
Abstract
Alzheimer’s disease (AD) is one of the major age related neurodegenerative diseases whose pathology arises due to the presence of two distinct protein aggregates, viz., amyloid-β plaques in extracellular matrix and tau neurofibrillary tangles in neurons. Multiple factors play a role in AD pathology, which includes familial mutations, oxidative stress, and post-translational modifications. Melatonin is an endocrine hormone, secreted during darkness, derived from tryptophan, and produced mainly by the pineal gland. It is an amphipathic molecule, which makes it suitable to cross not only blood-brain barrier, but also to enter several other subcellular compartments like mitochondria and endoplasmic reticulum. In this context, the neuroprotective effect of melatonin may be attributed to its role as an antioxidant. Melatonin’s pleiotropic function as an antioxidant and neuroprotective agent has been widely studied. However, its direct effect on the aggregation of tau and amyloid-β needs to be explored. Furthermore, an important aspect of its function is its ability to regulate the process of phosphorylation of tau by affecting the function of kinases and phosphatases. In this review, we are focusing on the pleiotropic function of melatonin on the aspect of its neuroprotective function in tau pathology, which includes antioxidant function, regulation of enzymes, including kinases and enzymes involved in free radical scavenging and mitochondrial protection.
INTRODUCTION
Alzheimer’s disease (AD) is an age-associated neurological disorder with progressive neurodegeneration and dementia. It is hallmarked by the presence of intracellular deposition of protein aggregates of microtubule-associated protein tau and extracellular senile plaques made up of amyloid-β (Aβ) peptides. The formation of both these protein aggregates occurs due to multiple factors, which include post-translational modifications of tau and oxidative stress in neurons [1]. Various therapeutic strategies have been designed in order to ameliorate the protein aggregates and to restore the neuronal function with limited success [2–4]. Most of the drugs targeted that are found effective against inhibition of aggregates or able to dissolve preformed aggregates have various limitations. One of the biggest limitations of these drugs is their inability or limited ability to cross the blood-brain barrier (BBB), even though they are found to be effective in vitro. Thus, a molecule, which is effective against toxic aggregates, should also be highly available to neurons by crossing the BBB. Melatonin is a methoxyindole secreted by pineal gland in the absence of light. First identified in the bovine pineal gland tissue, it was initially believed to be involved only in hormonal functions like regulation of circadian rhythm, sleep homeostasis, vaso-activity, and development of reproductive glands [5]. Apart from being the hormone that regulates the sleep wake cycle, melatonin also functions as a free radical scavenger, an antioxidant, and immuno-modulatory agent (Fig. 1) [6], as well as promotes differentiation and proliferation of neuronal cells [7]. Antioxidant properties of melatonin are connected with its neuroprotective activity in several degenerative disorders [8, 9]. The origin and progression of neurodegeneration is a complex process and depends on a myriad of factors. One of the cause of neurodegeneration is oxidative stress, which results from an imbalance between free radical formation and antioxidative mechanisms [10]. Melatonin is found to be a potent antioxidant as well as it plays a key role in the regulation of other proteins involved in the protection against cellular stress and apoptosis [11, 12], and it is notable that the melatonin production decreases with age and is found to be lowered in age associated neurological disorders. The administration of melatonin in age-associated neurological disorders has been found to improve cognitive functions and sleep [13, 14]. As melatonin is produced in absence of light, the effect of prolonged light exposure in rats and its implications on neuronal function has been studied [15]. Tau undergoes other modifications such as acetylation, methylation, sumoylation, ubiquitination, and truncation, which modulate the properties of tau in terms of its microtubule binding and aggregation propensity. Also, the level of monoamine oxidase and superoxide dismutase increased, indicating oxidative stress. Aβ plaques, which are formed and deposited in extracellular regions of neurons, are also known to get ameliorated by administration of melatonin. When melatonin was supplemented simultaneously with light exposure, molecular, and behavioral impairments are mildly arrested. However, melatonin fails to ameliorate the Aβ pathology in Tg2576 mice when administered at the later stages of disease development. Melatonin treatment started at 14 months in Aβ plaques bearing Tg2576 mice showed no effect in relieving oxidative damage as well as removal of existing Aβ plaques even at elevated levels of plasma melatonin. Further deposition of Aβ plaques were continued to be observed in cortical and hippocampal region even after 4 months of melatonin treatment [16]. In contrast, it is found to be effective in suppressing the Aβ aggregates when administered in Tg2576 mice after 4 months of disease development [17]. Effect of melatonin on α-synuclein aggregation has also been studied extensively. Amphetamine (AMPH) induced α-synuclein toxicity was found to be attenuated in dopaminergic SK-N-SH cells. It also prevented the decrease in mitochondrial complex I level and phosphorylated tyrosine hydroxylase levels after AMPH treatment [18]. The expression level of α-synuclein was found to be reduced after the administration of melatonin in 4-day-old postnatal rat [19]. In another study, melatonin was found to inhibit α-synuclein fibrillization and destabilized α-synuclein aggregates [20].

