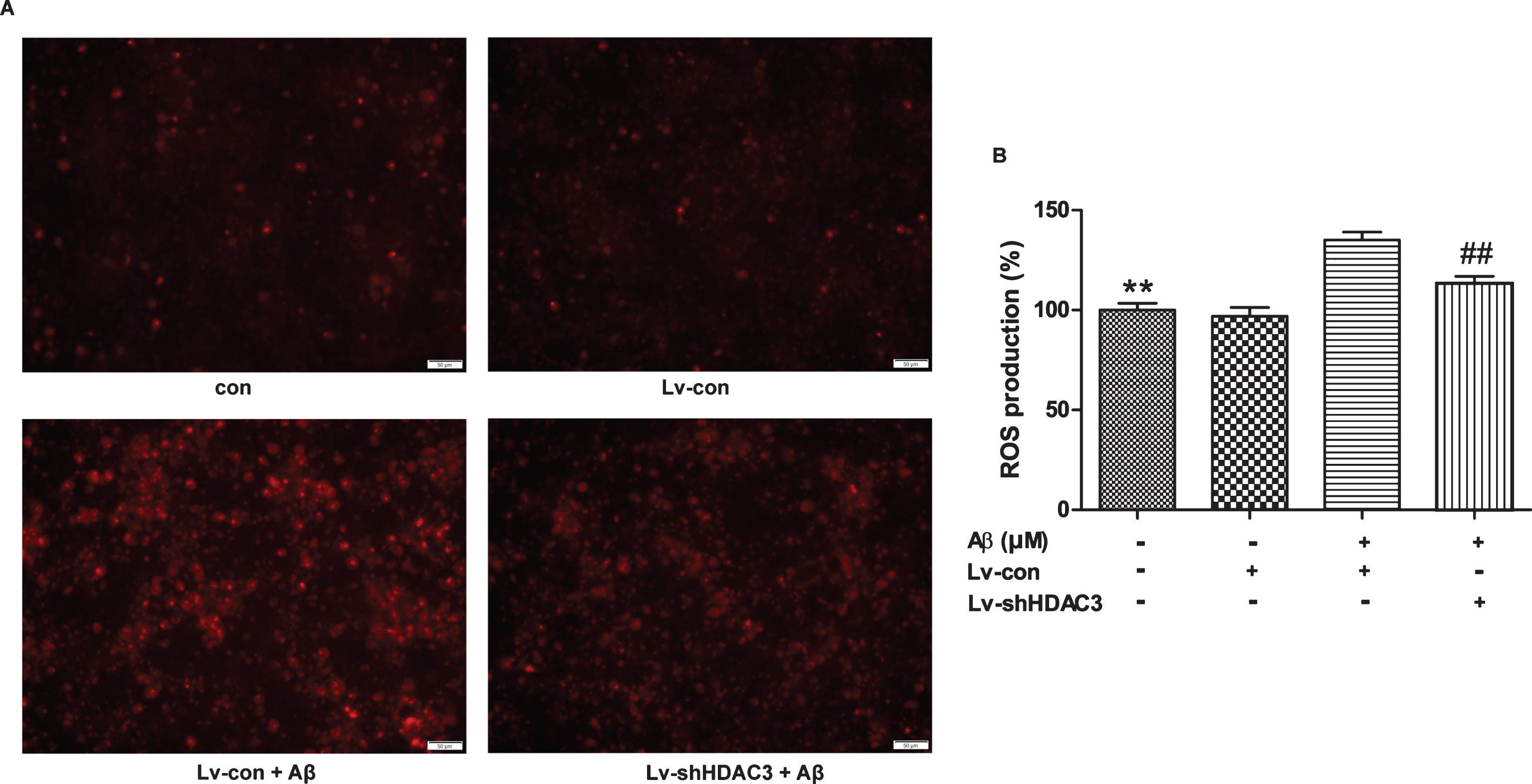
HDAC3 inhibition reduced ROS production in Aβ42-treated neurons. A) ROS production was measured by fluorescence in the neurons infected with lenti-shHDAC3 and treated with Aβ42 (n = 4–6 per group). Scale bar = 50 μm. B) Quantitative analysis of DHE staining normalized to control neurons. **p < 0.01 versus control, # #p < 0.01 versus Lv-shHDAC3.
