
Abstract
As the primary mediator for synaptic transmission, AMPA receptors (AMPARs) are crucial for synaptic plasticity and higher brain functions. A downregulation of AMPAR expression has been indicated as one of the early pathological molecular alterations in Alzheimer’s disease (AD), presumably via amyloid-β (Aβ). However, the molecular mechanisms leading to the loss of AMPARs remain less clear. We report that in primary neurons, application of Aβ triggers AMPAR internalization accompanied with a decrease in cell-surface AMPAR expression. Importantly, in both Aβ-treated neurons and human brain tissue from AD patients, we observed a significant decrease in total AMPAR amount and an enhancement in AMPAR ubiquitination. Consistent with facilitated receptor degradation, AMPARs show higher turnover rates in the presence of Aβ. Furthermore, AD brain lysates and Aβ-incubated neurons show increased expression of the AMPAR E3 ligase Nedd4 and decreased expression of AMPAR deubiquitinase USP46. Changes in these enzymes are responsible for the Aβ-dependent AMPAR reduction. These findings indicate that AMPAR ubiquitination acts as the key molecular event leading to the loss of AMPARs and thus suppressed synaptic transmission in AD.
INTRODUCTION
Alzheimer’s disease (AD), the most common form of dementia, is defined by two pathological hallmarks: extracellular amyloid-β (Aβ) plaques and neurofibrillary tangles. Aβ is a peptide derived from successive cleavage of amyloid-β protein precursor (AβPP), by the action of β- and γ-secretases. An increasing amount of evidence has shown that dysregulation of synaptic function including basal transmission and synaptic plasticity is the crucial pathology in the early stage of AD [1], which eventually leads to memory loss and other cognitive deficits. Therefore, it is important to understand the cellular and molecular processes linking Aβ signaling to synaptic malfunction.
AMPA glutamate receptors (AMPARs) mediate most of the excitatory synaptic transmission in the brain. A large number of studies have established that the primary cellular mechanism employed in synaptic plasticity, the cellular process underlying learning and memory, is the alteration of AMPAR abundance in the postsynaptic domain [2–4]. Cognitive and psychological functions and behaviors are fundamentally affected by alterations in glutamate receptors. An increase in synaptic AMPAR levels and activity leads to long-lasting synaptic potentiation with an improvement in learning and memory. Conversely, a reduction in glutamate receptor expression is associated with the deterioration of mental faculties, such as memory loss, seen most notably in AD. In line with this, altered AMPAR synaptic accumulation has been shown to be a crucial pathological feature of AD, particularly during its early phase [5]. An important neuropathology in AD is a reduction in synaptic strength and dysregulation in synaptic plasticity including long-term plasticity [6] and homeostatic synaptic plasticity [7]. Ultimately, alterations in synaptic transmission and plasticity result, at least in part, from alterations in AMPAR trafficking and/or turnover [3]. Indeed, a downregulation in AMPAR levels and their synaptic localization is often observed in AD conditions including animal models and in human patients [8–12]. However, the molecular mechanisms leading to AMPAR reduction in AD remain unclear.
Ubiquitination is the most important posttranslational modification in the regulation of protein trafficking and turnover. The conjugation of a ubiquitin-chain to a target protein acts as a tag, signaling a membrane protein for internalization and proteasome-mediated degradation. Structurally, ubiquitin is an 8.5 kDa, 76 amino acid polypeptide that forms a compact structure with an exposed carboxy-terminal tail containing a diglycine motif that can be covalently ligated via an isopeptide bond to lysine (K) residues on a target substrate. Our previous work has shown that AMPARs are subjected to ubiquitination mediated by the E3 ligase Nedd4 [13], and the process can be reversed by the deubiquitinase USP46 [14]. Following ubiquitination, surface AMPARs will interact with EPS15, an adaptor protein that interacts with the ubiquitin-chain via its ubiquitin-binding motif, for internalization [15].
In this study, we show that in cultured rat cortical and hippocampal neurons, incubation with Aβ causes an upregulation in AMPAR ubiquitination and an increase in AMPAR internalization. As a consequence of ubiquitination, Aβ treatment leads to AMPAR internalization and a reduction in total AMPAR levels due to facilitated receptor degradation. In line with increased levels of AMPAR ubiquitination, incubation of neurons with Aβ results in increased expression of the E3 ligase Nedd4 and decreased levels of the DUB enzyme USP46. Manipulation of these ubiquitination-related enzymes blocked Aβ-induced AMPAR reductions. In addition, using brain tissue from AD patients we found an increase in AMPAR ubiquitination and a decrease in total AMPAR abundance. Therefore, our findings demonstrate that, via regulation of ubiquitination enzymes, Aβ incubation triggers AMPAR ubiquitination, followed by receptor internalization and degradation.
MATERIALS AND METHODS
Antibodies and reagents
Antibodies and reagents were obtained from the following commercial sources: The protein synthesis inhibitors cycloheximide (CHX) and anisomycin were purchased from Sigma-Aldrich and prepared in sterile water. All drugs were prepared in higher concentration stock solutions, stored in aliquots at –20°C, and thawed only once prior to use to preserve their potency. The GluA1 N-terminal mouse antibody (Millipore, MAB2263, RRID: AB_11212678) was purchased from Millipore, and GluA1 C-terminal rabbit antibody was homemade. Tubulin (Sigma-Aldrich, T3950, RRID: AB_477576) and GAPDH (Sigma-Aldrich, WH0002597M1, RRID: AB_1841801) antibodies were purchased from Sigma-Aldrich. Ubiquitin (Abcam, ab7780, RRID: AB_306069), HA (Abcam, ab1424, RRID: AB_301017), USP46 (Abcam, ab88795, RRID: AB_2043162), and Nedd4 (Abcam, ab14592, RRID: AB_301364) antibodies were purchased from Abcam.
Synthetic Aβ1 - 42 was purchased from Invitrogen and prepared according to the manufacturer’s instructions. Aβ oligomers were prepared as previously described [7]. In brief, the peptide was dissolved in phosphate-buffered saline (PBS) at 200μM and incubated at 37°C for 24 h. Samples were aliquoted, stored at –20°C, and thawed once directly prior to use.
Neuron culture, HEK cell culture, and transfection
Primary cultured cortical and hippocampal neurons were prepared from embryonic day 18 (E18) rat (Charles River Laboratories, Wilmington, MA) embryos of either sex as previously described [7, 17]. All animal procedures were in compliance with the policies of the Institutional Animal Care and Use Committee (IACUC) at Boston University. Briefly, neurons were dissociated from hippocampus or cortex and cultured on poly-L-lysine-coated (100μg/ml) dishes containing plating medium (DMEM containing 10% fetal bovine serum, 5% horse serum, 31 mg L-cysteine, and 1% penicillin/streptomycin/L-glutamine). Plating medium was then replaced by feeding medium (Neurobasal medium supplemented with 1% HS, 2% B-27 and 1% P/S/G) the day after cell plating. Neurons were maintained in feeding medium with FDU (10μM) supplemented at day 8 in vitro (DIV8) to suppress glial growth until experimental use. Human embryonic kidney (HEK) 293T cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin. HEK cells were split into 6-well plates (1 million/well) or 6 cm dishes (2 million cells/well) to grow overnight prior to transfection. Transfections in cultured neurons and HEK cells were performed with Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. The following plasmids were used: EGFP, GFP-AβPP770, HA-Ubiquitin, GFP-GluA1, USP46, Nedd4, HA-Ubi-K63, HA-Ubi-K48.
siRNAs were used to knock down Nedd4 expression [15].
