
Abstract
The role of PCSK9 in Alzheimer’s disease (AD) is controversial. We compared cerebrospinal fluid (CSF) PCSK9 concentrations in 36 AD and 31 non-AD patients. CSF PCSK9 levels did not differ between AD and non-AD groups (2.80 versus 2.62 ng/mL). However, PCSK9 CSF levels were increased in AD and non-AD patients with other neurodegenerative process (non-AD ND, n = 20) compared to patients without neurodegenerative disorders (non-ND, n = 11): 2.80 versus 2.30 (p < 0.005) and 2.83 versus 2.30 ng/mL (p = NS), respectively. CSF PCSK9 were positively correlated with AD biomarkers (Aβ1-42, T-tau, and P-tau). PCSK9 concentrations in CSF are increased in neurodegenerative disorders rather than specifically in AD.
INTRODUCTION
Proprotein convertase subtilisin kexin type 9 (PCSK9) is a critical regulator of LDL-cholesterol (LDL-C) metabolism. Indeed, the canonical role of PCSK9 is to act as an endogenous inhibitor of the LDL receptor (LDLR) pathway [1]. Genetic studies demonstrated that Pcsk9 gain-of-function mutations are associated with autosomal dominant hypercholesterolemia and premature atherosclerosis [2], while Pcsk9 loss-of-function variants conversely lead to low levels of LDL-C and cardiovascular protection [3]. Briefly, after intracellular autocleavage, PCSK9 is secreted by the liver in the circulation, binds to the extracellular domain of the LDLR at the cell surface of the hepatocytes, and targets it to lysosomes for degradation [1]. Pharmacological inhibition of PCSK9 by PCSK9 inhibitors (i.e., human monoclonal antibodies directed against PCSK9) leads to a ≈ 60% reduction of LDL-C and a significant decrease of major cardiovascular events [4–6].
Beyond its role in the liver, an increasing body of evidence suggests that PCSK9 can also exert some extra-hepatic actions [7]. PCSK9 has been cloned as the ninth member of proprotein convertases in primary cerebellar neurons under apoptosis and was initially called NARC-1 (neural apoptosis-regulated convertase 1) [8]. PCSK9 seems to play a role in neurogenesis since its overexpression in a primary culture of embryonic neural progenitor cells stimulates the differentiation of cortical neurons [8]. While PCSK9 downregulates LDLR levels during brain development in mice [9], total PCSK9 knockout mice did not show signs of altered central nervous system (CNS) development [10]. Beside LDLR, PCSK9 also promotes the degradation of the apolipoprotein E receptor E2 (ApoER2) [11], which is involved in the regulation of neuronal apoptosis [12].
A more controversial issue is the role of PCSK9 on amyloid deposition and Alzheimer’s disease (AD). PCSK9 has been shown to modulate the disposal of BACE1 (beta-site amyloid precursor protein-cleaving enzyme 1), the enzyme involved in the generation of the amyloid β-peptides 1–40 and 1–42 (Aβ), with increased levels of BACE1 and total Aβ in the brain of PCSK9 knockout mice [13]. These results were, however, not confirmed in a subsequent study [14]. Recently, Zimetti et al. reported that PCSK9 levels in cerebrospinal fluid (CSF) were higher in AD than in non-AD subjects [15].
Here, we compared CSF PCSK9 in AD and non-AD patients (including subgroups of patients with (non-AD ND) and without (non-ND) neurodegenerative disorders) and investigated the correlations between PCSK9 and biomarkers of AD [Aβ1-42, total tau (T-tau), and phosphorylated tau (P-tau)] in CSF.
PATIENTS AND METHODS
Patients
CSF samples from patients with the three biomarkers in favor of biological AD (Aβ1-42 <700 ng/L, T-tau≥350 ng/L and P-tau≥60 ng/L) (n = 36) and age- and gender-matched non-biological AD subjects, with the three biomarkers normal (Aβ1-42 >800 ng/L, T-tau <350 ng/L, and P-tau <50 ng/L) (n = 31) were collected at the Neurological Memory Centre, Nantes University Hospital from April 2013 to May 2014. CSF studies were performed for routine biological diagnosis for patients presenting with cognitive impairment suspected to be AD or related disorders. All procedures followed were in accordance with the Helsinki Declaration of 1964, as revised in 2013. The biocollection was approved by the local ethical committee and the patients given their written informed consent.