HDAC3 inhibition attenuates spatial memory dysfunction in APP/PS1 mice
To confirm whether HDAC3 inhibition could improve spatial memory in 6-month-old APP/PS1 mice, MWM tests were performed. As shown in Fig. 4A, the cells infected with the GFP-tagged lentivirus, were mainly detected in the hippocampus, and the efficiency of HDAC3 inhibition in vivo was confirmed by western blotting (Fig. 4B, C, p < 0.01). The mean escape latency of Lv-shHDAC3-treated APP/PS1 mice was significantly decreased during the acquisition trial compared with that of the Lv-con-treated mice (Fig. 4D, p < 0.05). During the probe trial, HDAC3 inhibition significantly increased the number of target platform crossings (Fig. 4E, p < 0.01) and time spent in the target quadrant (Fig. 4F, p < 0.05) for the APP/PS1 mice. Furthermore, the latency to the target quadrant of the Lv-shHDAC3-treated group was significantly shorter than that of the Lv-con-treated group (Fig. 4G, H, p < 0.05), indicating that HDAC3 inhibition ameliorated spatial memory loss in the 6-month-old AD mice.
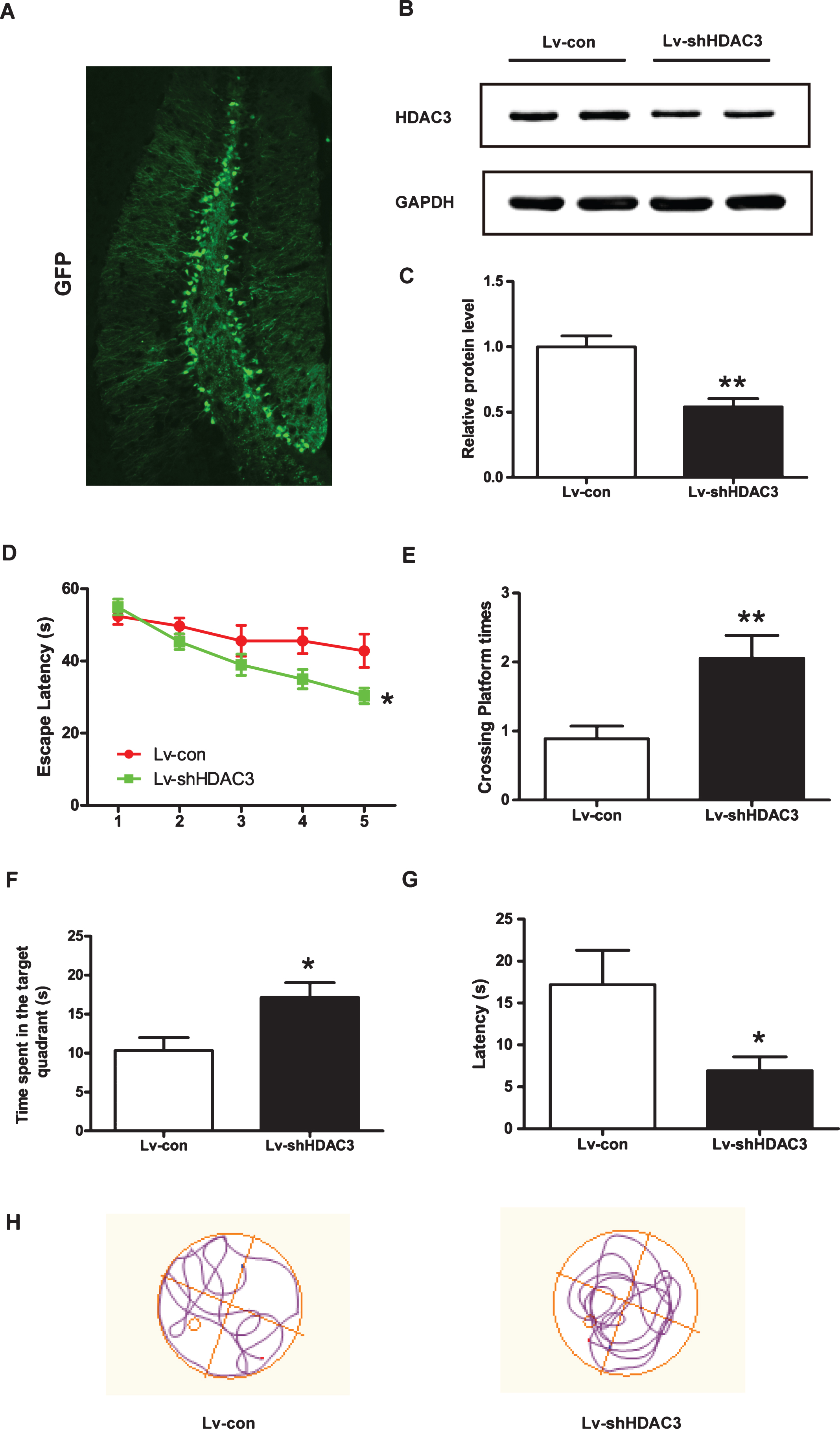
HDAC3 inhibition attenuated spatial memory dysfunction in 6-month-old APP/PS1 mice. After Lv-con or Lv-shHDAC3 was injected for 30 days, MWM tests were performed to assess the effects of HDAC3 inhibition on spatial memory in AD mice (Lv-con, n = 10; Lv-shHDAC3, n = 10). A) Representative image of Lv-shHDAC3-infected cells in the hippocampus of an APP/PS1 mouse. Scale