
Abstract
The role of ABCA7 in brain homeostasis and Alzheimer’s disease (AD) is currently under intense scrutiny, since it has been reported that polymorphisms in the Abca7 gene and a loss of function of the protein are closely linked to excessive accumulation of amyloid peptides and disturbed cholesterol homeostasis. The blood-brain barrier (BBB), which isolates the brain from the blood compartment, is involved in both of these processes. We therefore hypothesized that ABCA7 downregulation might affect cholesterol and amyloid exchanges at the BBB. Using siRNA and primary cultures of mouse endothelial cells purified from brain microvessels and seeded on Transwell ® inserts, we investigated the role of ABCA7 in cholesterol and amyloid exchanges across the BBB. Our results showed that a decrease in ABCA7 expression at the BBB provokes in vitro a reduction in ABCA1 expression and a decrease in APOE secretion. In vitro, these decreases reduce cholesterol exchange across the BBB, particularly for high-density lipoproteins and ApoA-I particles. When ABCA7 was absent, we observed a reduction in Aβ peptide basolateral-to-apical transport in the presence of ApoA-I, with non-significant changes in the expression levels of Rage, Lrp1, Abcb1, Abcc1, and Abcg2. Our study in murine BBB model highlighted a putative new role for ABCA7 in AD via the protein’s involvement in cholesterol metabolism and amyloid clearance at the BBB.
INTRODUCTION
In developed countries, Alzheimer’s disease (AD) is the most prevalent form of dementia in the elderly [1]. One of the pathological hallmarks of this neurodegenerative disease is the accumulation of amyloid-β peptides (Aβ) in brain parenchyma and cerebral blood vessel walls [2]. It has been clearly demonstrated that the majority of late-onset cases of AD are associated with several risk factors linked to cholesterol metabolism and transport [3, 4]. For example, the apolipoprotein E ɛ4 (APOE ɛ4) allele is the strongest genetic risk factor for sporadic, late-onset AD. The ApoE4 protein is the main cholesterol carrier in brain tissue and is thus involved in regulation of cholesterol metabolism in the brain. The APOE ɛ4 allele is also known to promote Aβ aggregation [5]. In contrast, the APOE ɛ2 allele is associated with protection against AD. Moreover, a high-cholesterol diet and thus hypercholesterolemia are associated with AD. In preclinical studies of murine AD models, the hypercholesterolemia induced by a cholesterol-enriched diet was associated with an elevated level of Aβ peptides in the brain [6], whereas Aβ deposition was lower when mice were treated with cholesterol synthesis inhibitors [7]. Thus, the level of cholesterol in the blood can modify Aβ peptide levels in the brain [6, 9]. These effects are seen despite the presence of the blood-brain barrier (BBB), which restricts the passage of several molecules (including cholesterol) from the circulation into the brain. The key structural components of the BBB are the brain endothelial cells (BECs), which are sealed together by tight junctions. Therefore, the passage of molecules across the BBB is facilitated by transporters that are expressed at the BECs’ luminal and/or abluminal membranes.
These transporters include members of the ATP-binding cassette (ABC) superfamily, which use ATP hydrolysis to transport a wide range of molecules against their concentration gradients and thus participate in the maintenance of the brain’s microenvironment [10]. At the BBB, ABCA1 contributes to cholesterol efflux [11, 12] by transferring lipids to apolipoproteins and initiating high-density lipoprotein (HDL) formation [13]. Other ABC transporters involved in the lipidation/maturation of lipoproteins are also expressed at the BBB [11, 14], although their exact roles require further characterization [15, 16]. Thus, ABC transporters are major players in cholesterol efflux and lipoprotein metabolism [13, 17], and confer the BBB with an important role in the regulation of brain cholesterol levels [18].
Interestingly, genome-wide association studies (GWASs) of AD have never identified single-nucleotide polymorphisms in members of the ABC transporter superfamily as possible genetic factors - albeit with the exception of ABCA7, for which loss of function is associated with an increased risk of developing AD [19, 20].
In the periphery, ABCA7 participates in cholesterol efflux to ApoE discs in kidney cells [21], although cholesterol release is not affected in macrophages from Abca7 knockout (KO) mice [22]. In the brain, Abca7 is strongly expressed in several different cell types but is particularly present in microglia [23]. It has been demonstrated that ABCA7 is involved in phagocytosis and in Aβ clearance by microglia [24]. In neurons, ABCA7 deficiency alters the lipid profile and promotes Aβ peptide production through increased BACE 1 expression [25]. Despite the fact that ABCA7 expression has been detected in an in vitro model of the BBB [14], its function at this barrier has not previously been studied.
By using RNA silencing, a mouse-based in vitro model of the BBB [26–28], the objective of the present study was to characterize the effect of ABCA7 deficiency on cholesterol and amyloid exchanges at the BBB.
Our in vitro results show that ABCA7 deficiency induced a decrease in ABCA1 and APOE expression, as well as a decrease in cholesterol efflux towards ApoA-I and HDL particles. When ABCA7 is downregulated, a decrease in Aβ peptide efflux across the BBB is observed in the presence of ApoA-I. Taken as a whole, our results highlight a role for ABCA7 in brain cholesterol metabolism and in Aβ peptide clearance across the BBB. The results may also explain (at least in part) the association between ABCA7 and AD.
MATERIALS AND METHODS
Chemicals
siRNA and 5X siRNA buffer were purchased from Dharmacon (Velizy-Villacoublay, France). Lyophilized siRNA was reconstituted in 1X siRNA buffer at a concentration of 5μM. ApoA-I and HDL were purchased from VWR (Fontenay-sous-Bois, France). Transfection reagent Ribocellin, ApoE2 and ApoE4 were purchased from Clinisciences (Nanterre, France). Lyophilized ApoE2 and E4 were reconstituted in sterile water at a concentration of 1 mg/mL. Lucifer yellow (LY), FITC-inulin and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (Lyon, France). [3H]-cholesterol (40–60μCi/mmol) was purchased from Biotrend (Köln, Germany) and [14C]-sucrose was bought from Perkin Elmer Life and Analytical Sciences (Waltham, MA, USA). Fluorescent Aβ40-Cy5 labeled was purchased from Phoenix Pharmaceuticals (Strasbourg, France).
