
Abstract
Neuronal amyloid-β (Aβ) accumulation plays an important role in the pathogenesis of Alzheimer’s disease (AD). The conformation and toxicity of Aβ are regulated by lipids on the plasma membrane. Previously, we found downregulation of Rolling Blackout (RBO) or phosphatidylinositol-4-kinase type IIIα (PI4KIIIα) reduces neuronal Aβ accumulation and associated neural deficits in a Drosophila model expressing Aβ42. In mammals, the homologs of RBO and PI4KIIIα were reported to form a plasma membrane-localized complex with a scaffold protein TTC7 and cytosolic protein Hyccin/FAM126A to tightly control the plasmalemmal level of phosphatidylinositol-4-phosphate. Here, we show genetic downregulation of Drosophila TTC7 and Hyccin also reduces neuronal Aβ accumulation and associated synaptic and motor defects as well as premature death in Aβ42-expressing flies, while overexpression of TTC7 and Hyccin produced the opposite effect. These results, together with our previous study, demonstrate that RBO/TTC7/PI4KIIIα/Hyccin regulate neuronal Aβ accumulation and associated neural deficits in the Drosophila model, further supporting the RBO/Efr3-PI4KIIIα complex as a potential therapeutic target for AD.
INTRODUCTION
Alzheimer’s disease (AD) is the most common neurodegenerative disease, with the progressive loss of learning and memory as the most prominent symptom, and accumulation of amyloid plaque and neurofibrillary tangle as characteristic pathological lesions, but currently without any preventive or disease-modifying drug or intervention. Since the identification of amyloid-β (Aβ) as the major component of amyloid plaque in the AD brain, enormous amounts of evidence, especially the genotype-phenotype relationship in familial AD patients, have been accumulated to support the causal-effect relationship between abnormal Aβ metabolism and AD [1–5].
For many years, there has been much focus on the inhibition of Aβ production and the removal of extracellular amyloid deposits in both patients and animal models. However, inhibition of Aβ production using γ-secretase inhibitors not only failed to improve the cognitive function but also worsened the decline in cognitive scores and led to other adverse events in AD patients [6]. Removal of extracellular Aβ plaques by immunotherapy against Aβ also failed to slow down the cognitive decline in AD patients [7]. In addition, clinical trials on a β-secretase inhibitor, which can robustly inhibit Aβ production without obvious side effects, were halted due to “virtually no chance of finding a positive clinical effect” according to an independent panel of experts (http://www.Alzforum.org). Besides the repetitive failures in the efforts targeting extracellular Aβ, clinical trials targeting tau, another major player in the pathogenesis of AD have also failed (http://www.Alzforum.org). With this situation, both the academic and pharmaceutical area of AD research should consider, or pay attention to, other abnormal aspects of Aβ metabolism in AD.
In the brain of AD patients and animal AD models, Aβ, particularly Aβ42, not only accumulates in the extracellular space as diffuse deposits and fibrillar plaques but also builds up intraneuronally in the early stages. Intraneuronal accumulation of Aβ may play an important role in the pathological changes including synaptic deficits, mitochondrial impairment, formation of amyloid plaques, and cell death in AD [8–13]. Intraneuronal Aβ accumulation may result from the internalization of extracellular Aβ. In cultured cells, Aβ internalization and intracellular accumulation involves several Aβ-binding proteins [10], including α7 nicotinic acetylcholine receptor (α7nAChR) [14], low density lipoprotein receptor-related protein 1 (LRP1), heparan sulfate proteoglycan (HSPG) [15, 16], and receptor for advanced glycation end products (RAGE) [17]. However, the direct contribution of these proteins to the intraneuronal accumulation of Aβ in vivo remains to be substantiated.
It has been reported that Aβ is produced in many intracellular organelles in addition to the plasma membrane (PM), such as endoplasmic reticulum, Golgi, endosome, autophagosome, and lysosome [18–21]. Intraneuronal Aβ accumulation may also result from intracellular production and retention of Aβ. Supportively, a recent study revealed a distinct intracellular localization between presenilin 1 and 2 secretases and their differential contribution to a prominent pool of intracellular Aβ42. Presenilin 2 is restricted to late endosomes and lysosomes, and produces intracellular Aβ42, while disease-causing mutations in presenilin 2 elevates intracellular Aβ42. Presenilin 1 is distributed widely among the PM and intracellular organelles, and some disease-causing mutations in presenilin 1 shifts mutant presenilin 1 to late endosomes/lysosomes and increases intracellular Aβ42 [22]. This study not only firmly confirms a prominent pool of intracellular Aβ42 and its intracellular origin, but also underscores the importance of intracellular Aβ42 in the AD pathogenesis.