Clinical diagnosis
The diagnoses were retained from the medical records. They were made according to clinical criteria, before the results of CSF biomarkers. Patients fulfilled the diagnosis criteria for probable AD dementia with evidence of the AD pathophysiological process [16], frontotemporal lobar degeneration (FTLD) [17], primary progressive aphasia [18], progressive supranuclear palsy [19], corticobasal syndrome [20], Lewy body disease [21], or multiple sclerosis [22].
Biochemical analyses
CSF were obtained by lumbar puncture. The transport to the laboratory was performed within 1 hour and CSF samples were centrifuged at 2,100 g for 10 min at 4°C within 2 h. A part of CSF was used for routine analysis, including measurement of total cells, total protein, and glucose level. CSF and serum IgG and albumin levels were measured by immunochemical nephelometry method using Immage® 800 Analyzer (Beckman Coulter, Villepinte, France). In all patients, blood-CSF-barrier (B-CSF-B) dysfunction was determined by the CSF/serum albumin quotient (QAlb).
In the same time, CSF samples were aliquoted in polypropylene tubes of 2 mL and stored at –80°C until measurement of AD biomarkers were done. CSF Aβ1-42, T-tau, and P-tau were measured in duplicate using commercially available sandwich ELISAs Innotest, (Fujirebio, Les Ulis, France) following the manufacturer’s instruction. PCSK9 concentrations were assayed in duplicate using a commercially available quantitative sandwich ELISA assay (Circulex CY-8079; CycLex Co, Nagano, Japan) and following the manufacturer’s instructions, as previously described [23].
Statistical analysis
Results were presented as median (interquartile range), minimum, and maximum. Categorical variables were presented as counts (proportions). The Mann–Whitney-U test and Kruskal Wallis test were performed to test for statistical differences in continuous parameters between two groups or three groups. The χ2 or the Fisher exact test (based on expected frequency) were used to compare categorical variables between groups. A Bonferroni correction was used for multiple comparisons. Spearman’s correlations and linear regression were used to determine the association between PCSK9 levels and the biomarkers of AD in CSF. Statistical significance was set at p < 0.05 or p < 0.02 (with Bonferroni correction). Statistical analysis was performed using SAS, version 9.4 (SAS Institute, Cary, North Carolina).
RESULTS
Baseline characteristics of the patients
The baseline clinical and biological characteristics of patients are listed in Table 1. There was no significant difference between AD and non-AD patients for gender (≈60% female) and age (≈68 years). Non-AD patients were classified into the following two subgroups according to their final clinical diagnosis: non-AD neurodegenerative disorders (non-AD ND, n = 20) and non-AD non-neurodegenerative diseases (non-ND, n = 11). FTLD was the most frequent diagnosis in the non-AD ND group, while alcohol abuse and normal pressure hydrocephalus were the most common causes in non-ND group. Mean Mini-Mental State Examination (MMSE) score was <25 in all three groups. As stated in the methods section, AD patients had a typical AD neurobiomarker pattern, with a significant decrease in Aβ1-42 and a concomitant increase in T-tau and P-tau, whereas neurobiomarkers were normal in the non-AD group. There was no difference in the CSF neurobiomarker profile between the non-AD ND and non-ND subgroups. Markers of brain-blood barrier permeability, such as QAlb and IgG index all were comparable between all three groups.
Demographic data, clinical diagnosis, and biological parameters
AD, Alzheimer’s disease; MMSE, Mini-Mental State Examination; NS, not significant; CSF, cerebrospinal fluid; FTLD, Frontotemporal lobar degeneration; LBD, Lewy body dementia. Data are expressed as Mediane [Min-Max]; Nonparametric two-sided Kruskal Wallis test was applied to compare the three groups. *Nonparametric two-sided Mann Whitney test to compare AD versus other neurodegenerative process group (p < 0.001). αNonparametric two-sided Mann Whitney test to compare AD versus Non-degenerative group (p < 0.001).