Synthesis and functions of melatonin. The pineal gland is a pea-sized gland located in the epithalamus, near the center of the brain between two hemispheres. It is the major site for the synthesis of melatonin, other sites being skin, gut, lymphocytes, and platelets. Melatonin is synthesized from tryptophan in four steps involving hydroxylation, decarboxylation, acetylation, and methylation. The enzymes involved in these four steps are tryptophan hydroxylase, 5-OH tryptophan decarboxylase, serotonin N-acetyl transferase, and hydroxyindole O-methyl transferase to form 5-OH tryptophan, 5-OH tryptamine, N-acetyl Serotonin, and melatonin respectively. N-acetyl serotonin is its immediate precursor, whose synthesis in turn, is the rate limiting step for melatonin production. Melatonin is a pleiotropic molecule performing a wide variety of functions. It is mainly involved in the maintenance of circadian rhythm in mammals. As a metabolic regulator, it is involved in glucose and lipid metabolism in normal physiological conditions. Apart from this, it also functions as a free radical scavenger as well as shows anti-apoptotic and anti-inflammatory effects indirectly by regulation key players involved in these functions.
These findings suggest that melatonin could be effective in treatment only in earlier stages of AD and shows no or little effect in later stages. Thus, it is suggested to employ the properties of melatonin in combination with other potential drugs.
Current therapeutic strategies for AD targets either directly protein aggregates or focus on stabilizing the microtubule assembly and maintenance of cell homeostasis [21]. A multi-targeted approach would be beneficial for the effective treatment of AD.
SYNTHESIS AND FUNCTIONS OF MELATONIN
Melatonin is widely distributed across the plant and animal kingdoms, where it is involved in a variety of functions like pigmentation, cell proliferation, regulation of reproductive system, and maintenance of circadian rhythm. In the animal kingdom, melatonin is secreted mainly, but not exclusively, by the pineal gland [22–24]. Once formed, melatonin is not stored within the pineal gland but diffuses out into the capillary blood and cerebrospinal fluid. Melatonin is a hormone whose function depends upon the light and dark cycle. Melatonin secretion is triggered during the absence of light while it gets inhibited in its presence (Fig. 1). The signals to retina (absence of light) are transduced via retinohypothalamic pathway to the suprachiasmatic nucleus (SCN) of the hypothalamus. Melatonin release regulates the circadian rhythm, i.e., the maintenance of biological clock of the brain at the locus of suprachiasmatic nucleus [25]. Apart from the pineal gland, melatonin can also be synthesized and secreted by the skin, platelets, retina, and gut [26]. But the effect of its action on circadian rhythm comes primarily from pineal secretion since disturbances still exist after pinealectomy [27]. Age-associated neurological disorders often have the symptoms of changes in circadian rhythm and sundowning effect due to the imbalance in pineal-derived melatonin levels. This indicates the physiological importance of melatonin in neuronal health. Melatonin is synthesized from tryptophan when the stimulus of dark is perceived by retina, activating the SCN to secrete norepinephrine, which acts on the endoplasmic reticulum of pinealocytes to release Ca2+ into the cytosol. Further, increased cytosolic concentration of Ca2+ activates adenyl cyclase to produce cAMP, which in turn activates protein kinase A (PKA) through binding (Fig. 2). PKA acts on the rate-limiting step of melatonin synthesis, i.e., by activating serotonin N-acetyl transferase to produce melatonin from its immediate precursor N-acetyl serotonin [28].
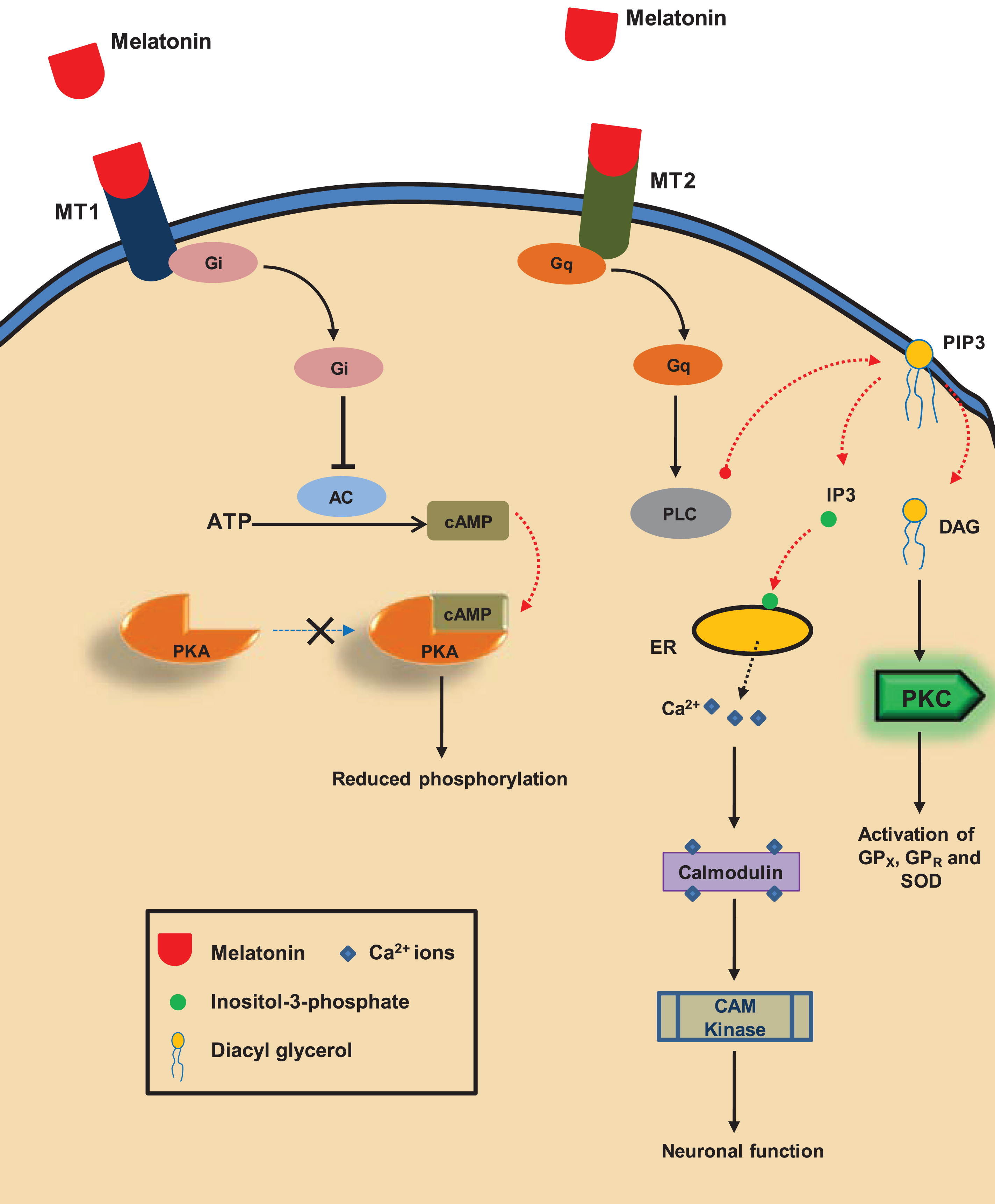