Aβ generated intracellularly can be transferred to the PM and released out of the cell by membrane trafficking between intracellular organelles and the PM [23–25]. On the PM, Aβ either produced on the PM or coming from intracellular organelles, or from the extracellular space, can interact with acidic phosphoinositides (e.g., phosphatidylinositol (PI), phosphatidylinositol-4-phosphate (PI4P), and phosphatidylinositol-4,5-phosphate (PI4,5P)), undergo structural conversion from random coil to β-sheet and form Aβ assemblies or oligomers [26–30]. Oligomeric Aβ has a much higher affinity to the PM than monomeric Aβ and are much more difficult to leave the cell [31, 32], therefore plasmalemmal phosphoinositides may regulate the internalization and retention of Aβ, and in turn regulate intracellular accumulation of Aβ.
Indeed, previously we found that in Aβ42-expressing fruit flies, exhibiting age-dependent intraneuronal accumulation of Aβ and associated neural deficits [30, 33– 35], genetic or pharmacological inhibition of phosphatidylinositol-4 kinase type III alpha (PI4KIIIα, cytosolic protein) and PI4KIIIα-interacting membrane protein Rolling Blackout (RBO) reduced intraneuronal accumulation of Aβ and associated neural deficits, likely via decreasing Aβ42 oligomerization in/on the PM [30]. In yeast and mammalian cells, PI4KIIIα, Efr3 (RBO homolog), TTC7 (a scaffold protein), and Hyccin (a cytosolic protein) interact with each other to form a PM-localized complex to tightly control the plasmalemmal level of phosphatidylinositol-4-phosphate (PI4P) [36–39]. TTC7 and Hyccin are well conserved in Drosophila (flybase). To further test and confirm the PI4KIIIα or PI4KIIIα complex as a potential target for reducing intraneuronal Aβ42 and treating AD, we investigated the effect of genetic manipulation of TTC7 and Hyccin on the intraneuronal accumulation of Aβ and its associated neural deficits.
MATERIALS AND METHODS
Animal strains and genetics
Drosophila stocks were cultured on standard medium at 24– 26°C. After pupation, the adult flies were cultured on standard medium and entrained into a 12 h light/dark cycle at 25°C if not otherwise indicated.
The following Drosophila lines were used: [Gal4]A307 (lab stock, used to drive the expression of UAS-transgene in the giant fiber (GF) system and other components of the nervous system), [UAS-Aβ42] (gift from Dr. Crowther at Cambridge University, transgenic lines for expressing arctic mutant human Aβ42 and is located on the 3rd chromosome), P[lacW] l(2)k14710k15603 (a strain with a P-element insertion in the l(2)k14710 gene, which is the Drosophila homolog of the tetratricopeptide repeat domain 7 gene, TTC7, in mammals), bin3EY09582 (a strain with a transgene containing the P[EPgy2] construct that causes overexpression of endogenous TTC7 in the presence of Gal4, and is named here as TTC7OE), [UAS-h-hyccin] (a transgenic strain for expressing the full coding sequence of human Hyccin in the presence of Gal4, and is named here as h-hyccinOE, please refer below for the construction of the UAS-h-hyccin transgene), and [UAS-hyccinRNAi] (VDRC ID 104577, a strain for expressing ds-RNA targeting the Drosophila hyccin or FAM126A, CG6406, in the presence of Gal4, and is named here as hyccinRNAi).
Flies containing these transgenes or mutations were back-crossed to an isogenic wild-type fly (w1118) of pure genetic background and then crossed to one or another to obtain the following groups:
Ctrl: [Gal4]A307/+;
Aβ42: [Gal4]A307/+;[UAS-Aβ42]/+;
hyccinRNAi: [Gal4]A307/[UAS-hyccinRNAi];
Aβ42 ;hyccinRNAi: [Gal4]A307/[UAS-hyccinRNAi];
[UAS-Aβ42]/+;
h-hyccinOE: [Gal4]A307/+;[UAS-h-hyccin]/+;
Aβ42 ;h-hyccinOE: [Gal4]A307/+;[UAS-h-hyccin]/
[UAS-Aβ42];
TTC7-/+: [Gal4]A307/P[lacW] l(2)k14710k15603;
Aβ42 ;TTC7-/+: [Gal4]A307/P[lacW]
l(2)k14710k15603; [UAS-Aβ42]/+;
TTC7OE: [Gal4]A307/bin3EY09582;
Aβ42 ; TTC7OE: [Gal4]A307/bin3EY09582;
[UAS-Aβ42]/+;
Construction of [UAS-h-hyccin] transgenic flies
[UAS-h-hyccin] construct was synthesized according to the coding sequence of human hyccin/FAM126A cDNA with the addition of Not1 and Xba1 before the start and after the stop codon respectively, and subcloned into the Not1— Xba1 sites of pUASTattB (Shanghai Generay Biotech Co. Ltd.). [UAS-h-hyccin] construct was injected into the embryos of isogenic w1118 flies to generate transgenic flies.