Sample preparation and western blot
Human hippocampal tissues from control and AD patients were provided by National Institutes of Health (NIH) NeuroBioBank.
Transfected HEK cells or rat cultured cortical neurons used for western analysis were first rinsed with cold PBS and scraped from the culture dishes with RIPA lysis buffer [50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP40, 1% sodium deoxycholate (SDOC) and 1% sodium dodecyl sulfate (SDS)] containing protease inhibitors (Roche). After sonication, the lysates were centrifuged for 10 min at 13,000 rpm.
For the ubiquitination assay, the supernatant of 500μl in RIPA buffer was incubated with antibodies against GluA1 and protein A-Sepharose beads (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 15 h at 4°C. Immunocomplexes were washed three times with ice-cold NP-40 buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP40, 1% SDOC), resuspended in 2X Laemmli buffer, and denatured at 95°C for 10 min.
Prepared cell lysates or tissue lysates were subject to SDS-PAGE. Proteins were transferred to PVDF immunoblotting membranes (Bio-Rad) and probed for different targets with the stated antibodies. Immunoblots were visualized using a chemiluminescence detection system (GE Healthcare) and exposed to Fuji medical X-ray films (Fisher Scientific), scanned, and analyzed using the NIH ImageJ program.
Surface biotinylation assay
The surface biotinylation assay was conducted as described previously [18]. In brief, cortical neurons were first treated with Aβ or vehicle, washed with cold artificial cerebrospinal fluid (ACSF), and incubated in ACSF containing sulfo-NHS biotin conjugate (1 mg/ml, Pierce) for 10 min rocking at room temperature. Neurons were scraped into cold modified RIPA buffer containing protease inhibitors on ice and then rotated for 30 min after sonication at 4°C. After centrifugation, 1/10 supernatant of cell lysates was boiled with equal volume Laemmli 2X sample buffer. The rest of the supernatant was removed into a new tube containing 20μg (40μl) Avidin beads (Sigma, Oakville) and incubated overnight at 4°C. The beads were washed three times with 0.3% Triton-X-100 (Fisher Biotech) in PBS and boiled with equal volume Laemmli 2X sample buffer for western blot analysis.
Internalization assay
For AMPAR internalization assays, neurons were treated with Aβ or vehicle for 4 h and then incubated with GluA1 N-terminal antibody (1:300, Millipore) at 37°C for 10 min. Cells were rinsed twice with ACSF, and placed back into the culture medium containing Aβ or vehicle for 20 min. Neurons were then rinsed and fixed with 4% paraformaldehyde, followed by incubation with secondary antibodies conjugated to fluorophores of 488 nm (green) for 1 h to label the surface receptors. Neurons were then permeabilized with 0.3% Triton-X-100 in PBS for 10 min and incubated with another secondary antibody conjugated to a 555 nm fluorophore (red) for 1 h to visualize the internalized GluA1. After washing with PBS 3 times, neurons were mounted to microscope glass slides with Prolong Gold for subsequent visualization.
Immunocytochemistry
Transfected or treated hippocampal neurons were used for immunostaining analysis. Low-density (50,000/coverslip) hippocampal neurons were washed once in ACSF and fixed for 10 min in 4% paraformaldehyde. To stain the total protein, cells were permeabilized for 10 min in 0.3% Triton-X-100 (Fisher Biotech) in PBS, rinsed three times with PBS, and then subjected to a 1 h blocking (10% goat serum in PBS). Cells were incubated with primary antibodies (in 5% goat serum in PBS) for 2 h at room temperature, washed with PBS, and then incubated with Alexa Fluor-conjugated fluorescent secondary antibodies (Life Technologies) for 1 h. For surface staining, cells were immunostained under non-permeant conditions. Cells were then mounted to microscope glass slides with Prolong Gold anti-fade mounting reagent (Life Technologies) for visualization.
Image analysis
The immunostained coverslips were kept in the dark for at least 24 h before imaging. Using a Carl Zeiss inverted fluorescent microscope, neuron images were collected with a 63X oil-immersion objective (numerical aperture, 1.4). The exposure time of the fluorescent signal was adjusted manually to ensure that the signal intensity was within the full dynamic range by using a glow scale look-up table. Once an exposure time was established, it was employed throughout the imaging process for all samples. Neuron images were quantified using NIH ImageJ. For quantification, GluA1 puncta were first selected using a thresholding function, and the mean puncta size and intensity were automatically measured. GluA1 total intensity was calculated by multiplying the puncta mean intensity and the puncta size. Three dendritic segments from different branches were selected from each neuron. At least 10 neurons were analyzed for each condition.
Statistical analysis
All values are presented as mean±SEM. Data were evaluated statistically by using the Student’s t-test, one-way ANOVA, or two-way ANOVA followed by Dunnett’s Multiple Comparison Test. N indicates the number of independent experiments and samples in Western blots or the number of cells in immunostaining assays. p < 0.05 was considered to be statistically significant.
RESULTS
Aβ induces a decrease in the expression of total and surface AMPARs
To investigate the effect of Aβ on AMPAR expression, we treated DIV14 primary cortical neurons with Aβ (1μM) or vehicle for 24 h and 48 h. Western analysis showed that Aβ treatment caused an approximate 28% (24 h) and 53% (48 h) reduction in the protein amount of AMPAR GluA1 subunits compared to the control group (Fig. 1A, B). To examine the effect of Aβ on AMPAR abundance in dendrites, we performed immunostaining of AMPARs in Aβ treated hippocampal neurons. We found that incubation of Aβ significantly reduced GluA1 cluster intensity (0 h: 1.00±0.02; 24 h: 0.84±0.04; 48 h: 0.65±0.02. n = 30 cells for each condition) as well as cluster density (0 h: 8.69±0.61; 24 h: 6.07±0.39; 48 h: 4.75±0.50. n = 30 cells for each condition) (Fig. 1C-E). To examine the effect of endogenous Aβ produced by cells, we co-transfected hippocampal neurons with GFP and Aβ precursor AβPP770 [19] and immunostained GluA1 2-days after transfection. Consistent with Aβ treatment, neurons transfected with AβPP770 exhibited a marked decrease in fluorescence intensity of GluA1 compared to neurons transfected with pcDNA control (pcDNA: 1.00±0.10; AβPP770:0.81±0.06. n = 10 cells for each condition), which indicated a significant decrease in GluA1 expression (Fig. 1F, G).