CSF PCSK9 levels
There were no significant differences in CSF PCSK9 concentration between AD and non-AD patients (median [IQ25-IQ75]: 2.80 [2.34–3.63] versus 2.62 [1.73–2.98] ng/mL, p = 0.14) and between AD and the non-AD ND subgroup (2.80 [2.34–3.63] versus 2.83 [2.02–4.59] ng/mL, p = 0.90). A significant difference was only observed when AD group was compared to the non-ND group (2.80 [2.34–3.63] versus 2.30 [1.60–2.62] ng/mL, p = 0.005). In non-AD patients, CSF PCSK9 levels were increased in non-AD ND group compared to non-ND group but this difference did not reach statistical significance (2.83 [2.02–4.59] versus 2.30 [1.60–2.62] ng/mL, p = 0.04) (Fig. 1).

CSF PCSK9 levels in Alzheimer’s disease (AD), non-AD non neurodegenerative (non-AD ND), and non-neurodegenerative (non-ND) groups. Data are expressed as median (IQ25; IQ75) in ng/mL; Non-parametric two-sided Kruskal Wallis test was applied to compare the three groups (p = 0.03). Nonparametric two-sided Mann Whitney test with a Bonferroni correction (statistical significance: p < 0.017) was applied to compare AD versus non-AD ND (p = 0.90), AD versus non ND (p = 0.005), and non-AD ND versus ND (p = 0.04, NS). NS, not significant.
Correlation between CSF PCSK9 and AD biomarkers
As a next step, we investigated the correlations between PCSK9 and the neurobiomarkers of AD in the CSF (Fig. 2). PCSK9 was positively associated with both Aβ1-42 (p = 0.02) and P-tau (p = 0.0004), regardless of patients subgroups. In addition, PCSK9 was also positively associated with T-tau, but only in AD (p = 0.004) and non-AD ND (p = 0.009) groups. Of note, PCSK9 was neither significantly associated with markers of B-CSF-B permeability nor MMSE score.
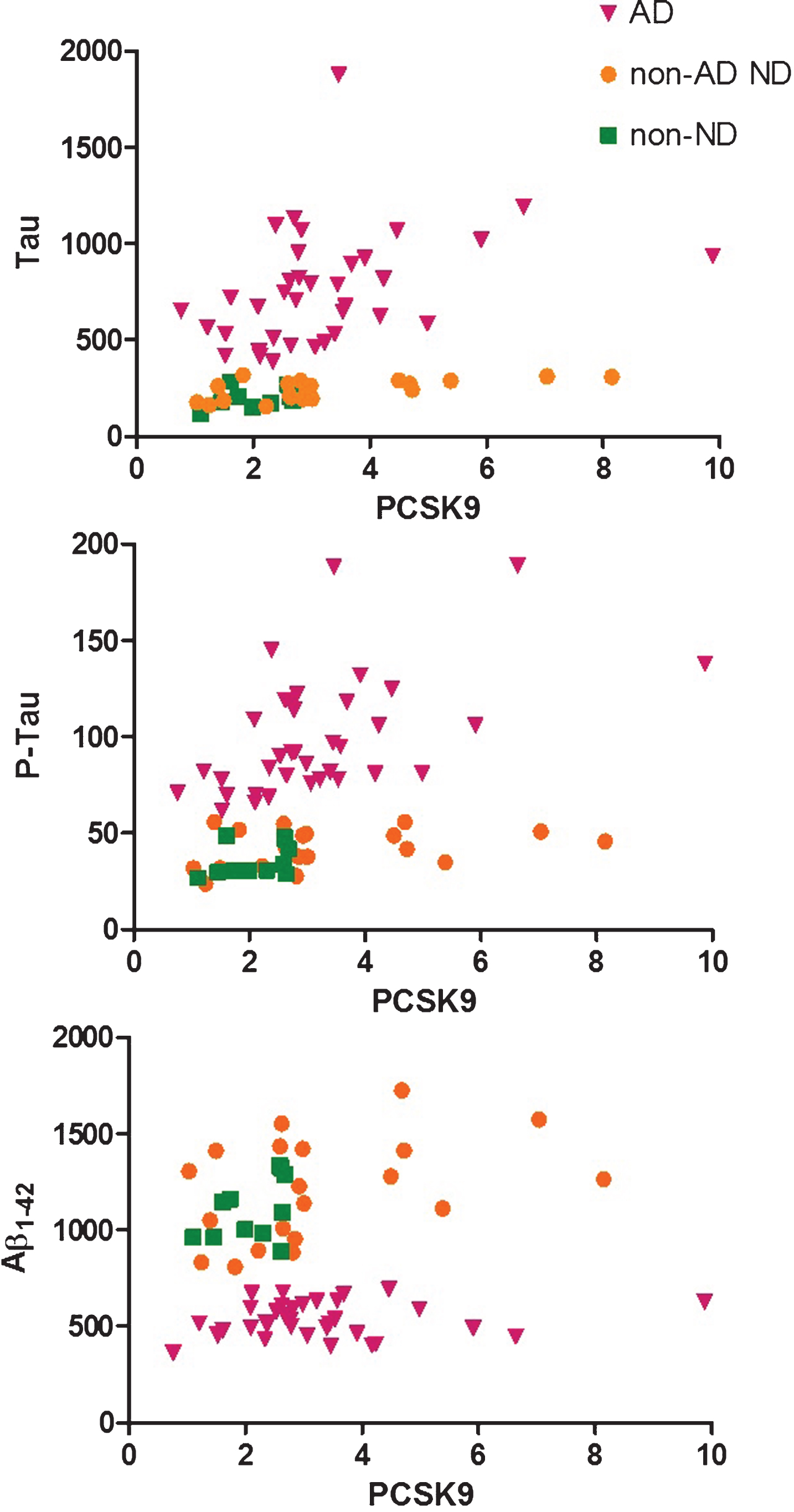
Tau, P-Tau, and Aβ1-42 levels (ng/L) in CSF as a function of PCSK9 levels (ng/mL) in Alzheimer’s disease (AD, triangles), non-AD non neurodegenerative (non-AD ND, circles), and non-neurodegenerative (non-ND, squares) groups. PCSK9 was positively associated with both Aβ1-42 (p = 0.02) and P-tau (p = 0.0004), regardless of patients subgroups, and with T-tau, but only in AD (p = 0.004) and non-AD neurodegenerative (p = 0.009) groups (Spearman’s correlations and linear regression).