Reverse transcription–polymerase chain reaction (RT-PCR)
To verify the downregulation or overexpression of TTC7, endogenous Drosophila hyccin or exogenous human hyccin genes in flies, [Gal4-elav.L]3 instead of [Gal4]A307 was used for pan-neuronal expression of Gal4. For RT-PCR, mRNA was isolated using the HiScript QRT SuperMix for qPCR kit (Vazyme) from 20 female flies of each group at 25-day age after eclosion. cDNA synthesis was performed using the 2xTaq PCR Mastermix (Vazyme). Analyzed products were assayed in triplicate, with each replicate including 10 samples per group taken from an independent experiment. The primers used for TTC7 in the flies were: forward primer: 5’-CTGAGTGAAGCAGCAAGCTCATTGA-3’ and reverse primer: 5’-TCACATAATACAGCCAAGCTCGAACAA-3’. The mRNA of GAPDH was used as an internal reference and its primers used as follows, forward primer: 5’-CGGAGTGTTCACCACCATTGACAA-3’ and reverse primer: 5’-ACGGTAAGATCCACAACGGAGACA-3’. The primers used for h-hyccin were: forward primer: 5’-CCTTAACTTACATGCCCAGTGTT-3’ and reverse primer: 5’-GACCTGATTGATGCTTCATTGGA-3’ (PCR product 1000 bp). For Drosophila hyccin, forward primer: 5’-GATCACCAAGAGCTTCGACTC-3’ and reverse primer: 5’-GAACATAGTCTCCCTCTGACTGAA-3’ (PCR product 700 bp).
Climbing ability assay
We used an automatic system for a highly replicable examination of the fly climbing ability [40]. Briefly, for each group of flies, 30 flies were equally divided and separately transferred into 3 testing vials. After being tapped down to the bottom of the testing vials, flies were allowed to climb up along the walls of the vertically placed testing vials and were videotaped. The height of each fly in each vial at the 7th second was measured for evaluating the fly’s climbing ability.
Longevity assay
One hundred flies from each genotype were equally separated into five vials containing standard fly food and dry yeast and cultured at 25°C. After 3 days, flies were transferred into vials with fresh food and dried yeast, and the dead flies were counted. Survival rates were analyzed using the Kaplan-Meier survival curve.
Electrophysiology test of synaptic transmission failure in flies
Evoked excitatory junctional potential (EJP) in the GF system was recorded intracellularly as described previously [33]. Briefly, an adult female fly at a certain age was mounted ventral side down on a glass slide with tackiwax (Boekel Scientific) under a dissection microscope. Before the recording, a reference electrode was inserted into the abdomen, two stimulating electrodes into the two eyes, and a recording electrode into one dorsal longitudinal muscular (DLM) cell. Intracellular penetration of the recording electrode into the muscle was monitored by a sudden potential drop of 40– 70 mV. Square wave electrical stimulation (0.2 ms pulse width, 100 Hz, 50 pulses generated by Electronic Stimulator (Nihon KOHODEN)) was applied to both eyes at the intensity of 5– 20 V (about 150% of the threshold stimulation intensity). The signal of the EJPs, whose success rate reflects the well-being of synaptic function, was recorded and amplified by NEUROPROBE AMPLIFIER model 1600 (A-M Systems) and digitized at 10 kHz by Digidata 1322A (Molecular Devices). Data was collected and analyzed with the pClamp software (version 9.2; Axon Instruments). All electrodes were glass electrodes and filled with 0.1M KCl. The recording environment temperature was 25°C.
Immunostaining and imaging
For whole-mount Aβ staining, the whole CNS of flies, including the brain and ventral ganglion, was dissected out in PBS and fixed with 4% paraformaldehyde in PBS for 45 min. Preparations were washed with PBS for 10– 15 min, repeated twice, and then treated with formic acid (70%) for 30 min to re-expose the epitope, followed by three washes with PBS-Triton X-100 (0.5%)-BSA (5%), 2– 3 h incubation with primary antibody (6E10), PBS wash, 2 h incubation with FITC-conjugated secondary antibody, preparation of CNS slice on glass and confocal imaging. Experiments were done at room temperature.