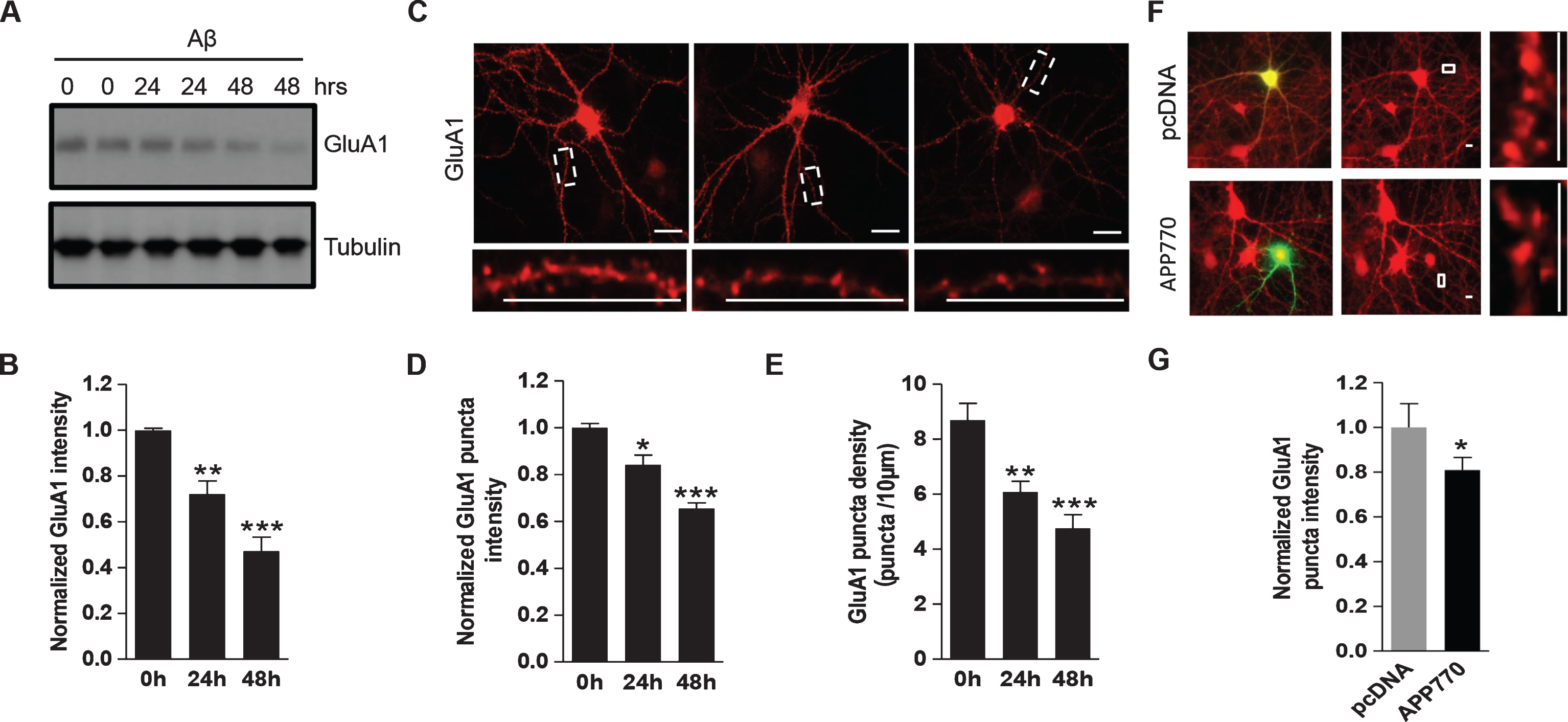
Aβ induces a reduction in AMPAR expression in neurons. A, B) Cultured cortical neurons were incubated with Aβ (1μM) for 24 h and 48 h. Western blot analysis showed that application of Aβ in cortical neurons caused a reduction in total GluA1 amount (n = 3 independent experiments). C-E) Hippocampal neurons were treated with Aβ and immunostained for total GluA1 after permeabilization. Aβ led to a reduction in both intensity (0 h: 1.00±0.02; 24 h: 0.84±0.04; 48 h: 0.65±0.02. n = 30 cells for each condition) and density (0 h: 8.69±0.61; 24 h: 6.07±0.39; 48 h: 4.75±0.50. n = 30 cells for each condition) of GluA1 clusters. F, G) Hippocampal neurons were transfected with AβPP770 or pcDNA as control and immunostained for total GluA1. GluA1 intensity was reduced in cells expressing AβPP770 (pcDNA: 1.00±0.10; AβPP770: 0.81±0.06, n = 10 cells for each condition). Green: GFP staining, indicating the transfected neuron; Red: GluA1 staining. *p < 0.05, **p < 0.01, ***p < 0.001, One-way ANOVA for B, D, and E; t-test for G. Scale bar represents 20μm.
Aβ incubation leads to internalization of surface AMPARs
We next investigated the effects of Aβ on the surface expression of AMPARs. DIV14 cortical neurons were incubated with Aβ for 24 h and 48 h, and AMPARs at the plasma membrane were isolated by surface biotinylation [20]. Western blot analysis showed that Aβ incubation caused a significant reduction in surface localized GluA1 (0 h: 1.00±0.02; 24 h: 0.61±0.05; 48 h: 0.58±0.05. n = 3 independent experiments) that was accompanied by a reduction in total AMPAR expression (0 h: 1.00±0.03; 24 h: 0.62±0.04; 48 h: 0.26±0.06. n = 3 independent experiments) (Fig. 2A, B).
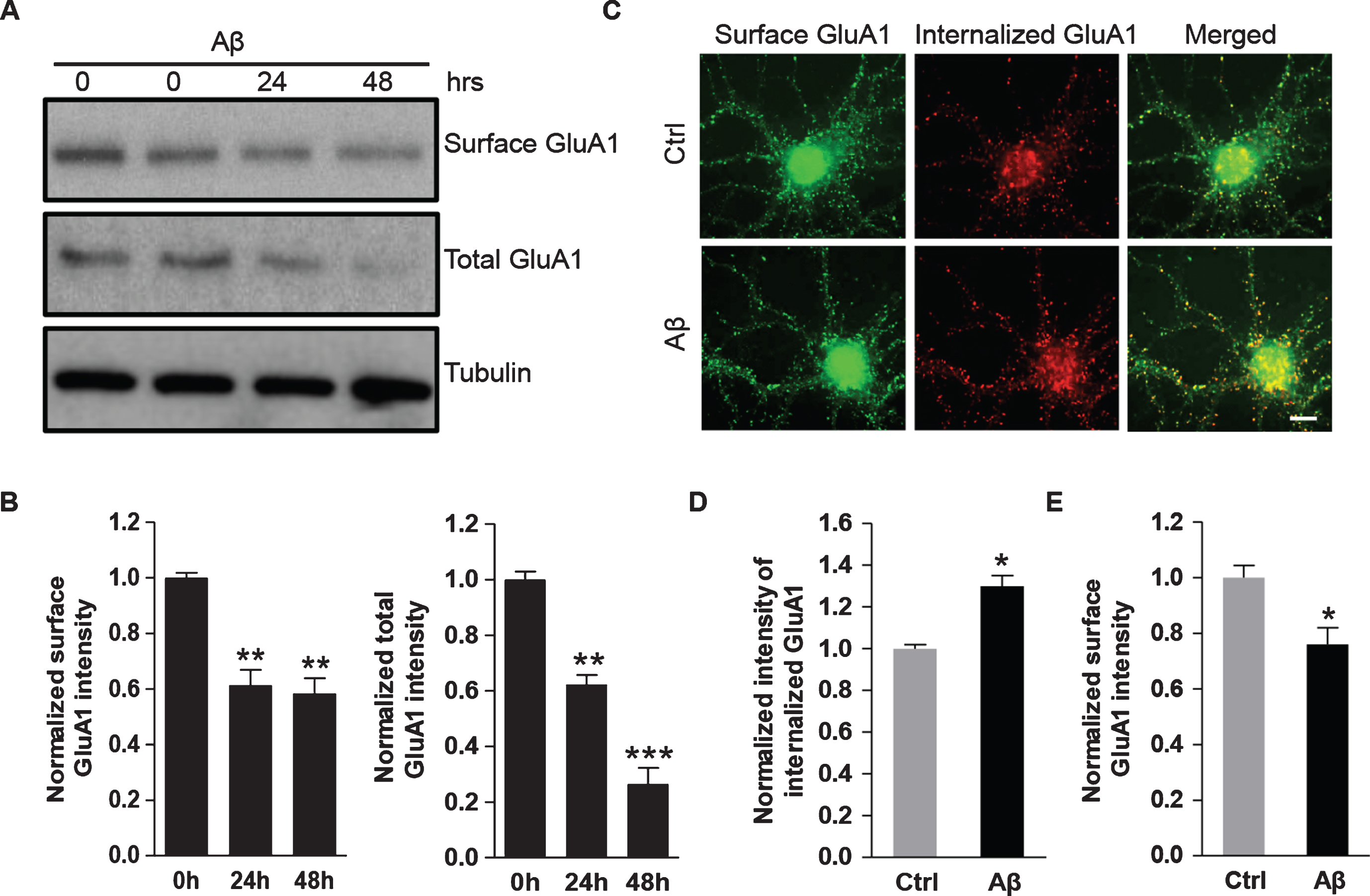
Aβ treatment leads to enhanced internalization and reduced surface expression of AMPARs. A, B) Cultured cortical neurons were incubated with Aβ (1μM) for 24 h and 48 h. AMPARs at the plasma membrane were isolated by surface biotinylation assays. Western blot showed that Aβ treatment significantly decreased both cell-surface GluA1 protein amounts (0 h: 1.00±0.02, 24 h: 0.61±0.05, 48 h: 0.58±0.05, n = 3 independent experiments) and total GluA1 protein amounts (0 h: 1.00±0.03, 24 h: 0.62±0.04, 48 h: 0.26±0.06, n = 3 independent experiments). C-E) Hippocampal neurons were treated with Aβ or vehicle for 4 h, and then subjected to immunostaining for surface GluA1. Aβ strongly induced the internalization of GluA1 (Ctrl: 1.0±0.02; Aβ: 1.3±0.05, n = 10 cells for each condition). Aβ drastically inhibited expression level of surface AMPARs (Ctrl: 1.0±0.04, Aβ: 0.76±0.06) *p < 0.05, **p < 0.01, ***p < 0.001, One-way ANOVA for B; t-test for D and E. Scale bar represents 20μm.