Cell culture reagents
Most reagents were purchased from Sigma-Aldrich: HEPES 1 M, amino acids 50X, vitamins 100X, tosyl-lysine-chloromethyl-ketone (TLCK), Hanks balanced saline solution (HBSS) 1X, dextran and basic fibroblast growth factor (bFGF). L-glutamine was obtained from Merck (Fontenay-sous-Bois, France). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Life Technologies (Illkirch, France). DNase I and collagenase/dispase were purchased from Roche Diagnostics (Meylan, France). Matrigel was purchased from BD Biosciences (Le Pont de Claix, France) and Transwell ® polyester membrane inserts (pore size: 0.4μm for all experiments) were obtained from Corning (Corning, NY, USA). Newborn calf serum (CS) was obtained from Integro B.V. (Zaandam, The Netherlands).
Animal experiments
In accordance with French legislation, the animal facility at the University of Artois has been approved by the local authorities (reference: B62-498-5). In compliance with European Union legislation (Directive 2010/63/EU), all procedures were approved by the local Animal Care and Use Committee (Comité d’Éthique en Expérimentation Animale du Nord-Pas-De– Calais; reference: C2EA 75) and the French Ministry of Research (reference: 2015090115412152). For in vitro experiments, 4- to 6-week-old C57BL/6 mice were provided by Janvier Labs (Le Genest-Saint-Isle, France).
Cell culture
The in vitro BBB model consisted of a primary monoculture of mouse brain capillary endothelial cells (MBCECs) seeded on filters coated with Matrigel ® [26–28]. Briefly, after removing the meninges and the outer vessels, cortices were homogenized in HBSS containing 10 mM HEPES and 0.1% BSA (WBB buffer) using a Dounce homogenizer. The resulting homogenate was mixed with 30% dextran (v/v, molecular weight 100,000–200,000) in HBSS and then centrifuged to discard neural cells. The pellet containing the vascular component was resuspended in HBSS and then filtered through a 59μm nylon mesh, in order to retain larger vessels on the mesh surface while capillaries passed through. The resulting capillary-enriched fraction was digested with collagenase/dispase (2 mg/mL) for 33 min at 37°C in WBB by supplemented with 10μg/mL DNase and 0.147μg/mL TLCK. Next, the resulting capillary suspensions were seeded on Matrigel ®-coated inserts (51,000 digested capillaries/cm2) and cultured in DMEM by supplemented with 15% CS, 2 mM glutamine, 1X amino acids, 1X vitamins, 50μg/mL gentamycin and 1 ng/mL bFGF. Remaining cellular debris and red blood cells were removed by washing with medium 24 h after seeding.
siRNA treatment
Twenty-four hours after seeding, endothelial cells were transiently transfected with 40 nM mouse Abca7 siRNA (treated condition, siABCA7 cells) or untargeted siRNA (the control condition) according to the manufacturer’s instruction (protocol D-044433-03-0005, GE Healthcare, Velizy-Villacoublay, France). First, the siRNA was incubated with 1X siRNA buffer and the transfection reagent Ribocellin for 15 min at room temperature (RT). Next, warmed cell culture medium (with no added antibiotic) was added to the solution. Lastly, cells were incubated with siRNA cell culture medium supplemented with 1 ng/mL bFGF for 60 h at 37°C in a 5% CO2 incubator. The medium was refreshed, and experiments were performed 24 h after.
BBB permeability measurements
Permeability to LY was used to check the MBCECs’ tightness, as previously described [29]. Filter inserts were transferred in a 12-well plate containing warm, HEPES-buffered Ringer’s solution (RH buffer, pH 7.4, HEPES 5 mM; NaHCO3 6 mM; NaCl 150 mM; KCl 5.2 mM; CaCl2 2.2 mM; MgCl2 + 6 H2O 1.2 mM), and a solution of LY (50μM) was added on the luminal side of the filter. Every 20 min for 1 h, the filter inserts were transferred into another well containing fresh RH. LY fluorescence was measured with a Synergy H1 multiplate reader (Biotek, Winooski, VT, USA). Lastly, the permeability coefficient was calculated as described by Cecchelliet al.
Immunostaining
MBCECs were rinsed twice with warmed CMF-PBS and then fixed for 1 min in cold methanol/acetone (v/v). After three washes with CMF-PBS, the filters were cut from the plastic insert and (to avoid nonspecific binding) incubated for 30 min in CMF-PBS with 10% normal goat serum. The cells were then incubated with the primary antibody (anti-ZO-1; dilution: 1/200; anti-claudin 5 (dilution: 1/100; both from Invitrogen, Cergy-Pontoise, France) diluted in CMF-PBS supplemented with 2% NGS for 1 h. After three washes in CMF-PBS with 2% NGS, the cells were incubated in the diluted secondary antibody (AlexaFluor 488/568; Invitrogen) for 60 min. Nuclei were stained with Hoechst 33258 reagent. Lastly, after a wash with CMF-PBS with 2% NGS followed by three washes with CMF-PBS, the cells were mounted using Mowiol reagent containing Dabco (Sigma-Aldrich). A Cool SNAP RS Photometrics camera (Leica Microsystems) was used to acquire the images, which were then processed using Adobe Photoshop software (Adobe Systems, San Jose, CA, USA).
mRNA extraction and reverse transcription-quantitative polymerase chain reactions
Twenty-four hours after the end of siRNA treatment and refreshment of the medium, MBCECs were rinsed twice in sterile cold phosphate buffered saline buffer and lysed using RLT lysis buffer (Qiagen, Valencia, CA, USA). Three filters were pooled for each condition. Total RNA was purified using an RNeasy total RNA extraction kit (Qiagen), according to the manufacturer’s instructions. Next, each sample’s purity and concentration were assessed by measuring absorbance at 260/280 nm using a Take 3 microplate reader (SynergyTM H1, Biotek). Reverse transcription was performed with 250 ng of mRNA and iScriptTM Reverse Transcription Supermix (Bio-Rad Laboratories, Marnes-la-Coquette, France), according to the manufacturer’s instructions. cDNA from each sample was amplified by real-time PCR using the SsoFastTM EvaGreen Master Mix kit (Bio-Rad Laboratories) and custom-designed primers. The sequences used for primer design and primer sequences are listed in Table 1. The amplification consisted of 40 cycles with an annealing temperature of 60°C, using a CFX96 thermocycler (Bio-Rad Laboratories). The specificity of amplification was assessed by melting curve analysis. mRNA levels were quantified using the comparative CT method and normalized against the reference genes Actb and Hprt.
