
Abstract
Alzheimer’s disease (AD) is characterized by accumulations of amyloid-β (Aβ42) and hyperphosphorylated tau proteins, associated with neuroinflammation, synaptic loss, and neuronal death. Several studies indicate that c-Jun N-terminal kinase (JNK) is implicated in the pathological features of AD. We have investigated in 5XFAD mice, the therapeutic effects of Brimapitide, a JNK-specific inhibitory peptide previously tested with higher concentrations in another AD model (TgCRND8). Three-month-old 5XFAD and wild-type littermate mice were treated by intravenous injections of low doses (10 mg/kg) of Brimapitide every 3 weeks, for 3 or 6 months (n = 6–9 per group). Cognitive deficits and brain lesions were assessed using Y-maze, fear-conditioning test, and histological and biochemical methods. Chronic treatment of Brimapitide for 3 months resulted in a reduction of Aβ plaque burden in the cortex of 5XFAD treated mice. After 6 months of treatment, cognitive deficits were reduced but also a significant reduction of cell death markers and the pro-inflammatory IL-1β cytokine in treated mice were detected. The Aβ plaque burden was not anymore modified by the 6 months of treatment. In addition to modulating cognition and amyloid plaque accumulation, depending on the treatment duration, Brimapitide seems experimentally to reduce neuronal stress in 5XFAD mice.
INTRODUCTION
Alzheimer’s disease (AD) is characterized by accumulations of amyloid plaques, made of amyloid-β peptide (Aβ42), accumulations of hyperphosphorylated tau proteins, associated with neuroinflammation, synaptic loss and neuronal death [1]. Aβ42 is produced through the sequential processing of the amyloid-β protein precursor (AβPP) by BACE1 (beta-site APP cleaving enzyme 1) and γ-secretase. Previous attempts to target Aβ42 by immunotherapy have not yet proven successful. Indeed, in most studies so far, treated patients did not show any cognitive improvement and postmortem brain analyses revealed an increased neuronal death [2 –4]. There is a need to find additional pharmacological targets, such as c-Jun N-terminal kinase (JNK). JNK is a serine-threonine Mitogen Activated Protein Kinase (MAPK), originating from three genes, JNK1, JNK2, and JNK3. The first two protein isoforms are ubiquitously expressed, whereas JNK3 is mainly brain-specific [5]. It has been shown that activated JNK (pJNK) accumulates in AD brains [6, 7]. We have shown that JNK3 levels are increased in AD cerebrospinal fluid, and correlated with cognitive decline of patients [6]. JNK can induce enhanced Aβ42 production, through the phosphorylation of AβPP on threonine 668 (Thr668), leading to an exacerbated amyloidogenic processing [8], and via the increase of BACE1 mRNA expression through the PKR-eIF2α pathway [9, 10]. JNK3 knockout in 5XFAD mice induced a decrease of Aβ burden, an increased number of neuron survival, and a cognitive improvement [11]. Inhibition of JNK with SP600125, a reversible ATP-competitive inhibitor, in AD mice APPswe/PS1dE9, showed comparable results, with cognitive improvement, reduction of Aβ burden, tau hyperphosphorylation, and inflammation [12]. But the use of SP600125 in humans is limited by its weak specificity. Indeed, targets of SP600125 include the MAPK kinases MKK4 and MKK7, p70 ribosomal protein S6 kinase, AMPK (AMP-activated protein kinase), DYRK1A (Dual specificity tyrosine-phosphorylation-regulated kinase 1A), and CDK2 (Cyclin-dependent kinase 2) [13].