Since AMPARs undergo constant trafficking between the cell surface and the cytosolic domain via receptor membrane insertion and internalization [21], the Aβ-induced reduction of surface AMPARs could result from enhanced receptor internalization. To test this idea, we performed AMPAR internalization assays. The hippocampal neurons were first treated with Aβ or vehicle followed by an incubation with GluA1 N-terminal antibodies for internalization assays. The neurons were then fixed and incubated with the secondary antibody with green fluorescence to label the remaining surface GluA1. After permeabilization, the cells were incubated with a red secondary antibody to specifically label the internalized AMPARs. The results showed that a larger amount of AMPARs were internalized in cells incubated with Aβ compared the control group (Ctrl: 1.00±0.02, n = 10 cells; Aβ: 1.3±0.05, n = 10 cells) (Fig. 2C, D), strongly indicating a role for Aβ in stimulating AMPAR endocytosis. Consistently, Aβ led to a significant reduction of surface AMPARs (Ctrl: 1.00±0.04; Aβ: 0.76±0.06, n = 10 cells for each condition) (Fig. 2E).
Aβ increases the turnover rate of AMPARs
A stable level of AMPARs results from a balance between receptor synthesis and degradation, so the Aβ-induced reduction of AMPARs may result from elevated receptor degradation. To test this idea, we sought to determine the degradation rate of AMPARs. DIV14 cortical neurons were pre-incubated with Aβ or vehicle, and CHX (1μg/ml) was applied to block protein synthesis for different periods of time. We found that when protein synthesis was blocked by CHX, GluA1 showed a faster reduction rate with the presence of Aβ (Fig. 3A). The amount of GluA1 was reduced by approximately 30% at 9 h after CHX treatment in the controls, whereas approximately a 50% reduction was observed in cells exposed to Aβ and CHX (Fig. 3B). To further confirm this effect, we performed similar degradation assays with another protein synthesis inhibitor anisomycin (10μM). Again, a facilitated degradation was detected in the presence of Aβ. GluA1 protein levels were reduced by approximately 7% at 9 h after anisomycin incubation, but a nearly 50% reduction was found in neurons pre-incubated with Aβ (Fig. 3C, D).
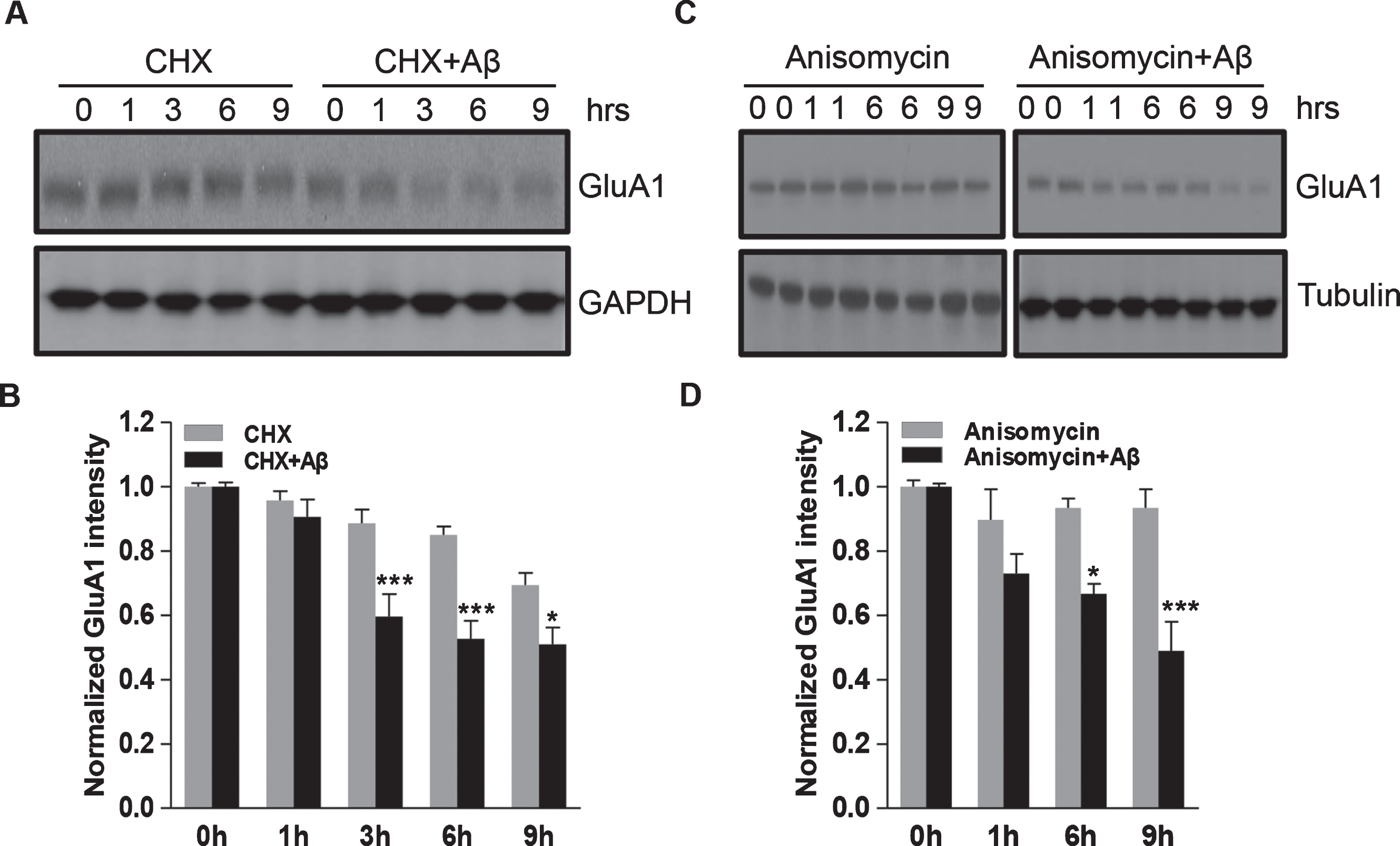
Aβ incubation results in a shortened half-life of GluA1 in cortical neurons. A, B) Cortical neurons were treated with CHX (1μg/ml) to block protein synthesis for 1–9 h, and the GluA1 protein levels at different time points were analyzed by western blot. Aβ treatment led to a reduction in GluA1 half-life (n = 3 independent experiments). C, D) Similar experiments were performed using anisomycin (10μM) to block protein synthesis. Western blot analysis showed a shortened GluA1 half-life in neurons treated with anisomycin (n = 3 independent experiments). *p < 0.05, ***p < 0.001, two-way ANOVA.