Functions mediated through melatonin receptors. The binding of melatonin to its receptors – MT1 (mel1a) and MT2 (mel1b) leads to a functional cascade, resulting in the activation of downstream regulators. MT1 is a G- protein coupled receptor (GPCR) associated with Gi protein. Melatonin binding to MT1 leads to the inhibition of adenyl cyclase, which ultimately leads to the down-regulation of PKA activity, which is a major kinase involved in tau hyperphosphorylation. MT2, mainly involved in regulation of circadian rhythm, mediates its function through the Gq protein. The cascade through MT2 leads to the dual function of PKC activation and Ca2+ release. Increase in the Ca2+ results in the activation of CAM kinase (through Calmodulin) and PKC regulates activity of other downstream regulators of metabolic and anti-oxidative function.
There is a wide range of functions that are served by melatonin (Fig. 1). Classically, melatonin is known for its effect on sleep/wake cycle in most of the mammals and as a reproductive regulator in seasonal breeders [29]. But, it can exhibit a plethora of functions in different sites either directly or indirectly by regulation of other factors. The antioxidant effect of melatonin has been established in a number of cellular and animal models, and it is found to be a more potent antioxidant than tocopherol (vitamin E) [30]. Being able to cross all biological membranes, it is widely distributed in all cell types and intracellular compartments. As a free radical scavenger, melatonin acts on reactive oxygen species (ROS) and reactive nitrogen species (RNS) to neutralize them and also plays a role in the activation of enzymes involved in antioxidant function [31]. The metabolic functions of melatonin have been implicated in diabetes and body weight regulation. It has been reported that the level of insulin and melatonin have inverse relationship. Reduced plasma melatonin is correlated with elevated insulin level. Also, in type 2 diabetes mellitus, the precursors for melatonin including tryptophan and serotonin are reduced in the pineal glands of Goto-Kakizaki rats, a model for type-2 diabetes mellitus [32]. The presence of melatonin receptors in islets of Langerhans, and its role in insulin secretion has also been shown [33, 34]. Melatonin is known to increase PI3K activity in a receptor-mediated manner. In skeletal muscles, melatonin is found to increase the uptake of glucose via IRS1/PI3K (insulin receptor substrate1/phosphatidylinositol-3kinasepathway). The mRNA analysis of pancreatic β-cells showed amplification product corresponding to MT1, indicating the presence of melatonin receptors in pancreas. It was indicated from mRNA analysis of pancreatic β-cells that the amplification product of mRNA corresponds to MT1 receptor [35]. Thus, it shows the direct effect of melatonin on insulin secretion. In another study, melatonin was found to relieve diabetes-induced oxidative stress [36]. Melatonin also showed significant effect on lipid metabolism, where it has a role in both lipogenesis and lipolysis. Studies on porcine oocytes in the presence of melatonin showed increase in the lipid droplet production required for their maturation and also the up-regulation of genes involved in lipid metabolism [37]. Most of the metabolic effects shown by melatonin are due to its ability to regulate the enzymes involved in said metabolism. For instance, it is reported to ameliorate serum fatty acid levels in alcohol-induced fatty liver disease in mice fed on high fat diet. The hypolipidemic effect of melatonin can be attributed to its ability to regulate AMP-activated protein kinase, which plays an important role in lipid metabolism [38]. It is also reported to modulate the epigenetic mechanism by upregulation of SIRT1, as well as p300 in different conditions. It has been observed that melatonin administration in SH-SY5Y cells increases acetylation of histones H3 and H4 [39, 40].