Brimapitide (or XG-102 or D-JNKI-1) is a cell-permeable JNK specific inhibitory peptide, already tested in clinical trials in other human diseases and in phase 1 [14]. We have demonstrated previously that Brimapitide is neuroprotective in vitro in Aβ42 neurotoxicity [15] and it has been tested in a mice ischemia model [16]. Brimapitide (22 mg/kg) was able to reduce phosphorylated AβPP in vitro [17], and can diminish cognitive impairment and Aβ burden in the AD mice TgCRND8 [18, 19]. But very little is known on the effect of Brimapitide on inflammation and apoptosis in AD transgenic mice. The goal of our study was to test the efficiency of Brimapitide with a lower dose (10 mg/kg) on AD neuropathology including inflammation and apoptosis in the more severe 5XFAD model with clear neuronal degeneration. 5XFAD mice exhibit Aβ42 deposition and plaque development starting from 1.5 months, earlier than TgCRND8 mice (3 months), an important cognitive impairment at 5-months (between 3 to 7 months for TgCRND8), and a more pronounced neuronal death starting from 6 months [20]. Mice were treated beginning at 3 months of age for 3 or 6 months with intravenous injections, not IP injections as previously carried out. In this severe model, our results confirm, in Brimapitide-exposed mice for 6 months, the reduction of cognitive decline, but also revealed a partial reduction of apoptotic and inflammatory markers, respectively caspase 3 and Il-1β, in treated animals. A decrease of the Aβ plaque burden, not Aβ42 production, was observed in 5XFAD mice treated for 3 months with Brimapitide. This effect was lost after 6 months of treatment.
MATERIAL AND METHODS
Mice
All experimental protocols used in this study were approved by the Animal Experiments Committees of the Transgenesis, Archiving and Animal Models (license D-45-234-6) and the Brain and Spine Institute (license A75-13-19) and were performed in accordance to the guidelines of the French Agriculture and Forestry Ministry for handling animals (decree 87849, approval number #5240.02). 5XFAD mice (Jackson Laboratory, ME, USA) and wild-type (WT) littermates, on a C57BL/6 genetic background were used. 5XFAD mice overexpress the K670N/M671L, I716V, and V717I mutations in human AβPP, and the M146L, and L286V mutations in human PS1.
Three-month-old male mice were used, n = 6–9 mice per group. Each mouse in the treatment group received Brimapitide peptide (Xigen SA, Switzerland) dissolved in saline (NaCl 0.9%, B. Braun) every 3 weeks at a dose of 10 mg/kg by intravenous injection in the caudal vein, for 3 or 6 months. Brimapitide is not an ATP-competitive inhibitor, such as SP600125, but a peptide inhibitor specifically targeting JNK binding domain (JBD). Brimapitide then prevents JNK from accessing its targets, such as c-Jun. All mice were sacrificed 3 weeks after the last injection. IC50 values for Brimapitide are respectively 0.76, 1, and 26μ M for JNK2, JNK3, and JNK1. Each control mouse received NaCl 0.9% (B. Braun, Germany) at the same frequency. The use of saline as control has been decided to exactly reproduce a future phase I study in humans in which the placebo group will receive a neutral placebo and not a tat-based control peptide. This is a preclinical study aimed at determining if at the dose of 10 mg/kg XG-102 can modulate brain lesions and cognitive decline in 5XFAD mice.
Behavioral tests
The spontaneous alternation Y-maze test was used to measure spatial and procedural working memory. The Y-maze has 3 equal arms of 27 cm length, 7 cm width, and 20 cm height. During 8 min testing, the spontaneous alternation between the three arms was measured. Percentage alternation corresponded to: number of triad entries/(total number of arms entered -2)×100.
The contextual fear conditioning test was used to evaluate associative memory. During training, mice were placed in the conditioning chamber for 2 min and then received, without prior signals, two foot shocks (0.7 mA, 2 s) at 1 min interval (Ugo Basile Shocker). Mice then stayed in the chamber for another minute. Fear conditioning was evaluated by scoring the number of freezing episodes for 3 min when the mice were placed back into the same conditioning chamber 24 h after training.
Brain tissue preparation
Mice were sacrificed following protocol previously described [21]. Briefly, mice were deeply anesthetized with a lethal dose of pentobarbital and intracardially perfused with cold PBS. Hippocampus and cortex from one hemi-brain were frozen. The other hemi-brain was placed in 4% paraformaldehyde (Sigma Aldrich, MO, USA) before paraffin inclusion.