DNA primers and accession numbers used for the qPCR experiments
Protein extraction and immunoblots
MBCECs were rinsed twice in cold CMF-PBS. The cells were scraped off with UT4 lysis buffer (urea 7 M, thiourea 4 M, 4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate) supplemented with a protease and phosphatase inhibitor cocktail (Sigma). After a freeze/thaw cycle, the cell lysate was centrifuged to eliminate cellular debris. The protein concentration was evaluated using Bradford’s method (Bio-Rad Laboratories) after dilution of the samples. Electrophoresis of 25μg (ABCA1) or 35μg (other proteins) of protein was performed in a 4–15% SDS-PAGE gel (Bio-Rad), followed by electrotransfer onto nitrocellulose membranes (GE Healthcare). Next, membranes were blocked in Tris-buffered saline with 0.1% Tween and 5% skimmed milk for 90 min at RT. Immunoblotting was performed by incubating membranes with dilutions of the appropriate primary antibody (anti-ABCA7; dilution: 1/1000; Proteintech, Manchester, UK; anti-ABCA1 [11, 31]; dilution: 1/1000; Abcam; anti-ABCG4 [32, 33]; dilution: 1/1000; Alpha Diagnostic International, San Antonio, TX, USA; anti-RAGE [34, 35], dilution: 1/1000, Abcam; anti-ABCB1 (C219) [12, 35], dilution: 1/400, GeneTex, Irvine, CA, USA; anti-ABCG2 (BXP-53) [32], dilution: 1/80, Santa Cruz Biotechnology, Dallas, TX, USA; anti-ABCC1 [36], dilution 1/20, Enzo Life Sciences, Farmingdale, NY, USA; anti-LRP1 [35, 37], dilution 1/200, Santa Cruz Biotechnology and anti-β-actin, dilution: 1/20000, Sigma) overnight at 4°C with slight agitation.
After rinsing, the membranes were incubated with horseradish-peroxidase-conjugated secondary antibodies (Dako and Millipore) for 1 h at RT. Immunoreactions were developed using an enhanced chemiluminescence kit (GE Healthcare) and revealed on chemiluminescence-sensitive films (GE Healthcare). The bands’ relative densities were measured using TotalLab TL 100 1D Gel Analysis software (Nonlinear Dynamics, Newcastle, UK). In these experiments, each deposit represented a pool of at least eight inserts per condition. Each experiment was performed at least four times.
ApoE detection
After 60 h of incubation with siRNA, cells were incubated at 37°C with serum-free medium. After 24 h, medium was collected and then centrifuged to eliminate cellular debris. Secreted proteins were precipitated with 20% trichloroacetic acid (Sigma). Cold acetone was added and the solution thus obtained was centrifuged. Protein pellets were washed twice with cold acetone, and the remaining acetone was air-dried. Proteins were resuspended in RIPA lysis buffer supplemented with protease/phosphatase inhibitors (Sigma). The protein concentration was evaluated using Bradford’s method (Bio-Rad Laboratories). Equal amounts of total secreted proteins (20μg) were loaded onto 4–15% SDS-PAGE gel (Bio-Rad Laboratories). Immunoblots were performed as described above, using anti-ApoE primary antibodies (dilution: 1/3000, Abcam). Each experiment was performed three times.
Cholesterol efflux studies
[3H]-cholesterol (0.5μCi/mL) was incubated with serum for 6 h at 37°C. Next, 2 mM glutamine, 1X amino acids, 1X vitamins and DMEM were added in order to obtain radiolabeled medium. Cells were incubated with this radioactive medium supplemented with siRNA solution and 1 ng/mL bFGF for 60 h. After two washes with DMEM/0.05% BSA, cells were then incubated for 24 h at 37°C with DMEM/0.05% BSA/bFGF. After this equilibration step, cells were rinsed twice, and cholesterol efflux was assessed for 7 h in the presence or absence of acceptors (20μg/mL HDL, 35μg/mL ApoA-I, ApoE2 and ApoE4). At the end of efflux experiments, media were collected and centrifuged (4 min, 4000 rpm, 4°C) in order to remove cellular debris. Lastly, after three washes with cold PBS, cells were lysed with PBS/1% Triton X-100. Radioactivity in the cell lysates and media was measured in a liquid scintillation counter (Tri-Carb 2100TR). The percentage of cellular cholesterol efflux was calculated from the disintegrations per minute (DPM) using the following equations:
percentage of luminal efflux = (DPMluminal)×100/(DPMluminal +DPMabluminal +DPMcell lysate)
percentage of abluminal efflux = (DPMabluminal)×100/(DPMluminal+ DPMabluminal +DPMcell lysate)
percentage of total efflux = (DPMluminal +DPMabluminal)×100/(DPMluminal+ DPMabluminal +DPMcell lysate)
Each assay was performed using three filters, and each experiment was run at least three times.
Quantification of cell cholesterol
After 60 h of incubation with siRNA, cells were incubated for 24 h at 37°C with serum-free cell culture medium. After this equilibration step, cells were rinsed with cold RH buffer and then lysed with 1 N NaOH. One aliquot of the cell lysate (from a single insert) was used for the protein concentration assay using Bradford’s method (Bio-Rad), and another was homogenized in chloroform/isopropanol/NP-40 (7:11:0.1) for 5 min with vigorous agitation, in order to extract lipids. After 15 min of centrifugation at 14000 rpm and 4°C, the organic phase was collected and then air-dried at 50°C for 2 h. The dried lipids were dissolved in the Cholesterol Reaction Buffer provided with a Cholesterol/Cholesteryl Ester Quantitation Kit (Calbiochem) by vortexing vigorously for 5 min. The reaction mixture was prepared according to the manufacturer instructions. Equal volumes of sample and reaction mixture were placed in a 96-well plate and incubated at 37°C for 1 h. Next, fluorescence was measured with a Synergy H1 multiplate reader (Biotek) at an excitation wavelength of 535 nm and an emission wavelength of 587 nm. The amount of cholesterol in each sample was determined with reference to a standard curve and then adjusted as a function of the protein level in each sample. We used three filters for each assay, and each experiment was run three times.
Amyloid-β peptide (Aβ) transport studies
Twenty-four hours after medium refreshment (i.e., following the end of siRNA treatment), MBCECs were rinsed with prewarmed RH buffer. Next, for apical-to-basolateral transport studies, filter inserts were transferred into 12-well plates containing 1.5 mL of RH/BSA 0.1% per well. A solution of RH/BSA 0.1% supplemented with Cy5-Aβ40 (10 nM), [14C]-sucrose (Perkin Elmer Life and Analytical Sciences, Waltham, MA, USA), FITC-inulin and (or not) ApoA-I (200 nM) was then added (or not) to the apical (luminal) compartment. For basolateral-to-apical transport studies, filter inserts were transferred into 12-well plates containing 1.5 mL of Cy5-Aβ40 FITC-inulin in RH/BSA 0.1%, and a solution of [14C]-sucrose supplemented (or not) with ApoA-I (200 nM) was added in the luminal compartment.