Aβ triggers AMPAR ubiquitination in both cortical neurons and HEK cells
Ubiquitination is one of the most important post-translational modifications in the control of protein abundance. The lysine residues in a target protein are conjugated with multiple ubiquitin (Ubi) units to form a poly-ubiquitin chain, which can be recognized by the proteasome for degradation. Our previous work demonstrated that AMPARs are subject to direct ubiquitination, which leads to receptor internalization and proteasomal degradation [17]. Therefore, we hypothesized that GluA1 ubiquitination contributes to the Aβ-induced AMPAR degradation. To test this idea, we treated cortical neurons with Aβ for 24 h and 48 h and probed the ubiquitination level of GluA1 using immunoprecipitation. According to the results, we found that the control neurons (0 h) had a low level of ubiquitination, while the neurons treated with Aβ showed significantly greater Ubi signals in a time-dependent pattern (Fig. 4A). Analysis showed that compared to the control, Aβ incubation led to a significant increase in GluA1 ubiquitination intensity (0 h: 1.00±0.03; 24 h: 1.52±0.06; 48 h: 1.57±0.09. n = 3 independent experiments) (Fig. 4B). To further examine the role of endogenous Aβ, we co-transfected HEK cells with HA-ubiquitin and GFP-GluA1, together with AβPP770 or pcDNA as a control, and used immunoprecipitation for a ubiquitination assay. Consistent with Aβ treatment, AβPP770 expression led to an increase in intensity of the GluA1 ubiquitination signal. Compared to the control, the GluA1 ubiquitination level was significantly increased by 66% in cells transfected with AβPP770 (pcDNA: 1.00±0.02; AβPP770:1.66±0.05, n = 3 independent experiments) (Fig. 4C, D). Also, the total amount of AMPARs was reduced in AβPP770-expressing cells, consistent with ubiquitination-dependent GluA1 degradation (Fig. 4C, D). To evaluate AMPAR ubiquitination in vivo, we also performed the AMPAR ubiquitination assay using hippocampal brain samples from human AD patients. Consistent with our in vitro results, AD brain samples showed increased levels of GluA1 ubiquitination (Ctrl: 1.00±0.06; AD: 1.72±0.27, n = 6 samples for each condition) together with a significant reduction in total AMPAR amounts (Ctrl: 1.00±0.13; AD: 0.42±0.17, n = 6 samples for each condition) (Fig. 4E, F). These results strongly support the idea that Aβ-induced AMPAR reduction in AD results from an enhancement in AMPAR ubiquitination.
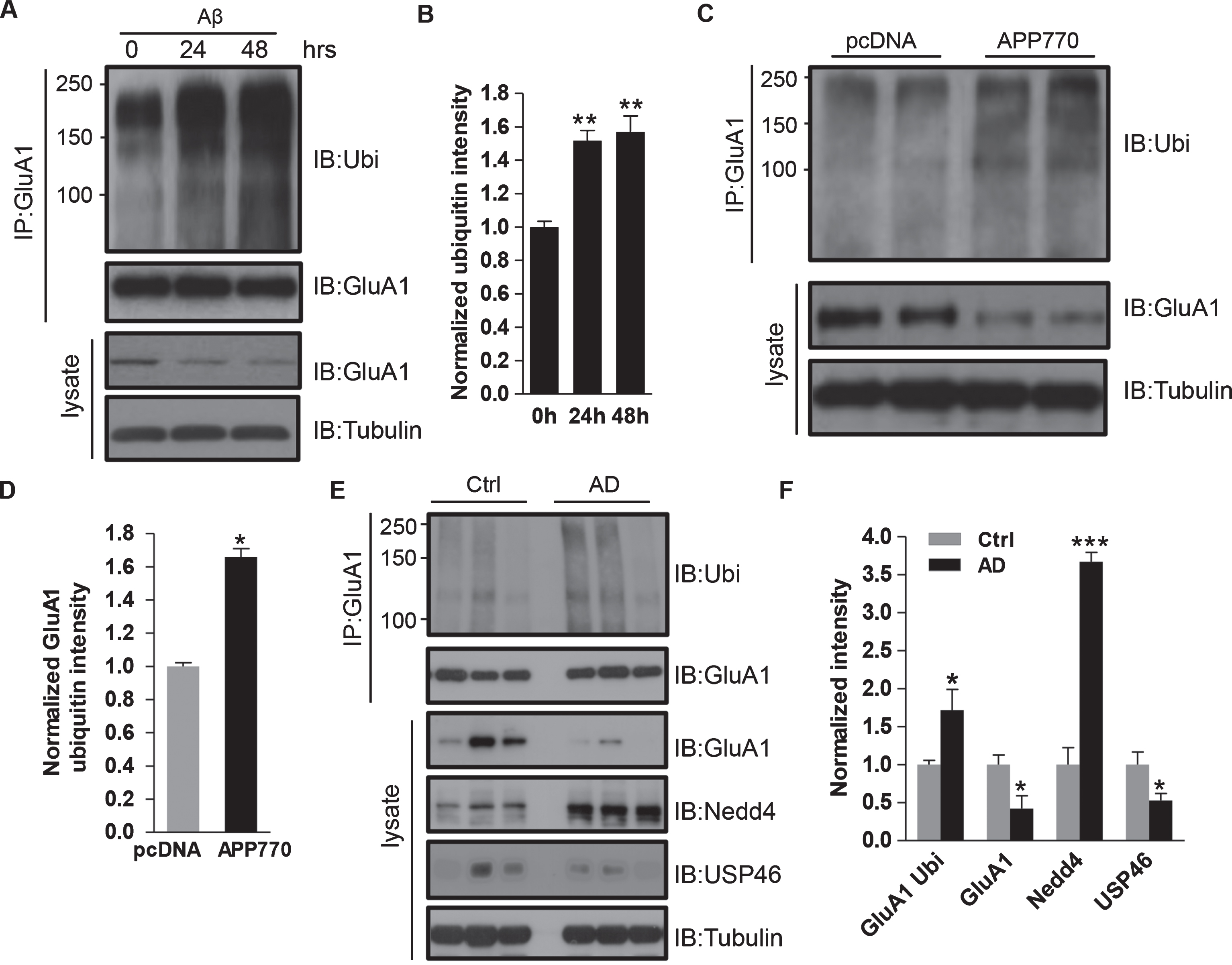
Aβ induces AMPAR ubiquitination. A, B) Cortical neurons were treated with Aβ for 24 h and 48 h, and GluA1 was immunoprecipitated for ubiquitin detection. Aβ incubation markedly increased GluA1 ubiquitination and its protein amount (0 h: 1.00±0.03; 24 h: 1.52±0.06; 48 h: 1.57±0.09, n = 3 independent experiments). C, D) HEK cells were co-transfected with GFP-GluA1 together with either AβPP770 or pcDNA. Ubiquitination assays showed that overexpression of AβPP770 led to a significant increase in GluA1 ubiquitination and a marked decrease in total GluA1 amount (pcDNA: 1.00±0.02; AβPP770:1.66±0.05, n = 3 independent experiments). E, F) Control and AD patient brain samples were analyzed by western blot. AD brains showed a decrease in total GluA1 (Ctrl: 1.0±0.13; AD: 0.42±0.17, n = 6 samples for each condition) and an increase in GluA1 ubiquitination intensity (Ctrl: 1.00±0.06; AD: 1.72±0.27, n = 6 samples for each condition). The expression level of Nedd4 was significantly enhanced (Ctrl: 1.00±0.23; AD: 3.67±0.12, n = 6 samples for each condition) and the expression of USP46 (Ctrl: 1.00±0.17; AD: 0.53±0.09, n = 6 samples for each condition) was evidently decreased in human AD patients as compared to control samples. Tubulin was used as loading control. *p < 0.05, **p < 0.01, ***p < 0.001, One-way ANOVA for B; t-test for D, F, and H.