Immunoblot
Brain samples were homogenized in RIPA buffer (Sigma Aldrich) containing 0.1μ M calyculin A, 1 mM Na3VO4 and a protease inhibitor cocktail (Sigma Aldrich), and then centrifuged at 15000 g for 10 min. Protein concentration on the whole cell supernatant was determined with Micro BCA Protein Assay Reagent Kit (Thermo scientific, Cergy-Pontoise, France) using manufacturer’s protocol. Immunoblots were performed following protocols previously described [21]. Briefly, protein samples were separated on Tris-glycine polyacrylamide gels (Bio-Rad, Nanterre, France) and transferred onto nitrocellulose membranes (GE Healthcare, Chalfont St. Giles, UK). Primary antibodies were directed against pAβPP [Thr668] 1:800 (RRID: AB_10831197), cleaved caspase 3 1:500 (RRID: AB_331440), p-c-Jun [Ser63] 1:500 (RRID: AB_2130162) (Cell Signaling, MA, USA), sAβPPα (IBL, Germany), ADAM-10 1:400 (RRID: AB_11215461), AβPP 1:1000 (RRID: AB_10694227), pJNK 1:400 (RRID: AB_310412), and GAPDH 1:5000 (RRID: AB_11211911) (Millipore, MA, USA), JNK full 1:500 (RRID: AB_675864), c-Jun 1:200 (RRID: AB_631263), and pBcl-2 [Ser87] 1:50 (Santa Cruz Biotechnology, TX, USA). IR Dyes 800 and 700 1:5000 (Azure Biosystems Inc., CA, USA) antibodies were used as secondary antibodies. All antibodies used are commercially available and validated by the companies that provide them. Protein bands were revealed using the Azure Biosystem (Azure Biosystems Inc.), quantified with the Multigauge software in arbitrary units (Fuji-film, Tokyo, Japan), and normalized on GAPDH protein level. Data of WT littermate mice saline-treated for 3 months were considered as the control group. Original uncropped and unadjusted blots can be found in the Supplementary Material.
Immunohistochemistry
Paraffined hemi-brains were sagitally sectioned on a microtome apparatus at 5μ m. Sections were processed for immunohistochemistry following previously used protocols [6, 22]. Anti-Aβ42 1:1000 (MOAB-2, Millipore), anti-Iba1 1:1000 (RRID: AB_839504) and anti-NeuN 1:100 (Wako Chemicals, VA, USA) were used as primary antibodies, and biotinylated anti-rabbit and anti-mouse (Vector Laboratory, CA, USA) as secondary antibodies. All antibodies used are commercially available and validated by the companies that provide them. The stain was visualized using diaminobenzidine. Quantification of staining (% area stained of total area examined) was performed by ImageJ 1.48v software (National Institutes of Health, USA). Seven non-overlapping images of the cortex and one image of the subiculum, taken at magnification x40, were captured from each slide of tissue for quantification.
Protein concentrations, activity analyses, and ELISA assays
Caspase 3 and BACE1 activity were measured using their respective Activity Assay kits (Abcam, Cambridge, UK). Aβ42 concentration was assessed in duplicate in RIPA-brain homogenates using Human Aβ42 ELISA kit (Invitrogen, CA, USA). IL-1β, IL-6, IL-10, TNFα, and INFγ cytokines concentrations were evaluated in duplicate in RIPA-cortex homogenates using a proper mouse kit (Bio-Plex Pro Mouse Th17 Panel A 6-plex, Bio-Rad, CA, USA) by LUMINEX analysis.
Statistical analyses
Two-way ANOVA analyses were performed (GraphPad Prism). When ANOVA was significant, a post-hoc multiple comparison tests with a Tukey’s test was performed (GraphPad Prism). For all comparisons, p values of 0.05 or less were considered to be statistically significant.
RESULTS
Brimapitide decreased JNK and c-Jun activation levels in mice brain
To validate the on target-engagement of Brimapitide, we have analyzed JNK activation using the ration phosphorylated JNK (pJNK)/total JNK and its activity using the phosphorylation levels of its main target c-Jun. WT littermates and 5XFAD mice showed increased JNK activation levels at 9 months versus 6 months (Fig. 1B): +68% and +65%, respectively, in the cortex, and +87% and +48% in the hippocampus (p < 0.001). A 6-month treatment with Brimapitide decreased JNK activation in the cortex of WT littermates and 5XFAD mice (–60% and –40% respectively, p < 0.001) and in the hippocampus (–44% and –23% respectively, p < 0.001 and p < 0.01) (Fig. 1B). We also observed a 30% decrease of JNK activation in the hippocampus of 5XFAD mice treated for 3 months with Brimapitide (p < 0.05).