Cells were incubated at 37°C with slight agitation. Transport was measured after 3 h. BBB permeability and non-receptor-mediated transport were respectively evaluated by measuring [14C]-sucrose permeability and FITC-inulin transport. Radioactivity was measured using a liquid scintillation counter, whereas fluorescence was measured using a Synergy H1 spectrophotometer (Biotek). The percentage passage was calculated in the presence and absence of cells, in order to determine the influence of the filter and the coating on transport. The results for inulin were used to normalize the Aβ results, as previously described [31]. The mass balance was also calculated, and the value had to be between 90% and 110%. Each assay was performed using three filters, and each experiment was run at least three times.
Statistical analysis
Results were expressed as the mean±SEM and analyzed using a Student’s T-test or a one-way analysis of variance. All statistical analyses were performed using Prism Software (GraphPad Software Inc., San Diego, CA, USA).
RESULTS
Abca7 knock-down does not affect the BBB’s integrity in vitro
First, we used an RNA silencing technique to downregulate Abca7 expression in our murine BBB model previously described [26–28]. In vitro, Abca7 knock-down resulted in a 58.7±4.7% reduction in mRNA expression and a 40±18% reduction in protein expression, relative to the control condition (Fig. 1A-C).
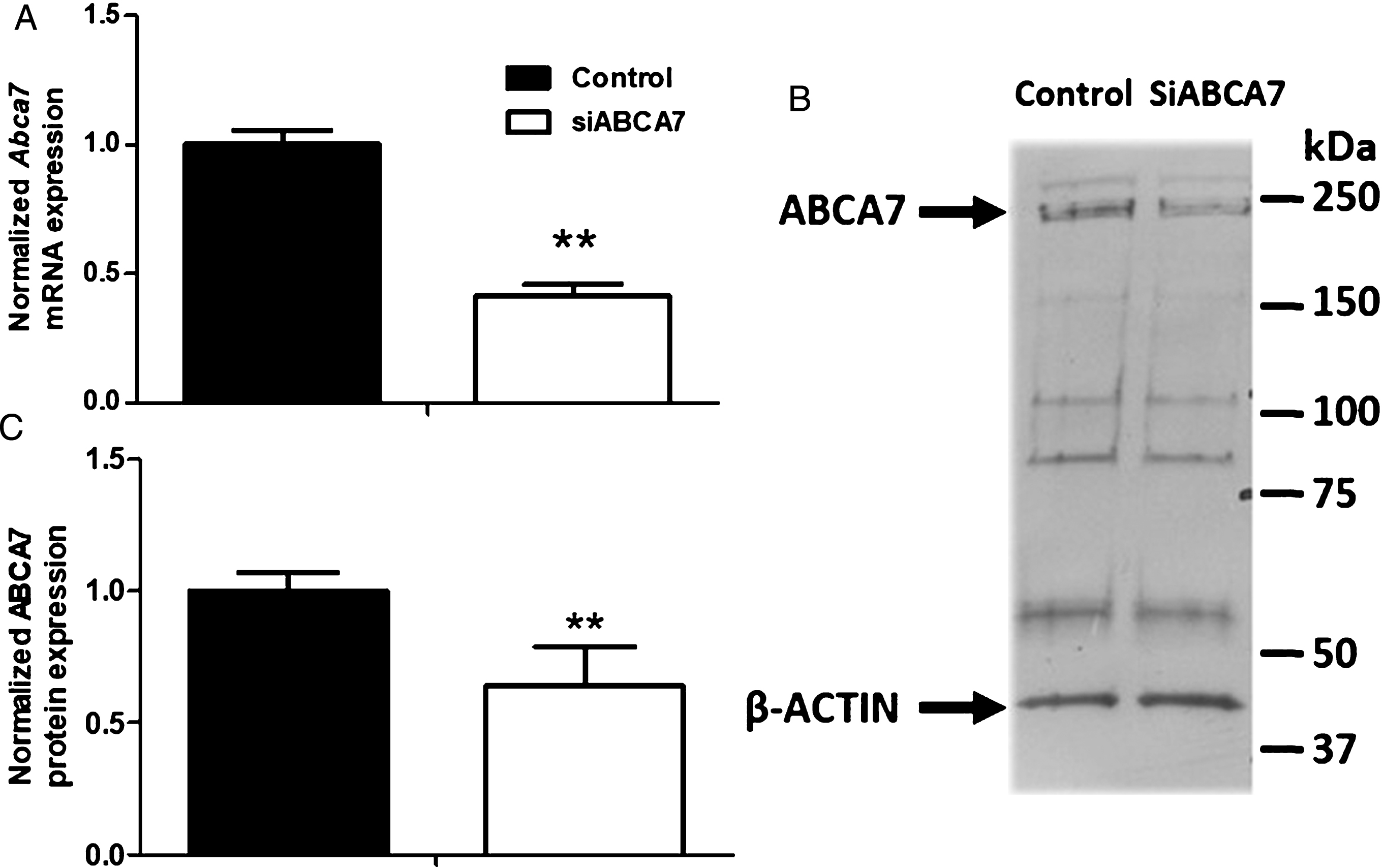
Effect of siRNA treatment on ABCA7 mRNA and protein expression. A) Transcriptional expression of Abca7 was assessed in MBCECs using the primers listed in Table 1. Each bar represents the level of mRNA expression normalized against the housekeeping genes Actb and Hprt, relative to the control condition (untargeted siRNA). Data are quoted as the mean±SEM of three experiments pooled from three filters. Statistical analysis: t-test, **p < 0.01. B) Protein levels were assessed by immunoblotting. ABCA7 detection gives unspecific bands as previously reported in mouse brain total extracts [24]. C) Quantification of ABCA7 expression. β-Actin was used as a loading control. Data are quoted as the mean±SEM of at least six experiments pooled from eleven filters. Statistical analysis: t-test,**p < 0.01.
We then evaluated the impact of Abca7 downregulation on the BBB’s integrity. As shown in Fig. 2A, the MBCECs’ paracellular permeability to LY was similar in the siABCA7 and untargeted siRNA conditions (0.41±0.08×10–3 cm.min–1 and 0.40±0.07×10–3 cm.min–1, respectively). The value in non-treated cells was 0.34±0.07×10–3 cm.min–1. Immunofluorescentlabeling of the tight junction proteins claudin-5 and ZO-1 showed strong, intact signals at the cell periphery (Fig. 2B) - suggesting that siRNA treatments did not affect the MBCECs’ integrity.