bar = 100 μm. B) Protein levels of HDAC3 were determined by western blotting after lenti-shHDAC3 injection (n = 4–6 per group). C) Quantification of the intensities normalized to GAPDH as a loading control was shown. D) In the acquisition trial, the escape latencies of the Lv-con- or Lv-shHDAC3-treated groups were recorded. In the probe trial, the number of platform crossings (E), the time spent in the target quadrant (F), and the latency to target quadrant (G) were analyzed. H) Representative image of the track plot for the probe test on day 6. *p < 0.05, **p < 0.01.
HDAC3 inhibition decreases apoptosis in the hippocampi of APP/PS1 mice
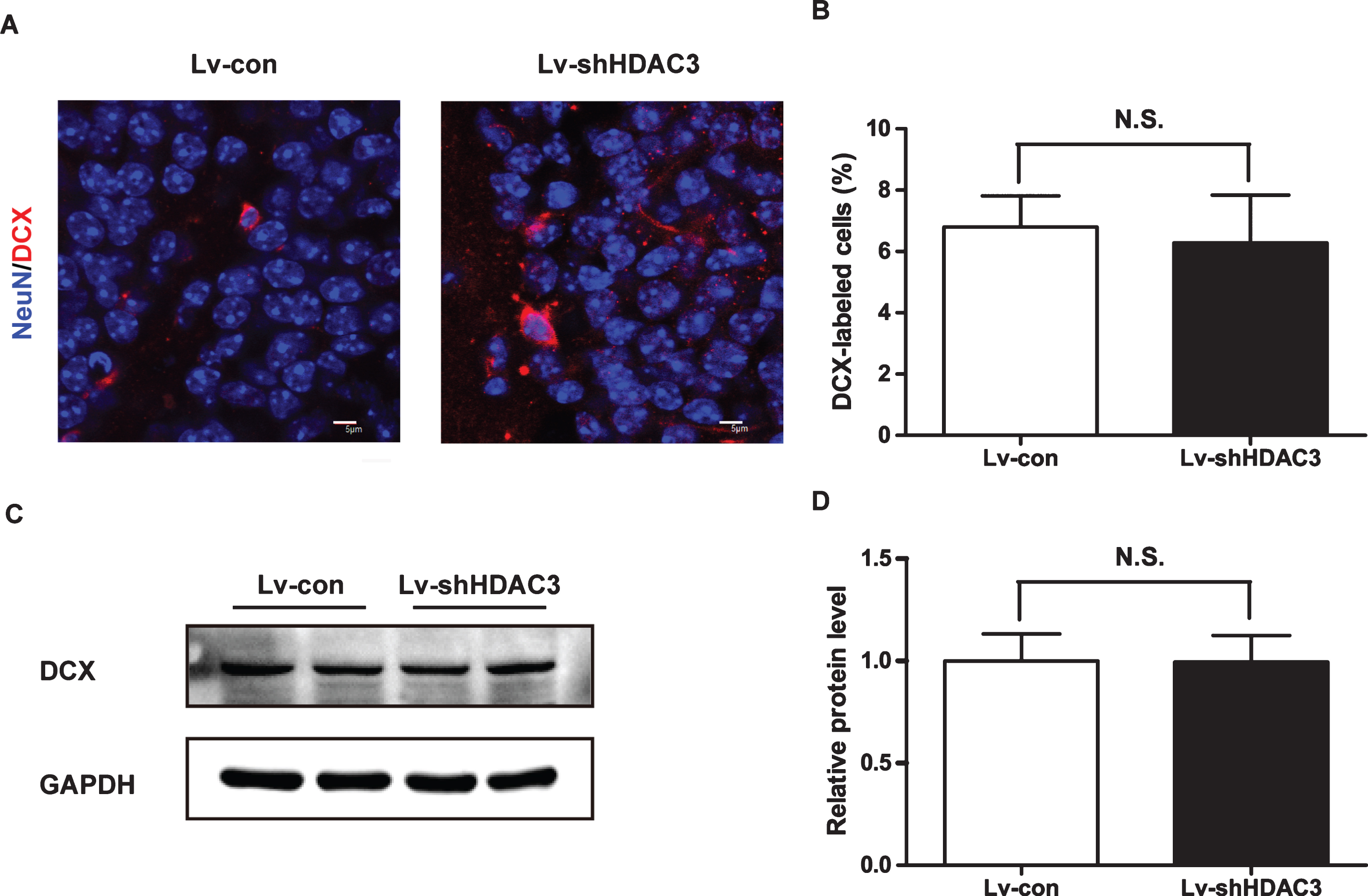
To investigate whether HDAC3 inhibition could protect the neurons from apoptosis in the hippocampi of APP/PS1 mice, TUNEL staining was performed. As demonstrated in Fig. 5A, the number of TUNEL-positive cells in the Lv-con-treated group was greater than that in the Lv-shHDAC3-treated group, which indicated that HDAC3 inhibition could reduce apoptosis in the hippocampi of APP/PS1 mice. Moreover, the surviving neurons stained by NeuN in the CA1 area of the hippocampus were significantly increased in the Lv-shHDAC3-treated group (Fig. 5B, C, p < 0.01). To confirm the anti-apoptotic effects of HDAC3 inhibition, the protein expression levels of Bax, Bcl-2 and NeuN were determined using western blotting. As shown in Fig. 5D, E, HDAC3 inhibition decreased the levels of Bax (p < 0.01); however, the expression levels of Bcl-2 and NeuN were increased (p < 0.01 and p < 0.05, respectively). To explore whether HDAC3 inhibition could affect neurogenesis, the levels of NeuN and DCX were determined by immunostaining and western blotting to analyze neuron proliferation in the dentate gyrus (DG) areas of APP/PS1 mice. As shown in Fig. 6, the levels of NeuN and DCX were not significantly changed in the DG areas of the Lv-shHDAC3 group, which indicated that HDAC3 inhibition did not affect neurogenesis in the hippocampus (p > 0.05).
HDAC3 inhibition decreased the apoptotic rate in the hippocampi of APP/PS1 mice. A) The apoptotic rate in the hippocampi of APP/PS1 mice was determined by TUNEL staining. Scale bar = 100 μm. B) The number of neurons in the hippocampi of the Lv-con or Lv-shHDAC3 group (n = 4–6 per group) was determined by NeuN (red) immunostaining, and was analyzed in (C). Scale bar = 50 μm. D) The hippocampi levels of Bax, Bcl-2 and NeuN were determined by western blotting (n = 4–6 per group). E) Quantification of the intensities normalized to GAPDH as a loading control. *p < 0.05, **p < 0.01.

HDAC3 inhibition did not affect neurogenesis in the hippocampi of APP/PS1 mice. A) DCX levels were determined by immunostaining (n = 4–6 per group). B) Quantitative analysis of DCX-labeled neurons. C) DCX levels were also determined by western blotting (n = 4–6 per group). D) Quantitative analysis of DCX normalized to GAPDH.