ROLE OF MELATONIN IN NEUROPROTECTION
Melatonin’s effect on mitochondrial function: Antioxidative effect and its implications on neuronal health
Mitochondria are the center for energy generation in cells by means of oxidative phosphorylation. The electron transport chain (ETC), which takes place in the inner mitochondrial membrane is coupled to oxidative phosphorylation to generate ATP. Nearly 95% of ATP produced in the cell comes from mitochondrial oxidative phosphorylation. The transfer of electrons through membrane-associated protein complexes (Complex I, II, III, and IV) is a well-regulated, stringent process. However, some of the electrons ‘leak’ and can generate free radicals by attacking other molecules and rendering them to become unstable radicals. Most of the molecular O2 is completely reduced in ETC to produce H2O. Still, some amount of partially reduced O2 produces ROS. Apart from ROS, generation of RNS also takes place in mitochondria [41, 42].
Melatonin can work effectively against free radical induced oxidative damage either by acting as a direct scavenger or by up-regulation of antioxidative enzymes [43, 44]. Melatonin and some of its derivatives like kynuramines are potent free radical scavengers. AFMK (N1-acetyl-N2-formyl-5-methoxykynuramine) is one such kynuramine produced enzymatically by indoleamine 2, 3-dioxygenase from melatonin or non-enzymatically by melatonin‘s reaction with free radicals like O2
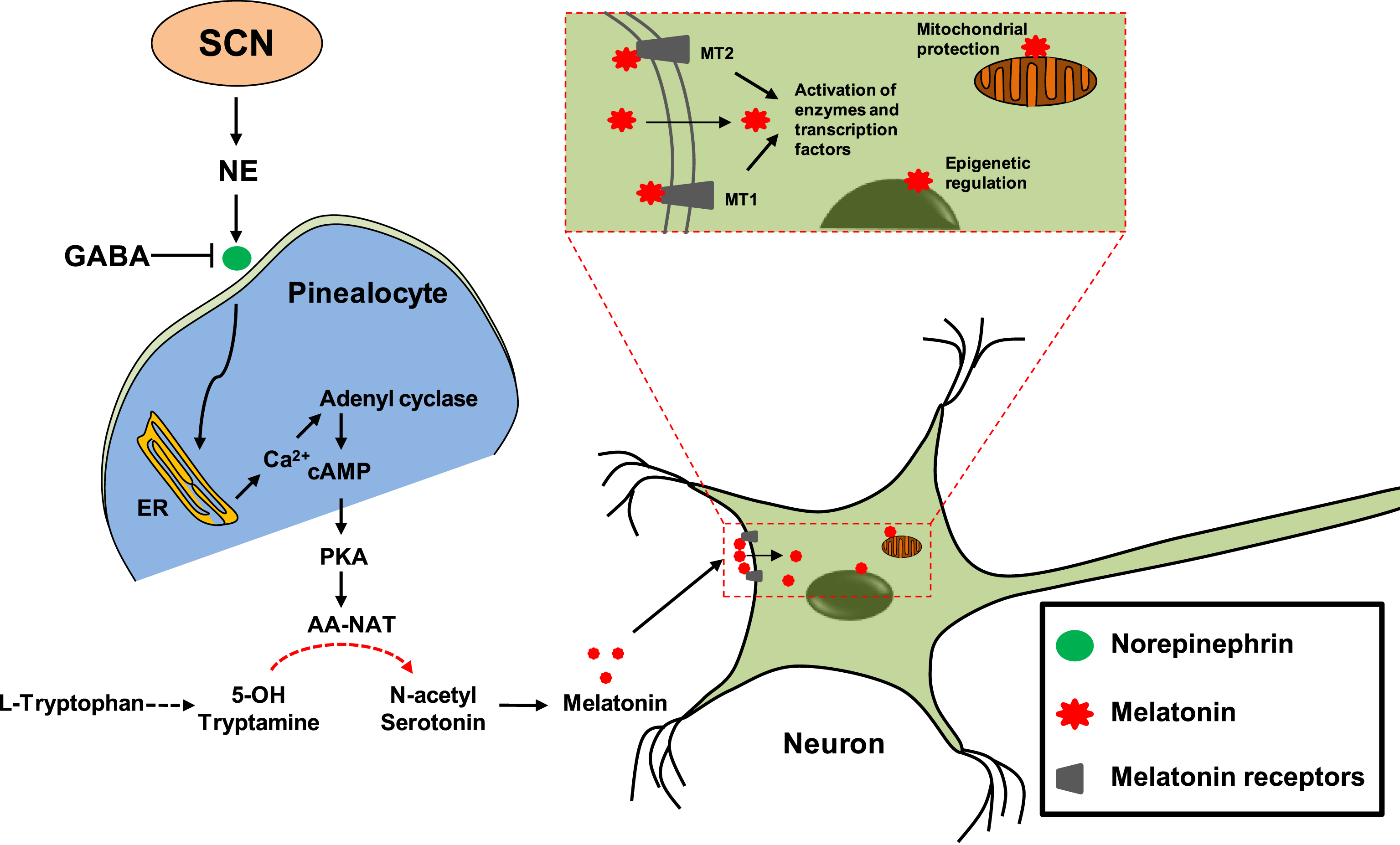
Pleiotropic effect of melatonin in neurodegenerative diseases. Secretion of NE (norepinephrine) from SCN (suprachiasmatic nucleus) acts on pinealocytes and leads to Ca2+ release from ER (endoplasmic reticulum) and the activation of adenyl cyclase to produce cAMP. GABA (Gamma amino butyric acid) inhibits the release of NE from SCN. cAMP binds to PKA (protein kinase A), which in turn activates AA-NAT (Aralkylamine N-acetyl transferase) or serotonin N-acetyl transferase. AA-NAT, is the enzyme required for the conversion of 5-hydroxy tryptamine to N-acetyl serotonin, which is the direct precursor of melatonin. Melatonin can act on MT1 and MT2 receptors to mediate a series of cascade for the activation of enzymes and transcription factors involved in neuronal development and protection. Melatonin, being a small amphipathic molecule, can cross-neuronal membrane and acts on mitochondria where it deals with oxidative damage. It is also able to cross nuclear membrane where it is involved in the epigenetic regulation of genes.
Nitric oxide synthase is the rate-limiting enzyme in nitric oxide synthesis. Over activity of this enzyme in stress conditions leads to RNS-mediated damage of cellular and mitochondrial membrane. Melatonin and N-acetyl-5-methoxykynuramine were found to reduce the activity of nitric oxide synthase [50, 51]. Also, all cells require reduced glutathione (GSH) to effectively neutralize the free radicals usually formed in the cytoplasm during numerous redox reactions. GSH also maintains the reducing cellular environment for normal functioning and regulation of cellular enzymes. Melatonin has been reported to stimulate the activity of γ-glutamylcysteine synthase, the rate-limiting enzyme involved in GSH synthesis, and thereby maintaining the cellular levels of GSH [52, 53]. It is also reported to regulate superoxide dismutase (SOD) and glutathione peroxidase levels in neuronal cell lines [54]. Enhancement of glutathione peroxidase activity by melatonin has also been reported in different studies [55, 56].