DISCUSSION
It has been recently shown that PCSK9 levels in CSF are increased in AD versus non-AD subjects [15]. In the present study, we demonstrated that CSF PCSK9 concentrations are not specifically increased in AD since there was no significant difference when compared to patients with neurodegenerative processes (mainly FTLD). In addition, we showed for the first time that CSF PCSK9 concentrations are positively associated with neurobiomarkers of AD, such as Aβ1-42, T-tau, and P-tau. Finally, the variations of CSF PCSK9 levels were not associated with changes in B-CSF-B permeability markers.
Despite some apparent discrepancy, our results are in accordance with those previously published by Zimetti et al. [15]. Indeed, the majority of the patients in their control non-AD group have non-neurodegenerative diseases (mainly psychiatric disorders and hydrocephalus) [15]. Accordingly, in our study CSF PCSK9 levels in AD patients were also significantly increased when compared to non-AD patients without neurodegenerative disease (non-ND group). We also find a clear trend for an increase of CSF PCSK9 levels in other neurodegenerative patients (non-AD ND), suggesting that the PCSK9 CSF level is linked to the neurodegenerative process but not specific for AD. Since the CSF biomarkers are strictly normal in this non-AD ND group, we exclude the participation of an AD process to the cognitive decline and to the increase of PCSK9. Importantly, CSF PCSK9 levels are not correlated with biomarkers of B-CSF-B permeability. Thus, increased CSF PCSK9 level seems to be a marker of neurodegeneration, not explained by an unspecific aggression of the CNS. However, it should be underlined that our sample size is small and does not allow to detect some small statistically significant difference, if it exists, between the AD and non-AD groups. Further studies in larger cohorts are required to clarify this point.