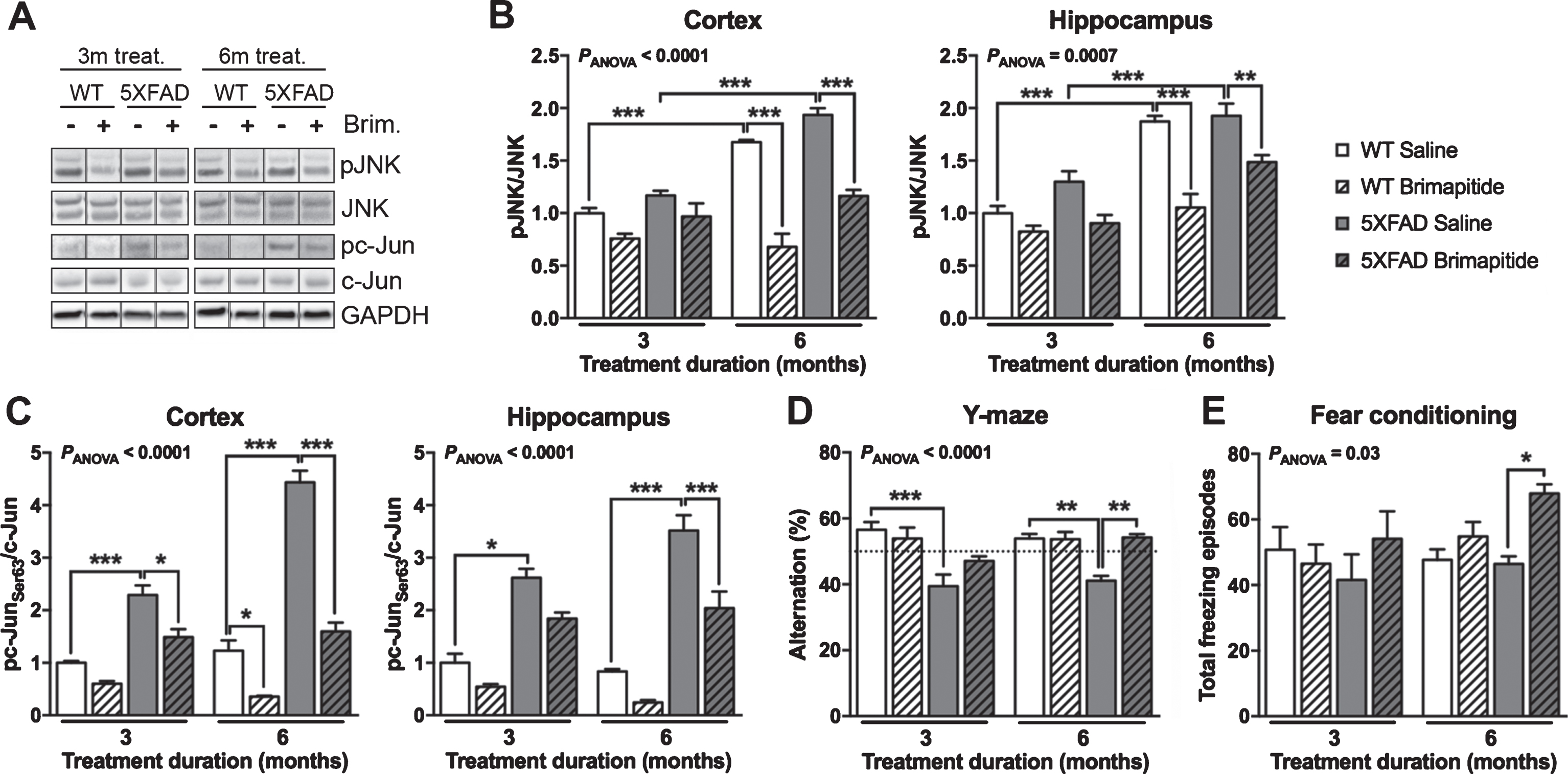
Cognitive abilities, JNK activation and activity in the brain of wild-type (WT) and 5XFAD mice. WT littermates and 5XFAD mice were IV treated every 3 weeks, for 3 or 6 months, with 10 mg/kg Brimapitide (Brim. +) or saline (Brim. -). A) Immunoblot analyzes of pJNK, JNK full, pc-Jun [Ser63], and c-Jun full in mice cortices. Each lane shows non-adjacent bands from the same blot. Respective histogram analyzes of JNK activation (B) and c-Jun activation (C) in the cortex and hippocampus of mice. D, E) Effects of Brimapitide treatment on memory in WT and 5XFAD mice. D) Y-maze: spatial and procedural working memory, percentage of alternation between the three arms. E) Fear conditioning: associative memory, number of freezing episodes. Data are means±SEM. Number of analyzed animals: 3–9. * p < 0.05, ** p < 0.01, and *** p < 0.001.