The effect of melatonin on mitochondrial complex activity has been shown in studies carried out on SAMP mice: SAMP8 (Senescence Accelerated Prone Mice) and SAMR1 (Senescence Accelerated Resistant Mice) [57, 58]. Complex I and III reflect the center where electrons are actively transported in ETC. After isolating the mitochondria from melatonin treated mice, it was observed that complex I and III activity significantly increases [57, 58]. Mitochondria are the center of oxidative stress generation in cells. Thus, targeting the free radical generation or underlying events of their generation is an important aspect of neuroprotection as oxidative stress accounts for neurodegeneration in AD [59, 60].
Direct interaction of melatonin with protein aggregates
Insoluble protein aggregates of both Aβ and microtubule-associated protein tau imparts neuronal toxicity in AD. Aβ gets accumulated in extracellular spaces after getting cleaved from its precursor transmembrane protein, APP (amyloid precursor protein), by γ-secretase while tau in its pathological form detaches from microtubules and self-associate to form neurofibrillary tangles [61, 62]. In order to reduce the protein aggregates-induced toxicity, it is important to target their aggregation. In this aspect, molecules or drugs, which can interact with protein aggregates and modulate their aggregation propensity by either altering their chemical properties, i.e. binding to specific amino acid residues or by weakening their self-association, can be helpful.
Apart from the antioxidant effects of melatonin, their direct interaction with protein aggregates is another way through which it can exert its neuroprotective function. Melatonin‘s role in dissolving Aβ aggregates has been studied. It was observed that it is able to interact with Aβ peptides and modulates its propensity for aggregation. Studies were carried out on Aβ aggregation in the presence or absence of melatonin to observe its effect on aggregation propensity and structural change [63]. NMR spectra for Aβ peptide with melatonin showed chemical shift dispersion indicating their binding and induction of local conformational changes [64]. To rule out the effect of melatonin as a free radical scavenger instead of its direct effect on aggregates, 5-hydroxy N-acetyl tryptamine (NAT), an analog of melatonin, or structurally unrelated N-t-butyl-α-phenylnitrone (PBN) were used as a control, both of which are potent free radical scavengers. In NMR, no chemical shift was seen for NAT, while in electron microscopy no Aβ fibril formation was seen with melatonin, which was evident with NAT and PBN [64]. The typical β-sheet structure seen in amyloid aggregates were observed to be reduced in the presence of melatonin. Similar observations were made for Aβ and melatonin interactions using ESI-MS and tryptic digestion of Aβ peptides after incubating with melatonin. It was observed that the hydrophobic region of Aβ peptide, involved in Aβ aggregates formation, is also involved in the interaction with melatonin [65]. Thus, the modulation of aggregates may occur through such hydrophobic interactions.