ELISA quantification of Aβ42
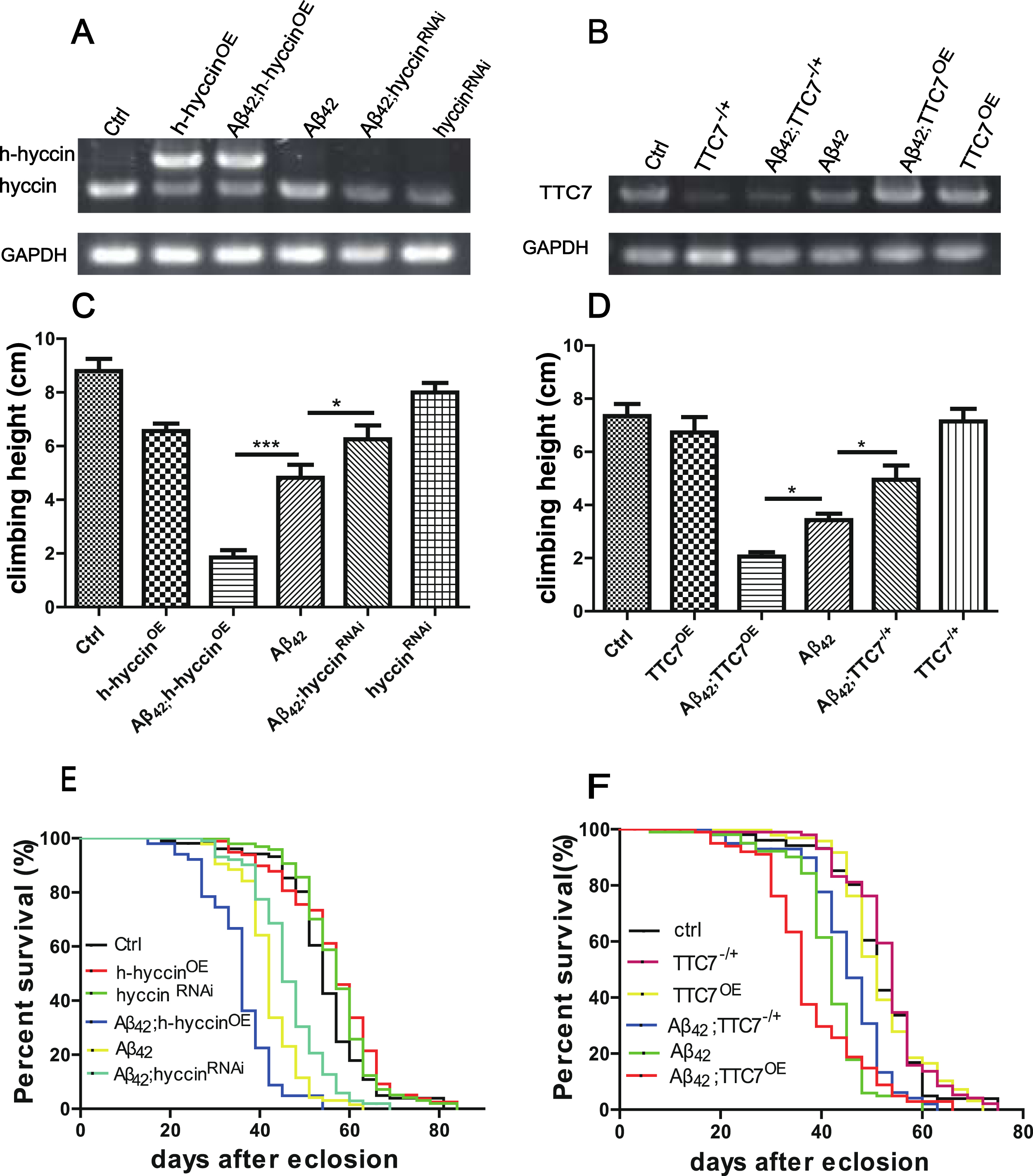
The dosage-dependent regulation of motor defect and premature death in flies expressing Aβ42 by hyccin and TTC7. A) Representative images showing the expression level of hyccin or h-hyccin in the Ctrl, h-hyccinOE, Aβ42;h-hyccinOE, Aβ42, Aβ42;hyccinRNAi and hyccinRNAi flies, repeated at least three times. B) Representative images showing the expression level of endogenous TTC7 in the Ctrl, TTC7-/+, Aβ42;TTC7OE, Aβ42, Aβ42;TTC7-/+ and TTC7OE flies, repeated at least three times. C,D) Quantitative analysis of the climbing ability by aRING assay in different groups of flies at the age of 24 days (C) or 28 days (D), minimum n = 30 flies per bar, one-way ANOVA using LSD test; *p < 0.05 and ***p < 0.001. E,F) The lifespan of different groups of flies, minimum n = 100 flies for each line, log-rank test, compared with Aβ42 flies, the p-value for Aβ42;h-hyccinOE, Aβ42;hyccinRNAi, Aβ42;TTC7OE and Aβ42;TTC7-/+ flies was less than p < 0.01.