Interestingly, a significant correlation between CSF PCSK9 and T-tau, a marker that reflects neurodegeneration, was observed in AD and neurodegenerative groups only [24]. Several authors have shown that high T-tau level predict a rapid progression in AD [25–27]. In a future study, it would be interesting to evaluate if higher PCSK9 level in CSF is associated with a worse cognitive evolution, arguing for a more aggressive neurodegenerative process.
A significant and positive correlation was observed between CSF PCSK9 and neurobiomarkers of AD: Aβ1-42 and P-tau in all the three groups. This finding may suggest that PCSK9 may interfere with Aβ production, as suggested by one previous study in mouse [13]. However, a Japanese study found no link between genetic Pcsk9 polymorphisms and AD risk [30]. Finally, the results of the prospective randomized controlled trial EBBINGHAUS with evolocumab, a PCSK9 inhibitor, did not find any changes in neurocognitive tests assessed using the Cambridge Neuropsychological Test Automated Battery toll and in patient-reported changes in memory and executive function, compared to placebo after a short median follow-up of 2.2 years [31], even in patients achieving a very low LDL-C levels (i.e., <0.5 mmol/L) [32]. It should be underlined, however, that PCSK9 monoclonal antibodies only block the extracellular form of PCSK9 in the circulation and do not alter intracellular PCSK9 in the CNS.
The mechanism by which PCSK9 could be involved in neurodegenerative processes is unknown. One can speculate on a proapoptotic effect of PCSK9 on neurons [12, 28], or alterations of lipid homeostasis in CNS, following a downregulation of LDLR, ApoER2, VLDLR, or LRP1 [11, 29], generating neuronal injuring. In addition, it has been recently demonstrated that brain endothelial-specific LRP1 deletion increases Aβ brain accumulation in mouse [33]. Beside AD pathogenesis, there are some pre-clinical data suggesting that PCSK9 overexpression stimulates the differentiation of cortical neurons [8]. Conversely, the silencing of PCSK9 expression in zebrafish eggs impaired the early CNS development, leading to an embryonic lethality between 2 to 4 days after fertilization [34]. However, PCSK9 KO mice did not show any signs of altered CNS development, suggesting a species-specific dependent action of PCSK9 on neurogenesis [10]. In accordance with a potential role for PCSK9 in CNS development, it has been shown recently that reduced PCSK9 plasma concentrations could serve as a biomarker for neural tube defects (spina bifida) in human [35]. These observations during CNS development could let us hypothesize an implication of PCSK9 in neuronal plasticity also later in life, after apparition of a neurodegenerative process, explaining its elevation in these conditions. However, a link between PCSK9 and neuroplasticity has never been demonstrated: although an elevation of PCSK9 in brain after ischemic stroke was found in mice, no elevation of markers of neurogenesis was associated, and the mice’s behavior as well as the extend of lesions were the same in PCSK9 KO and wild-type mice [9].
Our study has several limitations that should be underlined. Due to the retrospective design and the small sample, we cannot exclude that we have missed some correlations due to selection bias or lack of statistical power. We have only collected CSF samples and we were unable to perform some correlation with plasma PCSK9 and neurodegenerative diseases, although CSF PCSK9 concentrations are not correlated with plasma PCSK9 levels in a previous study performed in healthy volunteers [36]. The observational design of the study allows us to draw some associations but not causation, regarding PCSK9 role in neurodegenerative diseases.
In conclusion, we demonstrated that CSF PCSK9 levels are increased in neurodegenerative diseases rather than specifically in AD and are positively correlated with neurobiomarkers of AD. Additional studies are warranted to unravel the exact role of PCSK9 in neurodegenerative processes.