Aβ plaques are formed in the extracellular matrix while the neurofibrillary tangles made up of tau are present inside neurons. It was observed in case of Aβ fibrils that the interaction between melatonin and Aβ is hydrophobic, where it can bind to the region responsible for aggregation. Tau aggregates formed in AD also consist of hydrophobic patches through which they associate with each other. The conversion of highly hydrophilic tau protein monomers into insoluble hydrophobic aggregates involves charge neutralization by hyperphosphorylation. Most of the Ser or Thr residues that are phosphorylated in AD lies near to positively charged Lys residues, which gets charge neutralized after modification [66]. Melatonin can possibly bind to these regions and inhibit aggregation of tau but further studies are required to find out whether they can interact or not, and if they do, what is the nature of their interaction and its outcome [67, 68]. If melatonin is found to be interacting with tau as well as Aβ aggregates, it can be a good candidate drug for the treatment of AD associated neurodegeneration. Currently, there are no reports for the direct interaction of melatonin with tau and its effect on tau aggregation, and it would be interesting to explore the role of melatonin in this aspect.
Regulation of tau phosphorylation by melatonin
Pathological tau aggregates present in neurofibrillary tangles are found to be extensively hyperphosphorylated by a number of kinases [69]. Most of these are proline directed ser-thr kinases. GSK-3β is known to be involved in the hyperphosphorylation of tau. The effect of Aβ fibrils-induced toxicity on organotypic hippocampal slice cultures has been studied in Wistar rats [70]. It was observed that Aβ toxicity induced tau hyperphosphorylation in hippocampal neurons [71]. It was possibly due to increased activity of GSK-3β, which got reduced upon melatonin treatment [72, 73]. The regulation of GSK-3β activity by melatonin possibly occurs via activation of PI3K/Akt/GSK-3β signaling pathways [74]. The mechanism of attenuation of GSK-3β activity is not clear but its role in pathological stage dependent manner has been shown where melatonin administrations in older mice were found to relieve from GSK-3β-induced hyperphosphorylation and cognitive impairment [75]. In another study, the effect of melatonin on PKA activity was observed. PKA is a family of ser-thr kinases, which gets activated by binding to cyclic cAMP (cyclic AMP). It phosphorylates and regulates a number of cellular proteins, including tau. The activity of PKA increases in AD patients, implying its role in hyperphosphorylation [76]. Melatonin diminishes PKA activity through a receptor-mediated mechanism. On subjecting the Wistar rats to isopreterenol, which is specific PKA activator, there was an increased extent of tau hyperphosphorylation. PHF-1 and Tau-1 sites were most significantly phosphorylated which are also implicated in AD pathology. For the rats administered with melatonin prior to the PKA activity, hyperphosphorylation was reduced greatly [76]. Hyperphosphorylation of tau also induces oxidative stress, which is marked by increased SOD activity to fight against the peroxide radicals. Melatonin treatment further increases SOD activity to promote neuroprotection. Malondialdehyde (MDA) is a biomarker for oxidative stress, produced by the action of ROS on polyunsaturated fatty acids. The MDA level was found to increase along with Aβ-induced tau phosphorylation. On measuring the MDA levels in hippocampal extracts, melatonin treated rats showed decreased MDA indicative of its protective role in oxidative damage [76]. Effect of melatonin has also been shown in attenuation of wortmannin- or peroxynitrite-induced tau hyperphosphorylation [77–79]. Apart from PKA and GSK-3β, cyclin dependent kinase 5 (cdk5) is also known to be involved in tau hyperphosphorylation. Melatonin affects its activity by regulating its expression level as well as inhibiting the cleavage of p35 to p25, which forms a stable active complex with cdk5 [80]. Also, inhibition of melatonin synthesis in a Wistar rat model significantly increases cdk5 activity and tau hyperphosphorylation, which gets reduced after exogenous melatonin administration [81]. One of the derivatives of melatonin called melatonylvapromide has been found to be a potent inhibitor of phosphorylation as studied in APP transfected neuroblastoma N2a cells. Both melatonin and its derivative suppressed Aβ levels in N2a/APP cells, but the effect of its derivative was stronger. Phosphorylation was observed to be reduced at SMI33 and SMI34 epitopes of tau neurofibrils in melatonylvapromide-treated cells while melatonin exerts its effect only on SMI34 epitope [82].
In order to regulate the phosphorylated state of a protein, kinases and phosphatases work in a highly regulated and balanced manner. Hyperphosphorylation may occur either due to over-activity of kinases or lesser activity of their counterpart, i.e., phosphatases. The role of PP-2a (protein phosphatase 2a) being important in preventing hyperphosphorylation of tau has been reported [83–85]. Ischemic injury leads to reduction in PP-2a levels leading to imbalance in phosphorylation in a rat model. Melatonin has been reported to reinstate PP-2a levels back to normal [86]. In neuroblastoma cells N2a incubated with calyculin, an inhibitor of PP-2a, enhanced activity of GSK-3β and subsequent tau hyperphosphorylation was observed. Elevated level of MDA and decreased SOD in calyculin-treated cells implied oxidative stress. On incubating along with melatonin, the MDA levels were found to be reduced. Also, the level of phosphorylation at PHF-1 and Tau-1 sites decreased [87]. Thus, melatonin not only relieved oxidative stress but also modulates the phosphorylation of proteins.