Quantification of Aβ42 levels at 24 days after eclosion in the fly CNS was performed using the Aβ42 Human ELISA Kit (Invitrogen), according to the manufacturer’s instructions. To analyze Aβ42 levels in the fly CNS, intact brains of 20 flies per strain were dissected out in cold PBS and placed immediately into cold ELISA sample buffer supplemented with cocktail protease inhibitor (Calbiochem). Brains were homogenized thoroughly and incubated at room temperature for 4 hours. Samples were spun at 4,000 g, 4°C for 10 min and the supernatant collected for ELISA. Analyzed products were assayed in 3 replicates, with each replicate including 20 samples per group taken from an independent experiment.
Statistical analysis
SPSS (RRID: SCR_002865) and GraphPad Prism software were used for data analysis. The data is presented as mean±SEM if not specified elsewhere. The criterion for significant difference is p < 0.05 (*p < 0.05, **p < 0.01, and ***p < 0.001).
RESULTS
The effect of genetic manipulation of TTC7 and hyccin’s expression on the motor defect and premature death in Aβ42-expressing flies
The expression levels of TTC7 and hyccin, or transgenic human hyccin (h-hyccin), were analyzed in different groups of flies by RT-PCR. As shown in Fig. 1A, the mRNA level of endogenous hyccin was not changed in the flies with pure overexpression of Aβ42 (Aβ42 flies), but reduced in the hyccin-knockdown flies without and with the overexpression of Aβ42 (hyccinRNAi and Aβ42;hyccinRNAi respectively), when compared to that in the non-Aβ42 control flies (Ctrl). In the flies expressing human hyccin, without and with the overexpression of Aβ42 (h-hyccinOE and Aβ42;h-hyccinOE, respectively), the mRNA level of human hyccin was much higher than the mRNA level of endogenous hyccin in the Ctrl and Aβ42 flies. Similarly, as shown in Fig. 1B, the mRNA level of endogenous TTC7 was not changed in the flies with pure overexpression of Aβ42 (Aβ42), but reduced in the TTC7-partial knockout flies without and with the overexpression of Aβ42 (TTC7-/+ and Aβ42;TTC7-/+, respectively), and increased in the TTC7-overexpression flies without and with the overexpression of Aβ42 (TTC7OE and Aβ42;TTC7OE, respectively), when compared to that in the Ctrl flies.
Previously, we found that overexpression of Aβ42 in the GF system and some other neurons in the central nervous system produced an age-dependent decline of climbing ability and premature death in adult flies [30, 40]. Consistently, the aRING assay revealed that the climbing ability of the aged Aβ42 flies was reduced when compared to that in the Ctrl flies of the same age, and this Aβ42 expression-induced motor defect was ameliorated by the knockdown of hyccin, but enhanced by the overexpression of human hyccin (Fig. 1C). Similarly, partial knockout or overexpression of TTC7 suppressed or enhanced the motor defect induced by the expression of Aβ42 respectively (Fig. 1D). In the flies without Aβ42 expression, the alteration in the expression of hyccin or TTC7 largely did not change the motor ability (Fig. 1C, D).
Consistent with the effect on climbing ability, separate downregulation of hyccin and TTC7 extended, while separate overexpression of human hyccin and TTC7 further shortened the lifespan in the flies expressing Aβ42 (Fig. 1E, F), but without obvious effect in the non-Aβ42 flies (Fig. 1E, F).
The effect of genetic manipulation of TTC7 and hyccin’s expression on the synaptic failure in Aβ42-expressing flies
To investigate the effect of down- or upregulation of TTC7 or hyccin on the age-dependent failure of synaptic transmission in the flies expressing Aβ42 [33, 42], we examined the neural transmission through the GF system in vivo. Synaptic transmission was examined by intracellular recording of EJPs in the DLM cells under high-frequency brain stimulation (100 Hz, 50 pulses). Electrical brain stimulation at 100 Hz (total of 50 stimuli) was used to activate the GF system, and the evoked EJPs were intracellularly recorded in one of the DLM cells in flies at the age of 24– 28 days. Representative traces of recordings from different fly groups are shown in Fig. 2A and B. Consistent with our previous reports, expression of Aβ42 in the GF system caused synaptic transmission failure in aged flies, and this impairment was suppressed and enhanced by down- and upregulation of TTC7 and hyccin, respectively, without obvious effect in non-Aβ42 flies (Fig. 2A-D).