5XFAD mice showed a significant increase of brain c-Jun activation levels versus age-matched WT littermate mice, in the cortex (+129% and +260% at 6 and 9 months, respectively, p < 0.001), and in the hippocampus (+172% and +321% at 6 and 9 months, respectively, p < 0.001) (Fig. 1C). Brimapitide administration decreased c-Jun activation in the cortex, after a 3-month treatment in 5XFAD mice (–35%, p < 0.05) and a 6-month treatment in WT and 5XFAD mice (–71% and –64%, respectively, p < 0.05 and p < 0.001). In the hippocampus, the decrease was significant in 5XFAD mice treated for 6 months (–42%, p < 0.001, Fig. 1C).
Brimapitide reduced cognitive deficits in 5XFAD mice
It has been shown previously that 5XFAD mice exhibit a severe cognitive impairment starting from 6 months of age [20]. A significant decrease of the spatial and procedural working memory (Y-maze) of 6- and 9-month-old 5XFAD mice versus same-age WT littermate mice was noted in our study (–30 and –24%, respectively, p < 0.001 and p < 0.01) (Fig. 1D). 5XFAD mice treated for 6 months with Brimapitide had a 32% improvement of their working memory (p < 0.01). We did not find any difference in the average speed and the total distance travelled between the treatment groups and between genotypes. A significant improvement of the fear conditioning test (+46%, p < 0.05) was observed in 5XFAD mice treated with Brimapitide for 6 months (Fig. 1E). Brimapitide did not modify fear conditioning results in WT suggesting it is not a direct cognitive enhancer. Although no difference was noted in fear conditioning test between littermates and 5XFAD mice. Brimapitide only improved this test in 5XFAD mice.
Brimapitide reduced amyloid plaque load in 5XFAD mice
We next measured the Aβ load and metabolism in mice. In saline-treated 5XFAD mice, we observed a 69% increase of Aβ load in the cortex of 9-month-old versus 6-month-old mice (p < 0.001, Fig. 2A), and +29% augmentation of soluble Aβ42 concentration levels (ELISA, p < 0.001, Fig. 2B). Brimapitide induced a 30% significant decrease of Aβ plaque burden in the cortex of 5XFAD mice after 3 months of treatment (p < 0.05, Fig. 2A). No difference was detected after 6 months of Brimapitide treatment. Brimapitide seemed to have no effect on Aβ42 concentration level (Fig. 2B).