Role of melatonin in relieving inflammatory response and effect on cholinergic system
Neuronal inflammation is one of the important aspects of the neurodegenerative diseases. Inflammatory response arises due to the interplaying of a number of mechanisms, leading to a series of cascades. Complementary system, cytokines, and chemokines are all involved in the inflammatory response [88]. In AD, the degeneration of neurons due to the accumulation of Aβ plaque and neurofibrillary tangles triggers the signal to evoke an inflammatory response. Aβ aggregates induce the inflammatory response possibly through the activation of the complement system, as it was observed that the components of complement system are co-localized with both Aβ and tau aggregates [89]. The complement system component forms a covalent complex with tau and Aβ aggregates in order to get activated [90–92]. Also, the membrane attack complexes are formed in the vicinity of aggregates deposition, suggesting their role in evoking inflammatory response through the complement system [93, 94].
The role of melatonin as an anti-inflammatory agent has been well studied and documented. Melatonin exerts its function by modulating the various pathways involved in the inflammation. Cytokines comprising of interferons and interleukins are produced as an inflammatory response to tissue damage which generates a cascade of events leading to enhanced response in neighboring cells [95]. Melatonin has been shown to inhibit the production of pro-inflammatory cytokines like IL-6, IL-8, and TNF-α, which results in the suppression of inflammatory responses [96]. Melatonin also inhibits the activity of nitric oxide synthase and reduces malondialdehyde levels, which are associated with the inflammatory response. Melatonin is also known to reverse the chronic and acute inflammation [21, 97].
AD is also associated with decreased levels of neurotransmitter acetylcholine. The level of enzymes, acetylcholine transferase, and acetylcholine esterase, which are involved in synthesis and hydrolysis of acetylcholine, does not change until the later stages of AD [98, 99]. Melatonin has shown protective effect in the APP 695 transgenic mice having Aβ-induced reduction in acetylcholine transferase activity [100].
Blood-brain barrier permeability of melatonin and its implications
One of the important aspects of therapeutics targeted against neurodegenerative diseases is its ability to cross the BBB in order to exert its protective effect. Most drugs have a limited ability to cross this barrier. In this regard, the properties of melatonin make it a suitable candidate for therapeutic use against neurodegenerative diseases. Most of the molecules that cross the BBB are transported through either membrane transporter or trans-membrane diffusion [101, 102]. Melatonin, being a small ampiphilic molecule, is known to be able to cross all biological membranes including the BBB [103, 104]. The most plausible way for melatonin to pass through the BBB is trans-membrane diffusion. In order to pass through membrane, the candidate molecule should have a low molecular weight and high degree of lipid solubility. Melatonin fulfills these criteria for BBB transport. Most of the drug development strategies for the BBB are targeted for membrane transporters and mostly involve ‘Trojan horse’ approach where the drug is conjugated with a small molecule of high BBB permeability. But this approach has its limitation in terms of its binding to receptor and its pharmacological properties after conjugation [105]. But, since melatonin itself can be used as a potent drug, its conjugation with other molecules is not a prerequisite for its transport through the BBB.
Melatonin is produced naturally in the body, and thus its level of secretion limits its function as a neuroprotective agent, and its administration as a drug is required for its potent therapeutic action. One of the possible strategies to use melatonin as a drug against neurodegeneration is by combining it with some other potent agents effective against aggregation either in conjugation or combination.
CONCLUSION
The etiology of AD includes multiple factors and so are its pathological implications. So, in order to target AD, we need a multi-faceted approach for effective treatment. Melatonin’s role in neuroprotection is well studied. It is naturally produced by the pineal gland and has no side effects as a therapeutic agent. Its multifunctional behavior as an antioxidant, anti-inflammatory agent, free radical scavenger, enzyme regulator, epigenetic regulator and aggregation inhibitor makes it a good candidate as a therapeutic agent (Fig. 4). The effect of melatonin as aggregation inhibitor of Aβ has been studied but its role with respect to tau aggregates need to be explored. Its ability to cross the BBB can be employed to design a more effective conjugate drug. Further research is required to explore the possibilities of using melatonin effectively against AD. On the basis of previous studies, its multiple effects in neuroprotection can be understood, but requires a more potent therapeutic approach, which may involve its metabolites or combination with other neuroprotective drugs.