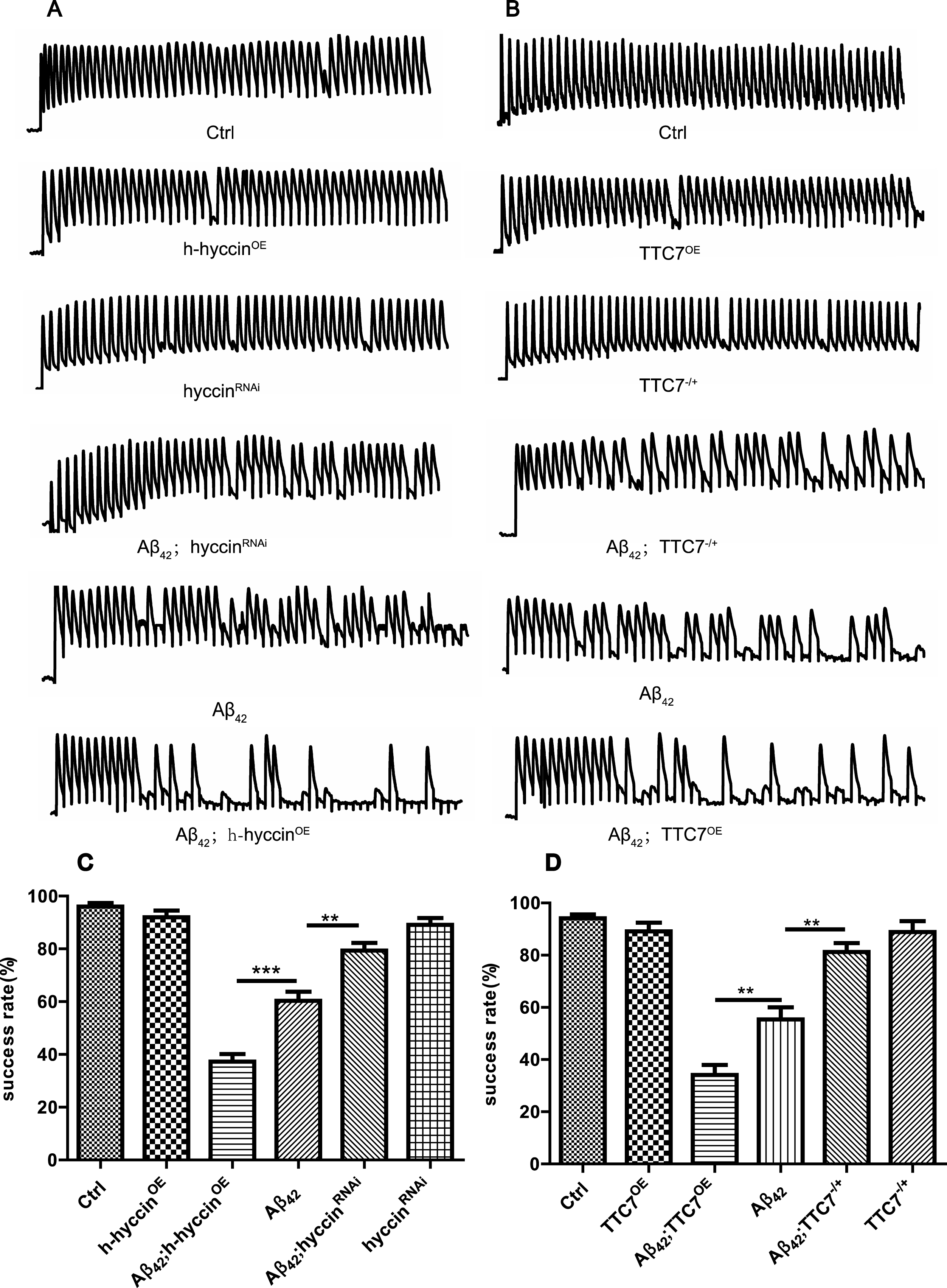
The dosage-dependent regulation of synaptic impairment in the flies expressing Aβ42 by hyccin and TTC7. Representative traces (A and B) and quantification (C and D) of the brain stimulation-evoked EJP recordings in the DLM cells of flies from different groups at the age of 24– 28 days, n = 6 flies per bar, one-way ANOVA using LSD test. *p < 0.05, **p < 0.01, and ***p < 0.001.
The effect of genetic manipulation of hyccin and TTC7’s expression on the intraneuronal accumulation of Aβ in Aβ42-expressing flies
Since the synaptic failure, motor impairment and premature death were caused by intraneuronal Aβ42 accumulation, we examined the effect of genetic manipulation of TTC7 and hyccin’s expression on the flies expressing Aβ42 by immunostaining. As expected, the intraneuronal accumulation of Aβ42 was decreased and increased by separate down- and upregulation of TTC7 and hyccin respectively (Fig. 3A, B), which are consistent with the findings in the behavioral and synaptic functional assays. Quantitative analysis of these results were obtained using ELISA to measure levels of human Aβ42 in the down- or upregulation of hyccin and TTC7 Aβ42-expressing flies (Fig. 3C and D, respectively). As expected, the Ctrl flies did not express any human Aβ42.