HDAC3 inhibition suppresses oxidative stress in the hippocampi of APP/PS1 mice
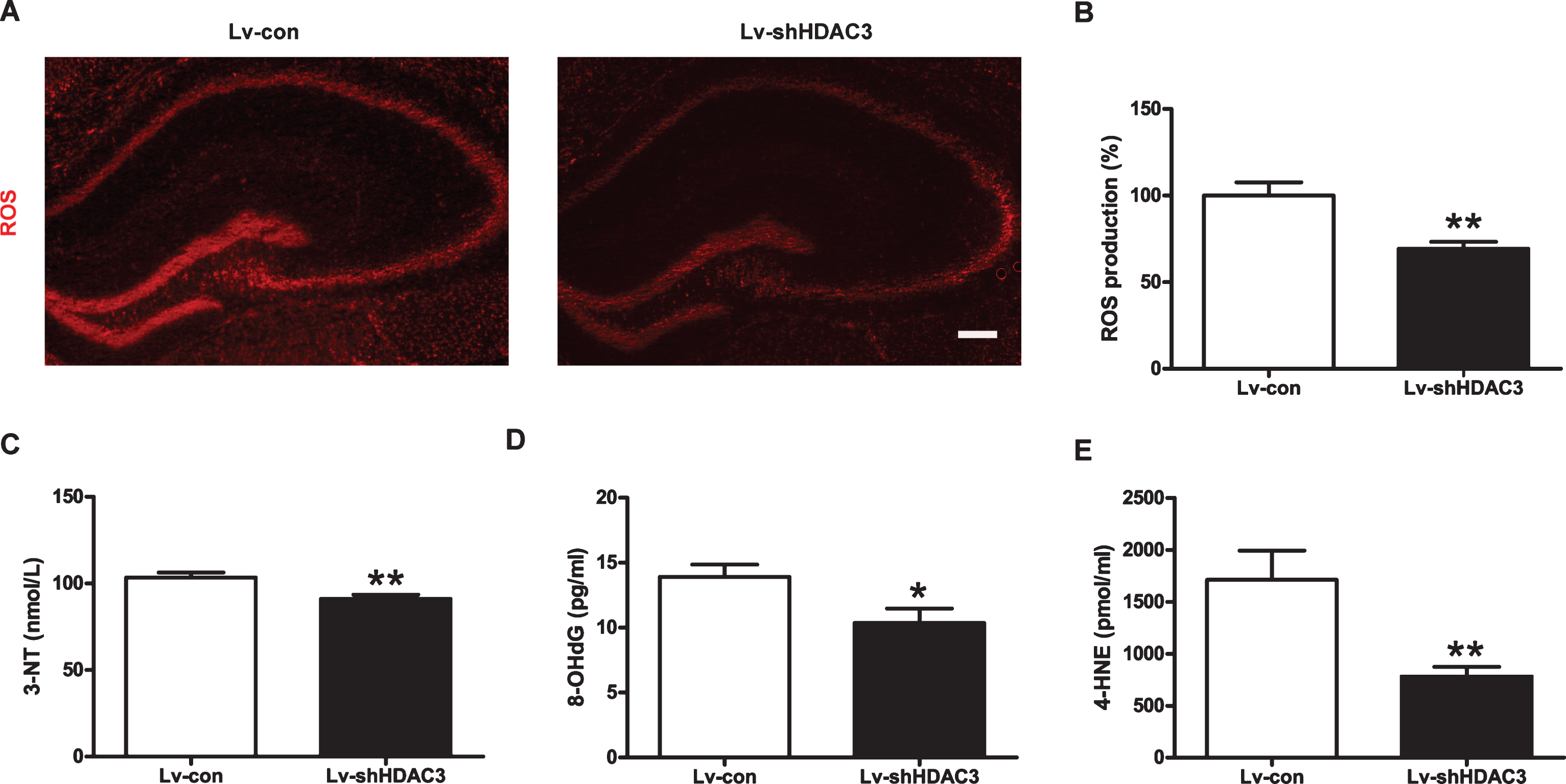
Increasing evidence shows that ROS overproduction plays a critical role in the pathologic process of AD [33]. As shown in Fig. 7A, B, the levels of ROS in the hippocampi of Lv-shHDAC3-treated mice were significantly decreased compared to those of the control group (p < 0.01). To further examine the oxidative stress in the hippocampi of AD mice, the levels of 4-HNE, 3-NT, and 8-OHdG were measured. As shown in Fig. 7C-E, the levels of 3-NT, 4-HNE, and 8-OHdG were significantly decreased in the hippocampi of AD mice after HDAC3 inhibition (p < 0.01, p < 0.05, and p < 0.01, respectively). These results demonstrated that HDAC3 inhibition could suppress oxidative stress in the hippocampi of APP/PS1 mice.