Effect of siRNA treatment on permeability and tight junction integrity in a monolayer of MBCECs. Endothelial cells were incubated with medium alone (Untreated), untargeted siRNA (Control) or siRNA targeting Abca7 mRNA (siABCA7) for 60 h. Twenty-four hours after the medium had been refreshed, permeability to the paracellular marker LY was evaluated (A). The bars represent the mean±SEM of five experiments, each performed with three filters per condition. Statistical analysis was performed using a one-way ANOVA followed by Dunnett’s correction for multiple comparisons. ns: non-significant. B) Tight junction integrity was also assessed by immunostaining of claudin 5 (red) and ZO-1 (green). Nuclei were stained with Hoechst 33258 reagent (in blue). Scale bar: 25μm.
Downregulation of Abca7 decreases expression of the transporters involved in cholesterol metabolism and lipid exchanges at the BBB
We then decided to investigate the expression of transporters and acceptors involved in cholesterol metabolism and lipid exchange at the BBB in siRNA-treated cells. In Abca7-depleted cells, mRNA expression levels of Abca1, Abcg4, and ApoE were low (by 53±5%, 34±4%, and 51±4%, respectively), whereas Abcg1 expression was not significantly different (Fig. 3A). The ABCA1 protein expression levels correlated with the mRNA expression levels (Fig. 3B). Indeed, a statistically significant 38±13% relative reduction in the ABCA1 protein level was observed. The ABCG4 protein level was not significantly different (Fig. 3C). Furthermore, the total secreted ApoE fraction was 45±27% lower but did not reach statistical significance (Fig. 3D). However, we observed that ApoE secretion was significantly lower in the abluminal media (by 63±17%) but not in the luminal compartment (18±44%).

Effect of Abca7 silencing on the expression of transporters involved in cholesterol release at the BBB. A) For transcriptional expression, each bar represents the level of mRNA expression normalized against the housekeeping genes Actb and Hprt, relative to the control condition. Primers are listed in Table 1. Data are quoted as the mean±SEM of three experiments pooled from three filters. Protein levels of ABCA1 (B) and ABCG4 (C) were also evaluated. Actb was used as a loading control. Data are quoted as the mean±SEM of at least four independent experiments pooled from eight filters. Secreted APOE protein was measured in media recovered from luminal and abluminal compartments (D). Data are quoted as the mean±SEM of three independent experiments pooled from nine filters. From A to D, statistical analysis: t-test, *p < 0.05, **p < 0.01, ***p < 0.001, ns, non-significant.
Thus, ABCA7 deficiency is associated with abnormally low expression of key players in HDL genesis. We therefore next assessed the ability of MBCECs to transfer cholesterol to apolipoproteins and lipoproteins.
Abca7 knock-down is associated with lower cholesterol release from the MBCEC membrane and greater cholesterol accumulation
The literature data indicate that human, porcine and bovine BBB cells release membrane cholesterol to ApoA-I, ApoE2, ApoE4, and HDL [12, 38]. We therefore investigated the MBCECs’ ability to efflux membrane cholesterol in basal conditions in the presence or absence of lipid acceptors. As shown in Fig. 4A, the presence of HDL, ApoE2, and ApoE4 was associated with significantly greater cholesterol release (6.8%, 6.3% and 7.9% respectively; i.e., up to 8-fold greater than the control value (0.8%) in the abluminal (brain side) compartment). The same trend was observed for cholesterol efflux to the luminal (blood side) compartment (Fig. 4B), which was promoted in the presence of each of the three lipid acceptors (11.8%, 14.6%, and 15.8% for HDL, ApoE2, and ApoE4, respectively, compared with the control value of 3.4%).