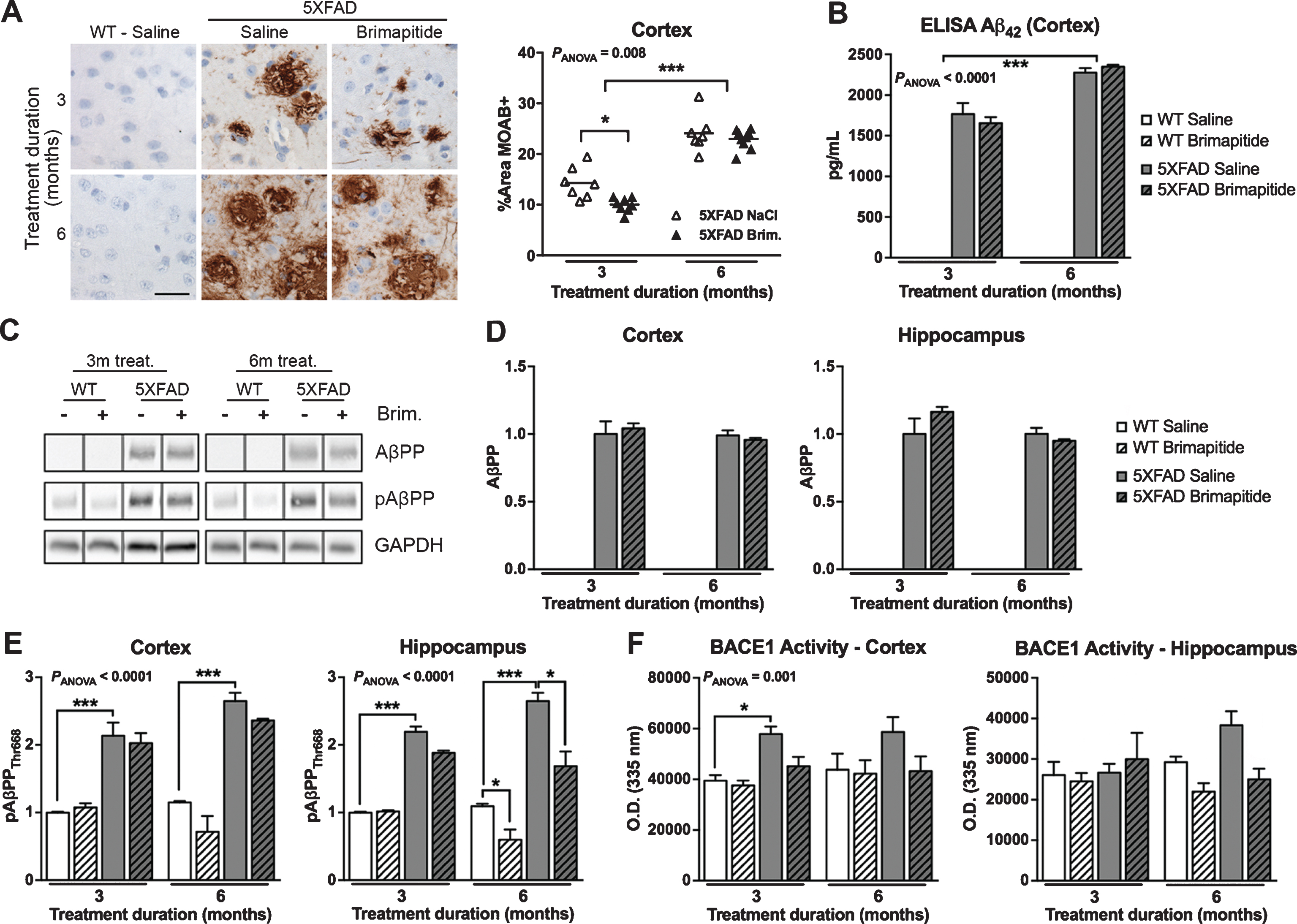
Amyloid burden in WT and 5XFAD mice. Mice were IV treated every 3 weeks, for 3 or 6 months, with 10 mg/kg Brimapitide or saline. A) Sagittal brain sections from 5XFAD mice treated with saline or Brimapitide, every 3 weeks, for 3 or 6 months incubated with an antibody against human Aβ42 clone MOAB, visualized by DAB staining, and micro photographed in the cortex. ImageJ quantification of Aβ42 labelling in the cortex (6≤n≥9). B) Histogram showing the concentration in pg/mL of soluble human Aβ42 peptide (ELISA method) in the cortex of WT littermates and 5XFAD mice treated with saline or Brimapitide. C) Immunoblot analysis of AβPP, and pAβPP [Thr668]. Each lane shows non-adjacent bands from the same blot. D, E) Corresponding histograms showing the level of human AβPP full protein (D), and the level of mice and human pAβPP [Thr668] (E) in the cortex and hippocampus of WT and 5XFAD mice treated with saline or Brimapitide. F) BACE1 activity analysis in mice cortex, and hippocampus. Data are means±SEM. Number of analyzed animals: 3–9. * p < 0.05, ** p < 0.01, and *** p < 0.001. Scale bar = 50μ m.