Aβ incubation regulates the expression of AMPAR ubiquitin enzymes in neurons
Protein ubiquitination and degradation are highly dynamic and reversible. The Ubi-activating enzymes, Ubi-conjugating enzymes, and Ubi-ligases (E3s) induce the ubiquitination of substrates, in which the E3s specifically recognize their substrates. On the other hand, the deubiquitinating enzymes (DUBs) remove the Ubi-chain from substrates and prevent further degradation of the substrates. Therefore, the Aβ-induced AMPAR ubiquitination increase could result from activation of the E3 ligase or suppression of the DUB activity. Our previous work has shown that the E3 ligase Nedd4 and the deubiquitinating enzyme USP46 are responsible for AMPAR ubiquitination and deubiquitination, respectively. We wondered whether these enzymes were involved in Aβ-induced GluA1 ubiquitination. To test this idea, we first examined the ubiquitination enzymes in human AD brain lysates. Interestingly, in the AD samples we found that the expression of Nedd4 was increased (Ctrl: 1.00±0.23; AD: 3.67±0.12, n = 6 samples for each condition) whereas the expression of USP46 was decreased (Ctrl: 1.00±0.17; AD: 0.53±0.09, n = 6 samples for each condition) (Fig. 4E, F). To further investigate the role of Aβ, we then treated DIV14 cortical neurons with Aβ over various periods of time and examined the expression level of Nedd4 and USP46. Consistently, we found that Aβ treatment resulted in a decrease in the amount of USP46 and an increase in Nedd4 expression (at 24 h, USP46:0.51±0.07; Nedd4:1.41±0.06, n = 3 independent experiments), suggesting that both enzymes are involved in Aβ-mediated GluA1 ubiquitination (Fig. 5A, B). To further confirm the role of Nedd4 and USP46 in Aβ-induced AMPAR ubiquitination, HEK cells were co-transfected with GluA1, Ubi, AβPP770 as well as USP46 or Nedd4. Indeed, in AβPP770-expressing cells, expression of Nedd4 enhanced GluA1 ubiquitination, whereas USP46 caused a marked reduction in receptor ubiquitination (Fig. 5C).
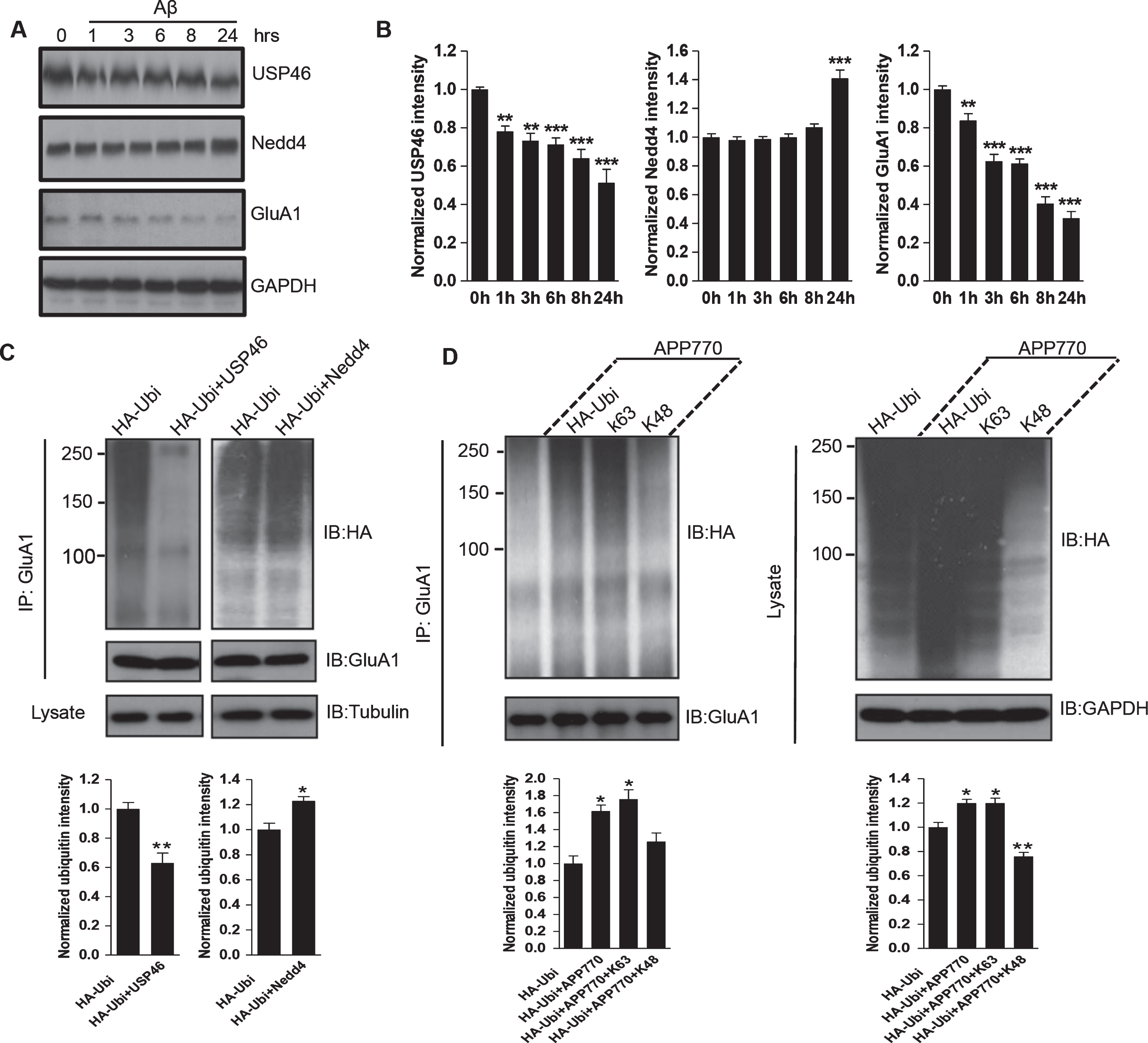
Role of ubiquitination enzymes in Aβ-induced AMPAR ubiquitination. A, B) Cortical neurons were incubated with Aβ at indicated time points and analyzed by western blot. Aβ application led to an increase in Nedd4 and a decrease in UPS46 in expression (at 24 h, USP46:0.51±0.07; Nedd4:1.41±0.06, n = 3 independent experiments). C) HEK cells were co-transfected with GFP-GluA1, HA-Ubi, AβPP770, together with either USP46 or Nedd4. Total GluA1 proteins were immunoprecipitated and probed with anti-HA antibodies. AβPP770-induced GluA1 ubiquitination was reduced by USP46 and increased by Nedd4. D) HEK cells were co-transfected with GFP-GluA1, AβPP770 (except the first lane), together with either HA-ubiquitin or mutant ubiquitin containing only one lysine (K63 or K48). K63, but not K48, was sufficient for GluA1 ubiquitination. *p < 0.05, **p < 0.01, ***p < 0.001, One-way ANOVA for B.