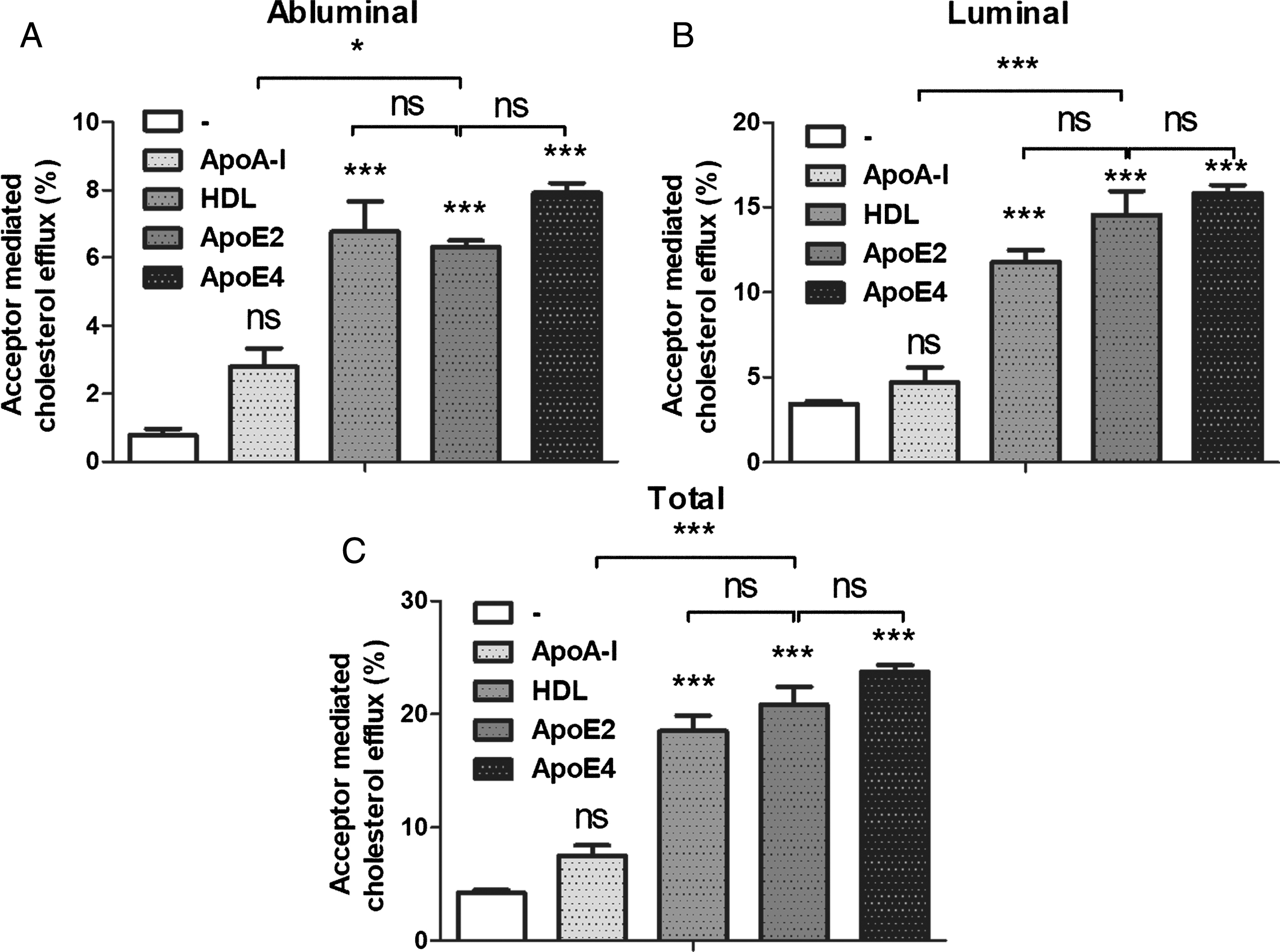
Basal cholesterol efflux from MBCECs. Cholesterol efflux was assessed for 7 h in the absence of cholesterol acceptor (DMEM/BSA 0.05%, the control condition) or in the presence of the acceptor ApoA-I (35μg/mL), HDL (20μg/mL), ApoE2 (35μg/mL) or ApoE4 (35μg/mL) in the abluminal (A) and luminal (B) compartments. By summing the levels of cholesterol efflux in the two compartments, we obtained the total cholesterol efflux (C). Data are quoted as the mean±SEM of 3 independent experiments representing 7 inserts. Statistical analysis: a one-way ANOVA followed by Bonferroni’s correction for multiple comparison, *p < 0.05, **p < 0.01, ***p < 0.001.
As a consequence, total cholesterol efflux was promoted in the presence of HDL, ApoE2, and ApoE4 (Fig. 4C). No significant differences between the three cholesterol acceptors wereobserved.
To investigate the impact of Abca7 downregulation on cholesterol efflux, we then investigated cholesterol release in siABCA7-treated cells. As shown in Fig. 5A and B, we observed significantly lower total cholesterol efflux in the presence of ApoA-I and HDL (32% and 20%, respectively, relative to the control). Cholesterol release to the luminal side was lower in the presence of ApoA-I, whereas cholesterol release to both sides was lower in the presence of HDL. These results are in line with those obtained in mouse macrophages in which Abca1 and Abca7 have beendeleted [39].

Effect of Abca7 knock-down on cholesterol release from MBCECs. As described above, cholesterol efflux was assessed for 7 h in the presence of various acceptors [ApoA-I (A), HDL (B), ApoE2 (C) and ApoE4 (D)] in control and SiABCA7 cells. Acceptors were added to the luminal and abluminal compartments. Data are quoted as the mean±SEM for at least 7 filters from 2 independant experiments. E) Effect of Abca7 knock-down on the cellular cholesterol level. Cholesterol content was assessed in MBCECs after siRNA treatment. Data are quoted as the mean±SEM cellular cholesterol level from 9 filters per condition (3 independent experiments). Statistical analysis: t-test, *p < 0.05, **p < 0.01, ***p < 0.001, ns, non-significant.
In the presence of ApoE4 and ApoE2, total cholesterol release did not significantly differ when comparing siABCA7 cells and the control (Fig. 5C and (D, respectively). However, when considering only the abluminal side, ApoE4-mediated cholesterol efflux was reduced by 16±2% in siABCA7 relative to the control, whereas ApoE2-mediated cholesterol release was similar in both conditions. Hence, Abca7 knock-down is associated with a relative decrease in cellular cholesterol efflux; this suggests that lipids accumulate in siABCA7 cells, relative to controls.
We thus evaluated the level of cholesterol accumulation in cells. As expected, we found that ABCA7 knock-down promoted a 26±9% increase in cholesterol levels in MBCECs, relative to the control condition (Fig. 5E).
ABCA7 knock-down decreases basolateral-to-apical transport of Aβ peptide mediated by ApoA-I
Given that Abca7–/– mice show elevated levels of amyloid deposition in the brain [24, 41], we hypothesized that Abca7 downregulation in MBCECs might impact amyloid transport across the BBB. Therefore, apical-to-basolateral and basolateral-to-apical transport assays of Aβ peptides were performed in our in vitro BBB models, as previously described [12, 31].
As shown in Fig. 6A, ABCA7 downregulation did not directly impact the influx (upper graph) or the efflux of Aβ1 - 40 peptides (lower graph).