The protein level of human AβPP in 5XFAD mice was not modified in treated or non-treated 5XFAD mice (Fig. 2D). We found an increase of human and murine pAβPPThr668 in 5XFAD mice versus age-matched WT littermates in the cortex (+114% and +130% respectively, p < 0.001) and hippocampus (+120% and +142%, p < 0.001) (Fig. 2E). In the hippocampus of WT and 5XFAD mice treated for 6 months, Brimapitide induced a significant decrease of pAβPPThr668 level (–45% and –36%, respectively, p < 0.05).
A significant increase of BACE1 activity (Fig. 2F) was seen in the cortex of 6-month-old 5XFAD mice versus WT mice (+47%, p < 0.05) and Brimapitide did not significantly modify this activity.
Brimapitide reduced apoptotic markers in 5XFAD mice
Previous stereological analyses of 5XFAD brains stained with cresyl violet exhibited a clear cell death at 9 months of age in correlation with caspase 3 upregulation [20, 23]. Brimapitide efficacy was then assessed on several apoptotic markers. The mean level of cleaved caspase 3 (Fig. 3A, B) was increased in the brain of 6- and 9-month-old 5XFAD versus WT littermates: +75% and +40% in the cortex (p < 0.01 and p < 0.05). Brimapitide induced a 51% decrease of mean cleaved caspase 3 level in the cortex of 5XFAD mice treated for 6 months (p < 0.001). The activity of caspase 3 (Fig. 3C) was increased in the brain of 9-month-old 5XFAD versus same-age WT mice: +59% in the cortex (p < 0.001) and +207% the hippocampus (p < 0.001). The treatment with Brimapitide after 6 months decreased caspase 3 activity in 5XFAD mice: –38% in the cortex (p < 0.01) and –61% and hippocampus (p < 0.001).
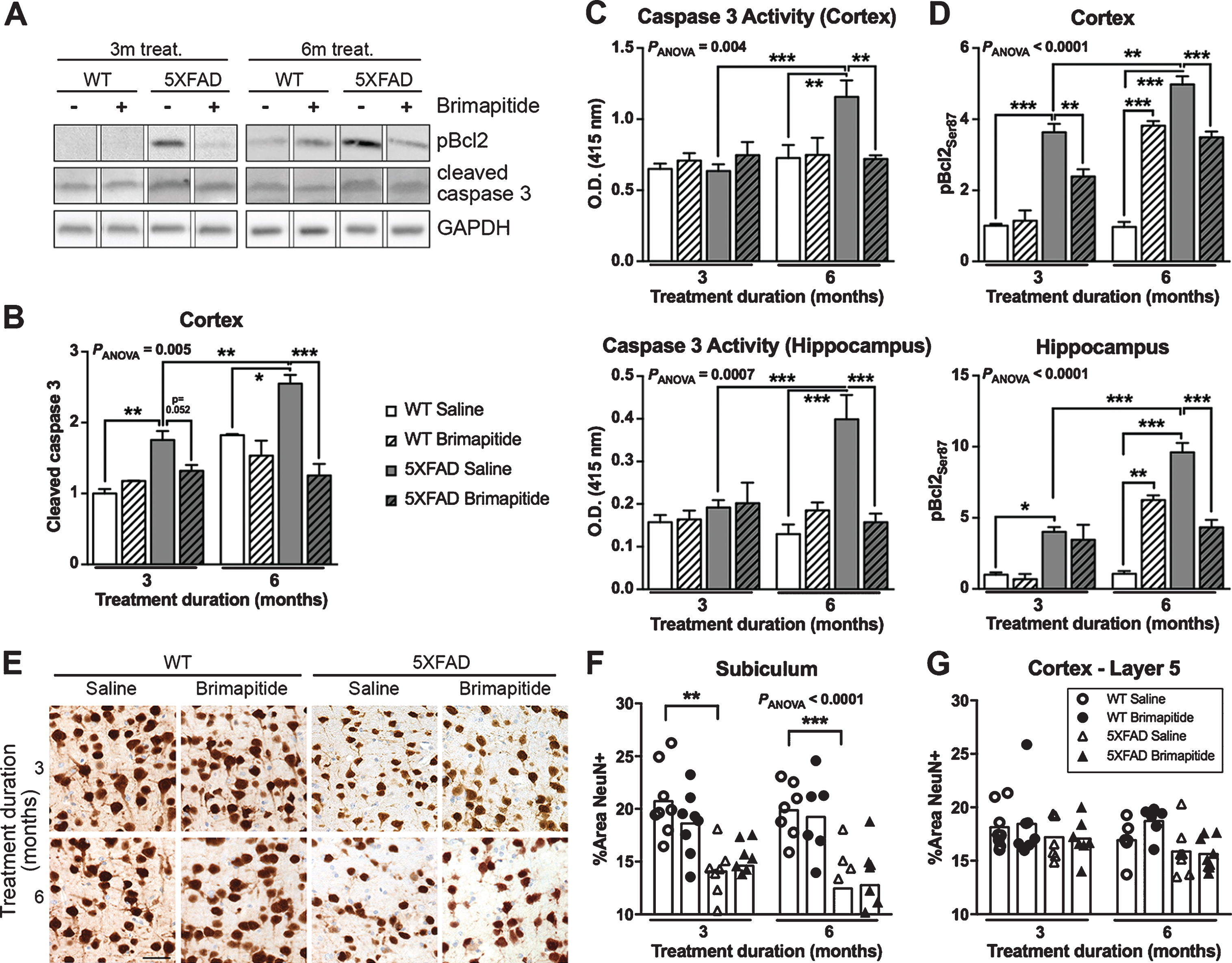
Cell death in WT and 5XFAD mice. Mice were IV treated every 3 weeks, for 3 or 6 months, with 10 mg/kg Brimapitide or saline. A) Immunoblot analyzes of cleaved caspase 3 and pBcl2 [Ser87] in mice cortex. Each lane shows non-adjacent bands from the same blot. B-D) Histogram of the immunoblot level of cleaved caspase 3 in the cortex (B), the level of caspase 3 activity (C), and the immunoblot level of pBcl2 [Ser87] in the cortex and hippocampus of mice (D). E) Sagittal brain sections from WT littermates and 5XFAD mice, treated