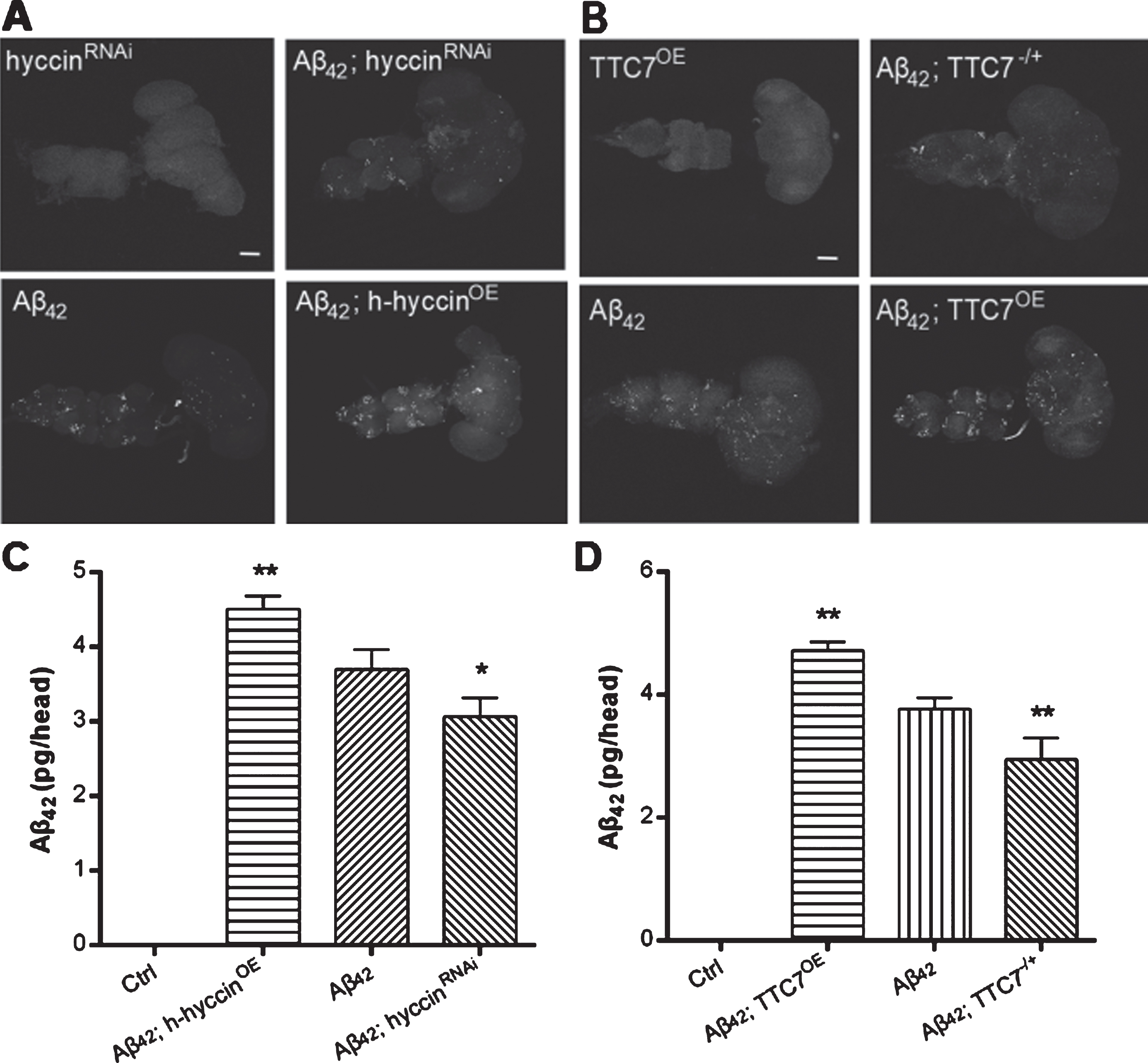
Representative images from different fly groups at the age of 24– 28 days. Immunostaining of Aβ42 in the whole CNS of flies, including the brain and ventral ganglion, of (A) hyccinRNAi, Aβ42;hyccinRNAi, Aβ42, and Aβ42;h-hyccinOE flies, as well as (B) TTC7OE, Aβ42;TTC7-/+, Aβ42 and Aβ42;TTC7OE flies. Scale bars indicate 100μm. Quantitative analysis of human Aβ42 levels, by ELISA, of the brains of flies expressing Aβ42 and with or without a knockdown or overexpression of (C) hyccin or (D) TTC7, n = 60 flies per bar, one-way ANOVA Tukey’s test was used for statistical significance; *p < 0.05, **p < 0.01. As expected, the Ctrl flies did not express human Aβ42.