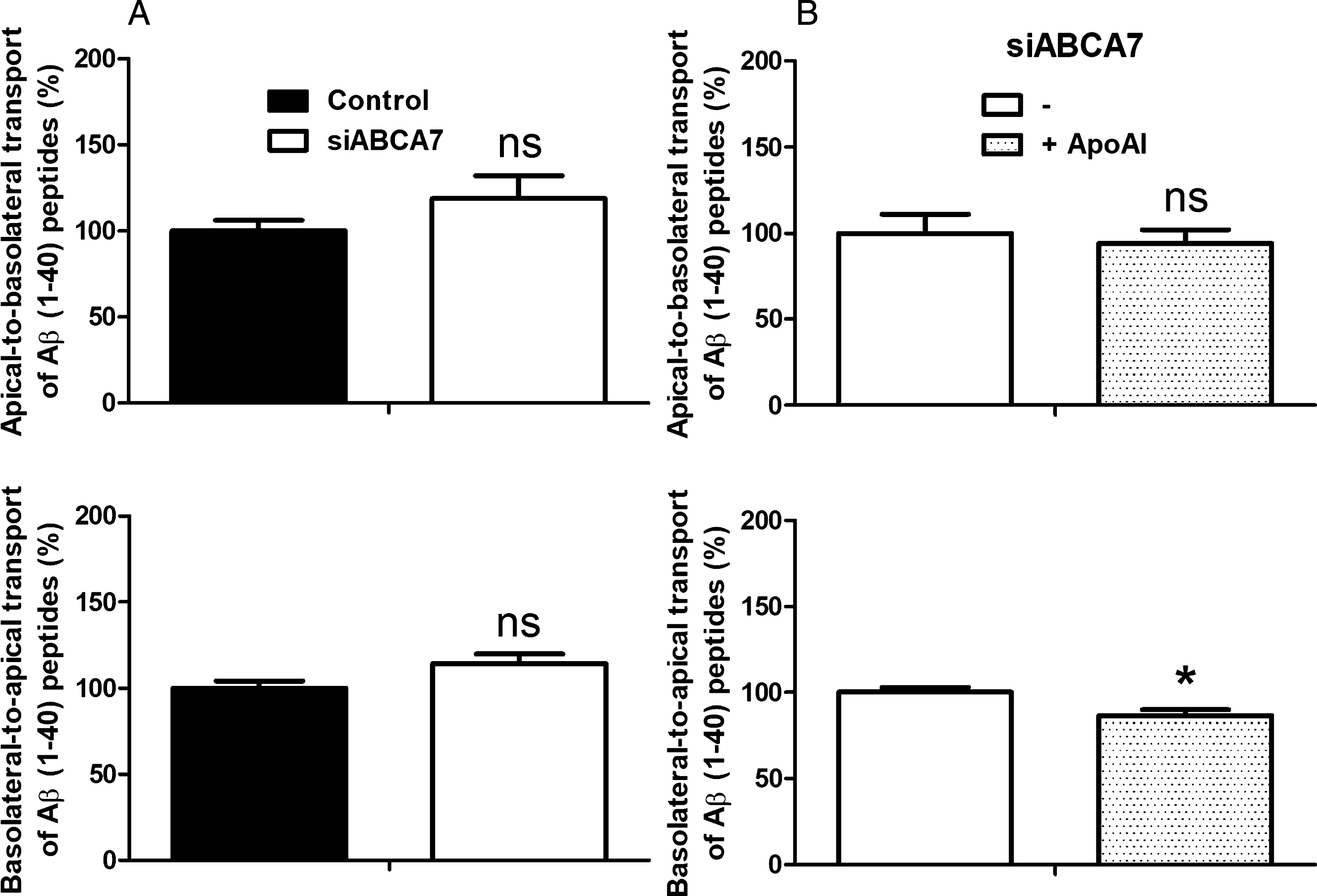
Effect of Abca7 knock-down and ApoA-I on Aβ peptide transport across the MBCEC monolayer. A) Aβ peptide transport was assessed for 3 h in MBCECs treated with untargeted siRNA (Control) or siABCA7. B) The impact of Abca7 downregulation was then investigated in the presence or absence of ApoA-I (200 nM, in the luminal compartment). Inulin was used as a marker of non-specific vesicular transport. Inulin passage was assessed in parallel, and the data were used to normalize the Aβ results. In addition to inulin, paracellular integrity was assessed using radiolabeled sucrose (data not shown) for each experiment. Data are quoted as the mean±SEM at least seven filters per condition (2 independent experiments). Statistical analysis: t-test, *p < 0.05, ns, non-significant.
We previously reported that the presence of ApoA-I in the luminal compartment promotes Aβ peptide efflux [42] and had observed in the present study that luminal ApoA-I lipidation is lower when ABCA7 is downregulated (Fig. 5A). Therefore, we next investigated Aβ peptide transport in siABCA7 cells in the presence of ApoA-I in the luminal compartment. When ApoA-I was present, we observed a slight but statistically significant relative decrease in basolateral-to-apical transport of Aβ (Fig. 6B, lower graph; 86.5±3.4% compared with the control condition in the absence of ApoA-I). No significant differences in apical-to-basolateral transport were observed (Fig. 6B, uppergraph).
To obtain additional data on the effect of Abca7 depletion on the expression of receptors and transporters found to be involved in Aβ clearance by us and others [12, 43–46], we used qRT-PCR and immunoblot assays to measure the mRNA and protein expression of Abcb1, Abcc1, Abcg2, Lrp1, and Rage. Downregulation of Abca7 was not associated with a difference in expression for any of these genes and proteins (Fig. 7). Despite our efforts, LRP1 and ABCC1 proteins were not detected, probably because they are expressed at very low level at the endothelial cells of the BBB as previously demonstrated[37, 48].

Effect of Abca7 silencing on the expression of receptors and transporters involved in Aβ peptide transport at the BBB. A) mRNA levels of key players of amyloid transport were measured by qRT-PCR. Each bar represents the level of mRNA expression normalized against the housekeeping genes Actb and Hprt, relative to the control condition. The primers used are listed in Table 1. Data are quoted as the mean±SEM of three independent experiments pooled from three filters. Statistical analysis: t-test; ns, non-significant. B) Protein levels were also evaluated and quantified. B-actin was used as a loading control. Data are quoted as the mean±SEM of at least four experiments pooled from eight filters.
Taken as a whole, these results suggest that ABCA7 downregulation could alter BBB cholesterol metabolism that in turn could affect amyloid clearance across this barrier.
DISCUSSION
We and others have demonstrated that the ABCB1 (also known as P-gp), ABCG2 (BCRP) and ABCC1 (MRP1) proteins expressed by endothelial cells of the BBB can restrict Aβ entry into the brain and can also mediate Aβ efflux into the blood stream [34, 50]. ABCA1, ABCG1, and ABCG4 are more involved in cholesterol release from cellular membrane, and control cholesterol homeostasis in the brain and lipid exchanges across the BBB. These observations suggested that the modulation of ABC transporter expression might restore cholesterol metabolism and decrease amyloid deposition in AD patients. Promising results were obtained in AD transgenic mice in which the elevation of ABCB1 expression (by administering pregnenolone-16α-carbonitrile [51] or caffeine [52]) promoted Aβ efflux across the BBB. Regarding cholesterol homeostasis, elevated ABCA1 expression in the AD mouse was associated with less amyloid deposition and less aberrant behavior [53–55]. Furthermore, we recently demonstrated that oxysterols (natural agonists of liver X receptors) were able to enhance both ABCA1 and ABCB1 expression— thus promoting cholesterol exchange and restricting Aβ influx across the BBB [31, 56]. Despite these interesting and promising results, however, little is known about the processes that lead to amyloid deposition and cholesterol imbalance in the central nervous system (CNS) of patients with AD. Interestingly, the only ABC transporter found to be involved in AD in GWASs is ABCA7; very few studies had focused on this protein until the publication of the GWAS results in 2011 [19]. More recent studies have tried to elucidate ABCA7’s role in the brain in health and during AD. For example, it was shown that ABCA7 does not have a significant role in the regulation of cell proliferation or neurogenesis in the adult mouse [57], and that the polymorphism identified in AD patients leads to a loss of function of ABCA7 [20]. Although Abca7–/– mice exhibit normal sensory abilities, neurological reflexes, and motor functions [58], it is now clear that Abca7-/- in AD transgenic mice promotes amyloid deposition. However, some researchers have explained this accumulation by the overproduction of Aβ peptides [21, 40] whereas others have suggested that a decrease in microglial phagocytosis was involved [24, 41]. Despite the fact that Abca7 is known to be expressed at the BBB [14], the effect of a decrease in Abca7 expression on amyloid and cholesterol exchanges across the BBB had not previously been investigated.