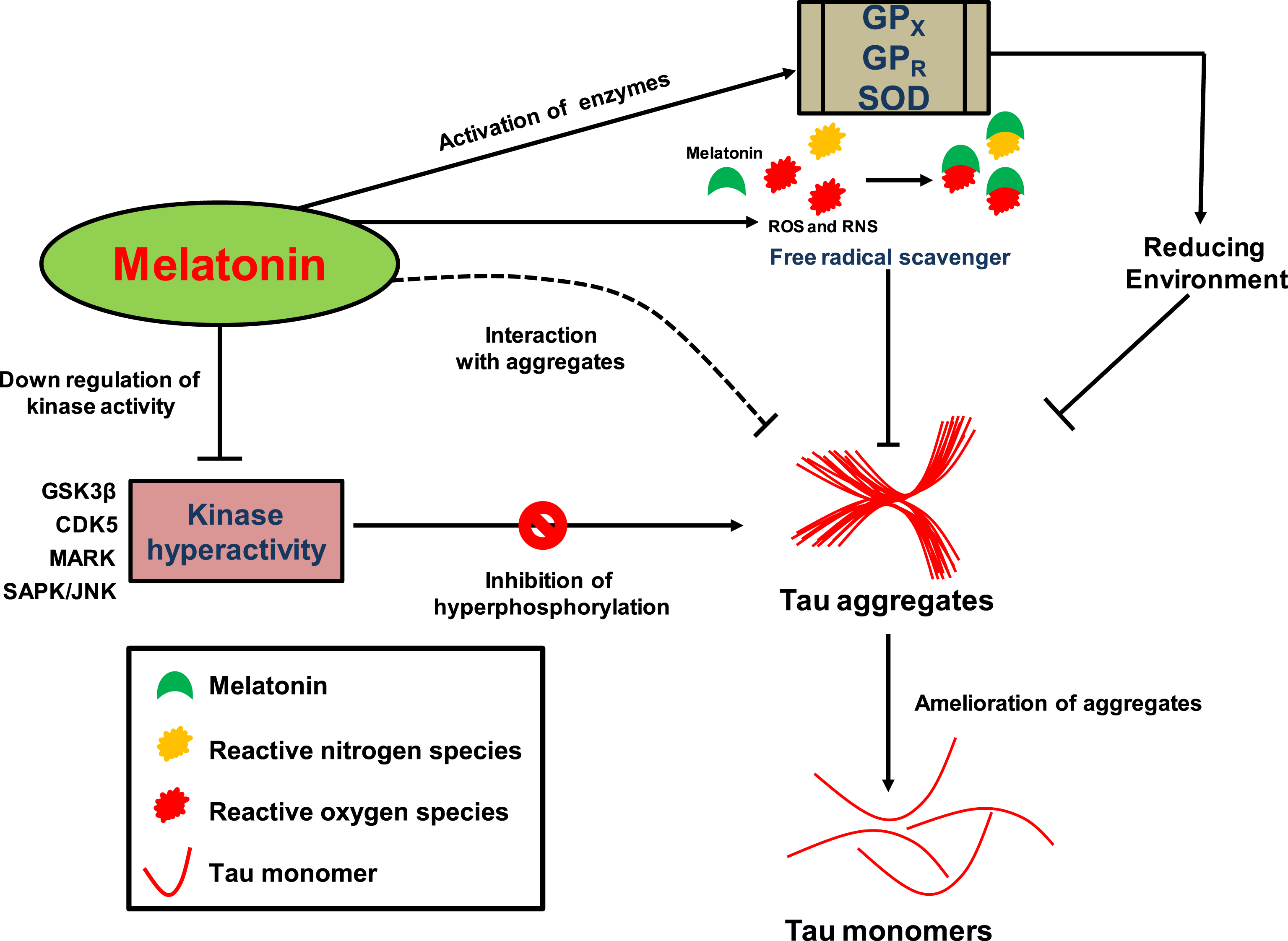
Possible model for the mode of action of melatonin against tau aggregation. The amelioration of tau aggregates by melatonin can be attributed to its pleiotropic effect. Activation of enzymes involved in antioxidant function like GPX, GPR, and SOD provides reducing cytoplasmic environment, which is preventive for aggregate formation. Melatonin itself can function as a free radical scavenger and is a precursor for more potent scavengers called Kynuramines, which exerts its effect on ROS and RNS. It can cross neuronal cell membranes and can interact with tau aggregates (dotted line), possibly by hydrophobic interaction, as reported for amyloid-β, and helps in inhibition/disassembly of aggregates. The down-regulation and inhibition of kinases involved in tau hyperphosphorylation like GSK-3β, cdk5, MARK (microtubule affinity-regulating kinases), and SAPK/JNK (stress-activated protein kinases/Jun amino-terminal kinases) may have a preventive role in tau aggregation resulting in reduced aggregate load inside neurons.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0900r2).
Footnotes
ACKNOWLEDGMENTS
Abhishek Ankur Balmik acknowledges the Shyama Prasad Mukherjee fellowship (SPMF) from Council of Scientific Industrial Research (CSIR), India.
This work was supported in part by grants from the Department of Science and Technology-Science and Engineering Research Board (DST-SERB, Young Investigator grant): SB/YS/LS-355/2013, Department of Biotechnology from Neuroscience Task Force (Medical Biotechnology-Human Development & Disease Biology (DBT-HDDB))-BT/PR/19562/MED/122/13/2016 and in-house CSIR-National Chemical Laboratory grant MLP029526.