Aβ induces K63 type AMPAR ubiquitination
A poly-ubiquitin chain is formed by a series of conjugations of ubiquitin molecules to lysine (K) residues or other Ubi units. There are 7 lysine residues, K6, K11, K27, K29, K33, K48, and K63, on a single Ubi protein that can form different types of ubiquitin-chains for protein ubiquitination. However, only two types of ubiquitin-chains are most commonly adopted in protein ubiquitination, i.e., K48 and K63 chains [22]. To investigate the types of ubiquitin-chains that form on AMPARs in the presence of Aβ, we co-transfected HEK cells with GFP-GluA1, AβPP770, and different HA-Ubi constructs in which most lysine residues are mutated to arginine with only the K48 (Ubi-K48) or K63 (Ubi-K63) lysine remaining. GluA1 was immunoprecipitated for the ubiquitination assay. According to the results, the cells expressing Ubi-K63 showed a high level of GluA1 ubiquitination, especially with the expression of AβPP770. In contrast, the AβPP770 expression failed to increase GluA1 ubiquitination with the presence of Ubi-K48 (Fig. 5D). Taken together, these results indicate that K63 conjugation is the main type used for Aβ-induced GluA1 ubiquitination, which is consistent with our previous findings in GluA1 ubiquitination [14].
USP46 and Nedd4 are necessary for Aβ-induced AMPAR ubiquitination
To confirm the direct involvement of both USP46 and Nedd4 in Aβ-induced AMPAR ubiquitination and degradation, neurons were transfected with USP46 or Nedd4 siRNA, together with GFP as a transfection indicator. One day later, neurons were incubated with Aβ or vehicle for 24 h and the surface GluA1 was immunostained with anti-GluA1 N-terminal antibodies under non-permeant conditions. We found that Aβ caused a significant decrease in GluA1 intensity in the GFP-transfected cells (GFP+Ctrl: 1.00±0.04; GFP+Aβ: 0.62±0.03, n = 15 cells for each condition) (Fig. 6A, C); however, in neurons with USP46 overexpression, only a modest change was detected in surface GluA1 immunointensity (USP46+Ctrl: 1.00±0.02; USP46+Aβ: 0.94±0.02, n = 15 cells for each condition) (Fig. 6A, C). Similarly, in neurons with Nedd4 siRNA knockdown, application of Aβ failed to produce a significant reduction in surface GluA1 (Nedd4 siRNA+Ctrl: 1.00±0.03; Nedd4siRNA+ Aβ: 0.96± 0.02; GFP+Ctrl: 1.00±0.05; GFP+Aβ: 0.70±0.04, n = 15 cells for each condition) (Fig. 6B, D). These findings strongly indicate that both the E3 ligase Nedd4, and the DUB enzyme USP46, are directly implicated in Aβ-dependent AMPAR ubiquitination. Nedd4 knockdown can fully rescue the Aβ effect, while the overexpression of USP46 can partially rescue the decrease in AMPAR levels. Taken together, our data indicates that the Aβ-induced AMPAR reduction results from an increased level of AMPAR ubiquitination mediated by Nedd4 activation as well as suppression of USP46.
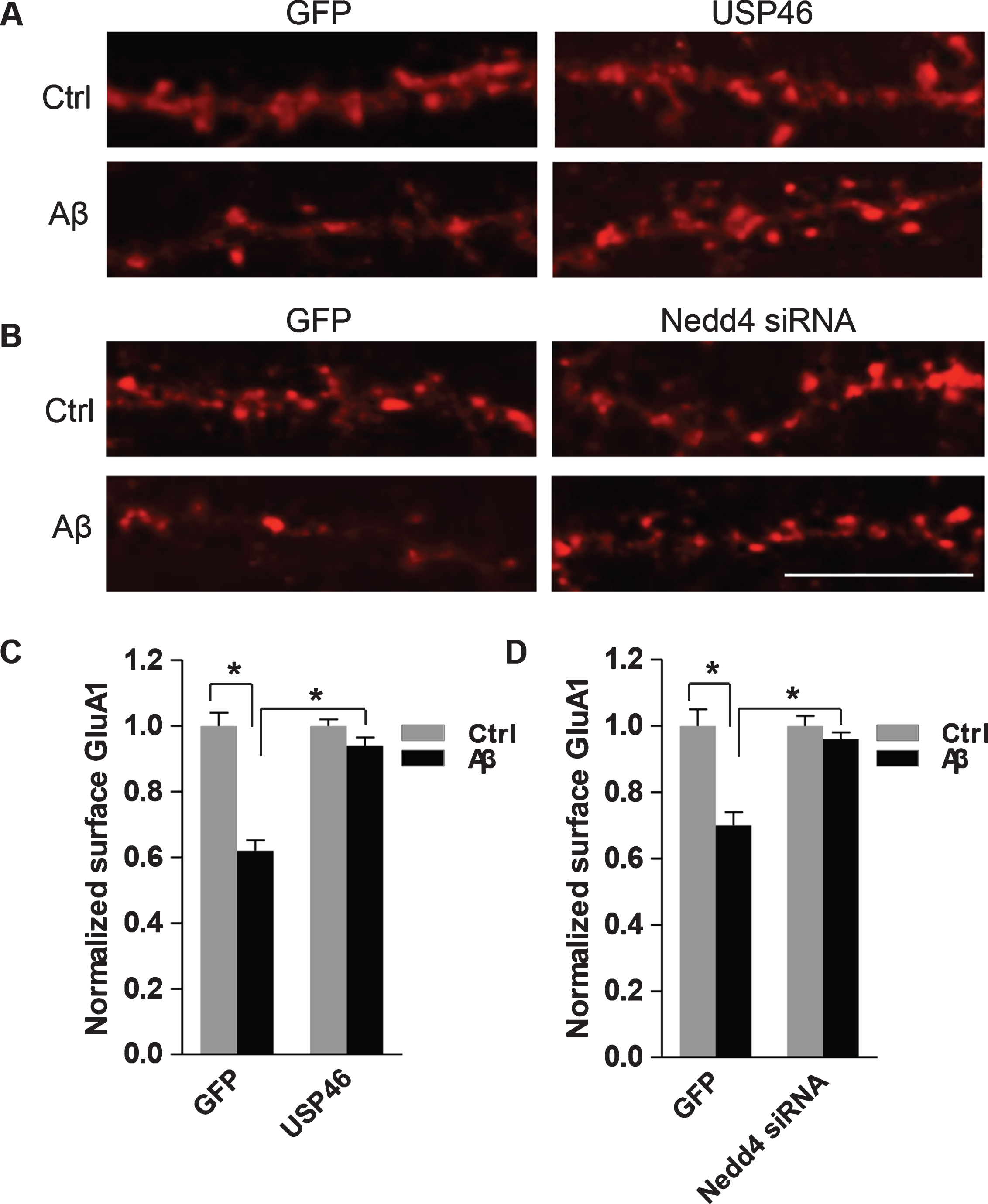
USP46 and Nedd4 mediate Aβ-dependent AMPAR downregulation. A) Hippocampal neurons were co-transfected with USP46 or (B) siRNA against Nedd4, together with GFP. 24 h after transfection, neurons were treated with Aβ or vehicle (Ctrl) for 24 h and surface GluA1 was immunostained with antibodies against the N-terminal of GluA1. C) Overexpression of USP46 (GFP+Ctrl: 1.00±0.04, GFP+Aβ: 0.62±0.03; USP46+Ctrl: 1.00±0.02: USP46+Aβ: 0.94±0.02, n = 15 cells for each condition) or (D) knockdown of Nedd4 (GFP+Ctrl:1.00±0.05, GFP+Aβ: 0.70±0.04; Nedd4 siRNA+Ctrl:1.00±0.03, Nedd4 siRNA +Aβ: 0.96±0.02. n = 15 cells for each condition) blocked Aβ-induced reduction of surface AMPARs. *p < 0.05, two-way ANOVA. Scale bar represents 20μm.
DISCUSSION
AMPARs and synaptic activity are the common substrates in the execution of normal brain functions as well as the pathogenesis of neurological diseases. A reduction in AMPAR expression and synaptic localization contributes to the weakened synaptic strength that is one of the early alterations in AD [5, 24]. However, the specific molecular mechanisms leading to an AMPAR reduction remain unclear. In this study, we show that ubiquitination of AMPARs is upregulated in the presence of Aβ in cultured neurons, as well as in the brains of AD patients. Consistent with enhanced ubiquitination, Aβ causes an increase in receptor internalization, a facilitated degradation and a reduction in total receptor amount. Under Aβ conditions, the elevated AMPAR ubiquitination is accompanied with increased expression of the AMPAR E3 ligase Nedd4 and decreased levels of the DUB enzyme USP46. In addition, the Aβ-induced AMPAR reduction can be rescued by Nedd4 knockdown or USP46 overexpression, indicating the involvement of both Nedd4 and USP46. These findings are consistent with two recent studies showing the role of Aβ in AMPAR ubiquitination and reduction [9, 25].