Here, we used RNA silencing to decrease Abca7 expression in an in vitro BBB model composed of primary MBCECs [26–28]. We did not observe any change in BBB integrity when Abca7 mRNA and protein expression were downregulated. Given that ABCA1 and ABCA7 display around 54% amino acid homology [42] and that ABCA7 generates HDL [21, 60], we hypothesized that cholesterol exchange across the BBB could be impacted by Abca7 downregulation. Indeed, we observed a relative decrease in cholesterol release (and particularly for HDL and ApoA-I) in this context. However, we cannot be absolutely sure that this relative decrease in cholesterol efflux is directly linked to ABCA7 downregulation because 1) ABCA1 mRNA and protein levels are also lower and 2) we previously demonstrated that ABCA1 is involved in cholesterol efflux to HDL and ApoA-I in human and bovine BBB models [12, 56]. We found no change in the expression of ABCG1, which has been recently identified as a key player in cholesterol metabolism at the BBB [38]. Abcg4 expression was significantly reduced in vitro when Abca7 is deleted but protein level was not affected. ABCG4 is exclusively expressed in the CNS and generates (along with ABCA1 and ABCG1) cholesterol rich-HDL [61]. However, this transporter is also involved (along with ABCG1) in the regulation of the intracellular pool of cholesterol in astrocytes and neurons [62]. Therefore, the downregulation of ABCA1 observed in the present study might also explain the decrease in cholesterol release to ApoA-I and HDL and thus the intracellular accumulation of cholesterol.
Interestingly, we also observed lower APOE expression and lower APOE secretion into the abluminal compartment (mimicking the CNS) of our BBB model. In the CNS, ApoE is required for lipoprotein uptake into neurons via the LDL family of receptors [63]. ApoE polymorphisms constitute the most common genetic factor involved in AD [64], and APOE mediates Aβ peptide elimination across the BBB. Downregulation of APOE secretion might lead to a reduction in Aβ transport across the BBB and therefore to greater accumulation of these peptides in the brain [64]. Noteworthy, ABCA1 deletion in AD mouse models is also associated with significant reductions in apoE level [65–67].
Our study is not the only one to investigate links between APOE, ABCA1, and ABCA7 regulations. Indeed, in Abca7–/– mice, Kim et al. reported that ApoE levels were not affected when compared with wild-type mice [22]. ABCA1 expression was not modulated in brain. However, in vitro, an upregulation of ABCA7 has been reported in Abca1-deficient mouse fibroblasts [68] whereas Abca7-deficiency in mouse macrophages increases ABCA1 expression [39]. Therefore, all these data suggest that links exist between ABCA7, ABCA1, and APOE but the molecular processes remain largely unknown; they might involve sterol regulatory element binding proteins or liver X receptors, which are key molecules regulating cholesterol and lipid metabolism [69, 70].
Taken as a whole, these data indicate that by decreasing APOE secretion and ABCA1 expression at the BBB, ABCA7 loss of function can significantly alter cholesterol exchange across the BBB and thus promote AD. This is consistent with a previous study reporting the contribution of ABCA7 in the cerebral lipid metabolism [25].
In addition to lipid metabolism, we also investigated the role of ABCA7 in amyloid transport across the BBB. When ABCA7 was downregulated, we did not observe any difference in amyloid exchange through the BBB in the absence of ApoA-I. However, as discussed above, we found that ABCA7 depletion was associated with a reduction in cholesterol release to ApoA-I; the latter is known to promote Aβ efflux across MBCECs [71]. Indeed, after adding ApoA-I to the luminal compartment of our experimental set-up, we observed a reduction in the basolateral-to-apical transport of Aβ peptide. Given that the expression of receptors and transporters known to mediate Aβ exchanges across the BBB did not change when ABCA7 expression was reduced, we suggest that the decrease in ABCA7 reported provokes a reduction of ABCA1 and APOE expression which in turn decreases lipoproteins lipidation and thus, reduces amyloid elimination across theBBB.
Lastly, by linking ABCA7 to cholesterol and amyloid exchanges across the BBB, our results 1) revealed novel pathways underlying the association between ABCA7 dysfunction and AD pathogenesis, and 2) suggest that this transporter may be a potential therapeutic target for curing or preventing AD. All these experiments were performed using an in vitro BBB model, therefore further in vivo experiments using wild-type and Abca7-/- mice should to be performed to confirm the relevance of these data and to confirm the links between ABCA7, BBB, and AD.
Footnotes
ACKNOWLEDGMENTS
This work was supported by internal grants from University of Artois (BQR-ABCA7, to FG) and from foundation France Alzheimer et Maladies apparentées (AAP 2013, #22, to FG). Y.L. has received fellowships from the Region Hauts-de-France. J.P.’s work was funded by grants from the following sources: Deutsche Forschungsgemeinschaft/ Germany (DFG PA930/9, DFG PA930/12); Leibniz Society/ Germany (SAW-2015-IPB-2); HelseSØ/ Norway (2016062); Norsk forskningsrådet/ Norway (246392, 247179 (NeuroGeM), 248772, 251290, 260786 (PROP-AD)); Horizon 2020/ European Union (643417 (PROP-AD)). NeuroGeM is an EU Joint Programme - Neurodegenerative Disease Research (JPND) project. The project is funded by the following organizations under the aegis of the JPND - www.jpnd.eu (CIHR – Canada, BMBF – Germany, NRF #247179 – Norway, ZonMW – The Netherlands). PROP-AD is an EU Joint Programme - Neurodegenerative Disease Research (JPND) project. The project is funded by the following organizations under the aegis of the JPND - www.jpnd.eu (AKA #301228 – Finland, BMBF #01ED1605- Germany, CSO-MOH #30000-12631 - Israel, NFR #260786 - Norway, SRC #2015-06795 - Sweden). This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement #643417 (JPco-fuND).