HDAC3 inhibition suppressed oxidative stress in the hippocampi of APP/PS1 mice. A) ROS levels in the hippocampi of APP/PS1 mice were determined by ROS assays (n = 4–6 in the Lv-con group, n = 4–6 in the Lv-shHDAC3 group). Scale bar = 200 μm. B) Quantitative analysis of DHE staining normalized to the control. Concentrations of 3-NT (C), 8-OHdG (D), and 4-HNE (E) in the hippocampi of APP/PS1 mice were measured by ELISA kits (n = 6 per group). *p < 0.05, **p < 0.01.
HDAC3 inhibition inactivates the Hippo signaling pathway in the hippocampi of APP/PS1 mice
The Hippo/MST1/2 signaling pathway plays an important role in modulating oxidative stress-induced cell death [16, 34]. To investigate the effects of the Hippo signaling pathway on the pathology of AD, the expression levels of c-Abl, p-MST1, and p-YAP were determined in the hippocampi of APP/PS1 mice by western blotting. As shown in Fig. 8A, B, the protein levels of c-Abl, p-MST1, and p-YAP were significantly increased in the hippocampi of AD mice compared with those in the hippocampi of WT mice (p < 0.05, p < 0.01, and p < 0.01, respectively). However, HDAC3 inhibition attenuated this signaling pathway, as demonstrated by significant reduction in the expression levels of c-Abl, p-MST1 and p-YAP (Fig. 8C, D, p < 0.05, p < 0.01, and p < 0.05, respectively).

HDAC3 inhibition inactivated the Hippo signaling pathway in the hippocampi of APP/PS1 mice. A) Protein levels of c-Abl, p-MST1/MST1, and p-YAP/YAP were detected in the hippocampi of APP/PS1 mice by western blotting (n = 4–6 per group). Quantification of the signal intensities of c-Abl (B), p-MST1/MST1 (C), and p-YAP/YAP (D) normalized to the control. E) C-Abl, p-MST1/MST1 and p-YAP/YAP levels in the hippocampi of APP/PS1 mice injected with Lv-con or Lv-shHDAC3 (n = 4–6 per group) were analyzed by western blotting. Quantification of the signal intensities of c-Abl (F), p-MST1/MST1 (G), and p-YAP/YAP (H) normalized to the control. *p < 0.05, **p < 0.01.
DISCUSSION
In the present study, we have demonstrated that HDAC3 is mainly expressed in neurons in the CNS and that HDAC3 inhibition significantly improves cell viability and attenuates ROS production in Aβ42-treated primary neurons. In addition, HDAC3 inhibition ameliorates spatial memory dysfunction in 6-month-old APP/PS1 mice by decreasing neuronal apoptosis in the hippocampi. The neuroprotective effect of HDAC3 inhibition might be associated with the reductions in oxidative stress and the regulation of the Hippo signaling pathway; these results are consistent with our previous findings showing that HDAC3 inhibition significantly decreases the amyloid plaque loads and Aβ levels in the brains of 9-month-old APP/PS1 mice [14]. Oxidative stress is reported to be a relatively early event in AD pathogenesis. In the brains of AD patients and models, markers of oxidative stress, including lipid peroxides, protein oxidation, DNA/RNA oxidation, and advanced glycation end-products (AGEs) are significantly increased by enhanced ROS generation and accumulation [35]. In the process of AD, redox balance is disturbed by the abnormal accumulation of Aβ, which might be related to mitochondrial dysfunction [36]. The accumulation of ROS also enhances Aβ generation, misfolding, and aggregation [37]. Furthermore, increasing evidence has shown that anti-oxidative therapy not only improves cognitive function and memory in AD mice, but also reduces the amount of Aβ deposition, indicating its potential neuroprotective effects in AD [38–41]. In our study, the levels of oxidative stress were decreased in primary cortical neurons and in the hippocampi of AD mice after HDAC3 inhibition, which might be associated with the enhanced spatial memory in Lv-shHDAC3-treated APP/PS1 mice.