In the presence of Aβ under AD conditions, AMPARs may be regulated in two stages. In the early phase, AMPARs are internalized from the synaptic surface, resulting in reduced surface localization of the receptor and suppression of synaptic transmission, without affecting the total receptor amount. This change in trafficking is regulated by an intracellular signaling cascade that may include protein phosphorylation mediated by PKA or CaMKII [24, 26] or local mitochondrial distribution and energy supply [27]. Following long-term or strong Aβ incubation, AMPARs become targeted for ubiquitination, leading to not only an elevated rate in receptor internalization but also receptor degradation. Thus, the fate of AMPARs shifts from a temporary and reversible surface reduction due to elevated internalization, to a persistent, terminal removal via ubiquitination-dependent degradation.
It is not clear whether the Aβ-induced ubiquitination selectively targets synaptic AMPARs, or affects the entire pool of AMPARs in the neuron. When AMPAR puncta, presumably synaptically localized receptors, were analyzed, we found that application of Aβ led to a marked decrease in GluA1-containing AMPARs in rat cortical neurons. Neurons transfected with AβPP770 or exposed to synthetic Aβ also displayed a decrease in GluA1 puncta density, consistent with spine withdrawal by Aβ or in AD [10]. Our previous study has indicated that the plasma membrane AMPARs, but not the cytosolic ones, are ubiquitinated [15]. Distinct from constitutive and recycling AMPAR trafficking under basal conditions, the internalization of ubiquitinated AMPARs are initiated via an adaptor protein EPS15 [15], which contains two ubiquitin-interacting motifs (UIM) at the C-terminal domain [28]. We have shown that interaction of EPS15 with the ubiquitin chain in AMPARs is responsible for AMPAR internalization [15]. It is possible that EPS15 is also involved in Aβ-dependent AMPAR internalization in AD.
Ultimately, the ubiquitination enzymes are responsible for Aβ-induced AMPAR ubiquitination. Consistent with a previous report [29], we found that Aβ incubation in neurons resulted in an upregulation in Nedd4, an E3 ligase responsible for GluA1 ubiquitination [30], together with a downregulation of the GluA1 DUB enzyme USP46 [31, 32]. The requirement for these enzymes was demonstrated by a blockade of the Aβ-mediated effect on AMPAR reduction after Nedd4 knockdown or USP46 overexpression. Consistent with this, a recent study also indicated a crucial role for Nedd4 in an Aβ-induced decrease in surface AMPARs [25]. Nedd4 has been shown to be involved in cognitive function and other neurodegenerative and neurological diseases [33–36]. Since both Nedd4 and USP46 [13, 14], as well as the proteasome [37], are localized at synapses, alterations of these enzymes by Aβ likely cause the synaptic AMPARs to be ubiquitinated and degraded locally within spines [38].
Our findings provide novel insights into the molecular mechanism regarding AMPAR dysregulation in AD. Although neurodegenerative disorders such as AD, Huntington’s disease, and Parkinson’s disease have multiple causative factors, these diseases share common mechanisms of neuronal cellular dysfunction, including excitotoxicity, aberrant protein deposition, mitochondrial impairment, and dysregulation of the ubiquitination system. Thus, our findings on Aβ-dependent AMPAR removal via ubiquitination may also have implications in other neurodegenerative diseases with high levels of Aβ including Down’s syndrome and Parkinson’s disease.
Footnotes
ACKNOWLEDGMENTS
We thank the National Institutes of Health (NIH) NeuroBioBank for providing human brain tissues. We thank James Gilbert, Margaret O’Connor and Margaret Hastings for assistance in manuscript preparation. This work was supported by NIH grant R01 MH079407 (HYM).