DISCUSSION
Together, the immunostaining, behavioral and electrophysiology results show that downregulation of TTC7 and hyccin prominently decreased the amount of intraneuronal accumulation of Aβ42, and as a consequence, ameliorated the enhanced decline of mobility and prolonged the lifespan of Aβ42-expressing flies. Overexpression of TTC7 and hyccin increased the amount of intraneuronal accumulation of Aβ42, concomitant with the deteriorating mobility and further shortened lifespan of Aβ42-expressing flies compared to the control Aβ42-expressing flies.
Recent studies suggest that the buildup of intracellular Aβ may be an early event in the pathogenesis of AD. Emerging evidence from transgenic animals and human patients indicate that Aβ can accumulate intraneuronally and cause severe neuronal and synaptic dysfunction and loss, preceding the formation of extracellular Aβ deposits, which contribute to disease progression. Aβ is produced as a monomer, but readily aggregates to form oligomers. Aβ oligomers have a much higher affinity to the PM and are much more difficult to leave the cell [31, 32], which is sufficient to initiate and propagate the AD pathology. Aβ aggregation has been shown to occur during interactions with lipid bilayers, for example, cholesterol and acidic phospholipids with the conformational transformation of Aβ from irregular coil to α-spiral and β-sheet [43]. This can facilitate the assembly of monomeric Aβ to generate a membrane-bound form of Aβ with seeding ability. Thus, the plasma and intracellular membranes have an important role in generating toxic Aβ species.
As biological lipid membranes can modulate both protein folding dynamics and rates of protein aggregation, different lipid compositions in different subcellular compartment membranes may have a role in Aβ aggregation. Phosphoinositides have been widely reported to be implicated in the AD pathogenesis. A depletion of PI and a reciprocal elevation of phosphatidylinositol phosphate (PIP) were recently observed in the parietal cortex of AD patients and found to correlate with disease development and progression [44]. Our previous studies show that RBO and PI4KIIIα insufficiency or pharmaceutical inhibition of PI4KIIIα, which can inhibit the production of PI4P on the PM, reduces the neuronal accumulation of Aβ and neural deficits in Aβ-expressing flies [30]. So, we propose that PI4P on the PM might play an important role in Aβ oligomerization and intracellular accumulation.
RBO is a PM-localized protein, whereas hyccin, PI4KIIIα and TTC7 are cytoplasmic proteins. TTC7, as the scaffold protein, can recruit PI4KIIIα from the cytoplasm to the PM, where TTC7 and PI4KIIIα will form an evolutionarily conserved complex with RBO. This complex can catalyze the phosphorylation of PI by PI4KIIIα at the 4-position of the inositol ring, generating PI4P on the PM. In addition, recent research has shown that the complex catalyzing the phosphorylation of PI to generate PI4P on the PM includes another cytoplasmic protein called hyccin [36]. Hyccin can work together with the scaffold protein TTC7 in recruiting PI4KIIIα from the cytoplasm to the PM and enhance the catalytic activity of PI4KIIIα.
By expressing Aβ42 in the GF system in Drosophila, with a tissue-specific driver, we generated an AD model for investigating the intracellular accumulation of Aβ, age-dependent alterations of synaptic function and motor ability [33–35]. Based on this model, we genetically reduced and overexpressed TTC7 or hyccin, which respectively ameliorated and deteriorated the intraneuronal accumulation of Aβ and age-dependent alterations of synaptic function and motor ability. In control flies, reduction and overexpression of TTC7 or hyccin did not significantly change the synaptic transmission, mobility, and lifespan. Based on these results, we further demonstrated the RBO-PI4KIIIα-TTC7-hyccin complex, which is responsible for the phosphorylation of PI to generate PI4P on the PM, plays an important role in Aβ oligomerization and intracellular accumulation, and this can affect the phenotype of Aβ42-expressing Drosophila. But the direct interaction of Aβ and PI4P on the plasma membrane at the single cell level still needs to be studied further. Nonetheless, the results in this study highlight the importance of intracellular Aβ in the AD pathogenesis and facilitating Aβ secretion as a novel therapeutic strategy for AD treatment. This therapeutic strategy would involve manipulating the RBO/Efr3-PI4KIIIα-TTC7-hyccin complex to reduce the neuronal accumulation of Aβ42 by facilitating Aβ42 secretion.