Histone deacetylation has been shown to play critical roles in memory function under both physiological and pathological conditions, and HDAC inhibition is considered to be a promising therapy for the treatment of AD [42, 43]. The memory enhancement caused by the inhibition of specific HDAC isoforms inhibition might be associated with an increasing in the chromatin accessibility of transcription-dependent memory processes. RGFP966 attenuates Aβ42-induced LTP impairment [13], and HDAC3 inhibition ameliorates spatial memory dysfunction in APP/PS1 mice, indicating that HDAC3 inhibition might be a potential method for treating AD [14]. Interestingly, the inhibition of HDAC3 activity by RGFP966 also enhances synaptic plasticity in aged rat hippocampi [44], which indicates that HDAC3 plays an important role in both normal aging and AD pathogenesis. Two types of nonselective HDAC inhibitors, suberanilohydroxamic acid (SAHA) and romidepsin have been approved by the US Food and Drug Administration for treating cutaneous T-cell lymphoma and peripheral T-cell lymphoma respectively [45, 46]. Thus, more specific and potent HDAC3 inhibition might be a potential strategy for treating AD. Moreover, HDAC3 has been shown to induce or inhibit oxidative stress, depending on the cell models and treatments. HDAC3 exerts protective effects against oxidative stress by inducing the expression of HO-1 [47]. HDAC3 also contributes to inhibiting H2O2-induced oxidative stress and cell death via PTEN-induced putative kinase 1 (PINK1) in dopaminergic neurons [48]. On the other hand, the ketone metabolite β-Hydroxybutyrate inhibits the expression of HDAC3 and suppresses oxidative stress in in vitro and in vivo models of spinal cord injury [49]. RGFP966 inhibits cardiac oxidative stress and attenuates diabetic cardiomyopathy in the hearts of type 1 diabetic mice [50]. Our data have shown that HDAC3 inhibition suppresses oxidative stress in the hippocampi of APP/PS1 mice and in Aβ42-treated neuron, which might contribute to its neuroprotective effects.
Increasing evidence has shown that c-Abl plays an important role in the pathogenesis of AD. The levels of c-Abl are increased in the brains of AD mice, and c-Abl phosphorylates tau at several tyrosine residues [21, 51]. Aβ treatment of neuronal cells and intrahippocampal injections of Aβ activate c-Abl, and the inhibition of c-Abl attenuates memory deficits and decreases amyloid plaques and tau phosphorylation in the brains of AD mice [21–23]. However, Aβ oligomers is not able to induce c-Abl activation in EphA4-knockout neurons, which indicates that EphA4 might be an upstream target of c-Abl [22]. Exibuprofen, an active enantiomer of ibuprofen, inhibits the c-Abl/p-CDK5 pathway and improves spatial memory in AD mice [52]. C-Abl also increases the expression and phosphorylation levels of HDAC2, a negative regulator of memory formation in AD patients and animal models [53]. It has been reported that c-Abl/MST pathways are associated with oxidative stress in the brain. Oxidative stress induces c-Abl-dependent MST1 phosphorylation, which protects it from ubiquitination-mediated degradation, and then facilitates neuronal death [16]. In addition, the c-Abl/MST2 pathway mediates oxidative stress-induced neuronal death [25]. Moreover, YAP, a nuclear transcription factor downstream of the Hippo pathway, accelerates Aβ35-induced PC12 cell apoptosis by inducing the translocation and activation of Bax [54]. Low-power laser irradiation induces YAP phosphorylation and inhibits Aβ35-induced cell apoptosis [55]. In this study, we have shown that the c-Abl/MST1/YAP pathway was activated in the hippocampi of 6-month-old APP/PS1 mice, and HDAC3 inhibition partially inactivated this pathway. Interestingly, c-Abl directly binds to and phosphorylates YAP, leading to the physical stabilization of YAP in response to DNA damage [56]. Whether YAP acts a direct downstream target of c-Abl in AD models remains to be further investigated.
Conclusion
In conclusion, the current study demonstrates that HDAC3 inhibition increases cell viability and inhibits ROS production in Aβ42-treated primary neurons and ameliorates oxidative stress and spatial memory deficits in APP/PS1 mice. HDAC3 inhibition also downregulates the c-Abl/MST1/YAP pathway in the hippocampi of AD mice, which indicates that HDAC3 inhibition might be a potential strategy for AD treatment.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the National Nature Science Foundation of China (81230026, 81630028, 81671055, 81200839, and 81300988), the National Key Research and Development Program of China (2016YFC1300500-504), the Natural Science Foundation (BE2016610) of Jiangsu Province of China, Jiangsu Province Medical Youth Talent (QNRC2016024), and Jiangsu Provincial Key Medical Discipline (ZDXKA2016020).