with saline or Brimapitide, incubated with an antibody against NeuN, visualized by DAB staining, and micro photographed in the subiculum. F, G) ImageJ quantification of NeuN positive neurons in subiculum (F) and Layer 5 cortex (G) of WT and 5XFAD mice. Data are means±SEM. Number of analyzed animals: 3–9. * p < 0.05, ** p < 0.01, and *** p < 0.001. Scale bar = 50μ m.
The level of pBcl2Ser87 (Fig. 3D) was increased in the brain of 6- and 9-month-old 5XFAD versus age-matched WT littermate mice: +263% and +415% in the cortex (p < 0.001), and +302% and +807% in the hippocampus (p < 0.05 and p < 0.001). Brimapitide decreased pBcl2Ser87 level in the cortex and hippocampus of 5XFAD mice treated for 6 months (–30% and –55% respectively, p < 0.001) and in the cortex of 5XFAD mice treated for 3 months (–34%, p < 0.01). For an unknown reason an increase of pBcl2Ser87 level in WT mice treated for 6 months with Brimapitide was found: +294% in the cortex (p < 0.001) and +490% in the hippocampus (p < 0.01) whereas caspase 3 levels were not modified.
The number of neurons stained with NeuN (Fig. 3E-G) was significantly decreased in the subiculum of 5XFAD mice versus age-matched WT mice (–29% for both 6- and 9-month-old mice, p < 0.01 and p < 0.001). We did not find any effect of Brimapitide treatment.
Brimapitide reduced Il-1β expression level
Reactive gliosis begins at 2 months of age in 5XFAD mice [20]. We analyzed Brimapitide effect on microglia and cytokine expressions. The load of microglia (Fig. 4A, B) was increased in the brain of 6- and 9 month-old 5XFAD versus age-matched littermate WT mice: +49% and +145% in the cortex (p < 0.01 and p < 0.001). We found a 61% decrease of microglial load in the cortex of WT mice treated with Brimapitide for 3 months (p < 0.001) but Brimapitide did not significantly alter microglial load in 5XFAD mice although a tendency was observed after 3 months of treatment. Using LUMINEX method, a 31% decrease of IL-1β expression level (Fig. 4C) was detected in the cortex of 5XFAD mice treated by Brimapitide for 6 months (p < 0.05). No modification of cytokines levels including Il-6, IL10, TNFα, IFNγ were noted between treated and non-treated mice.
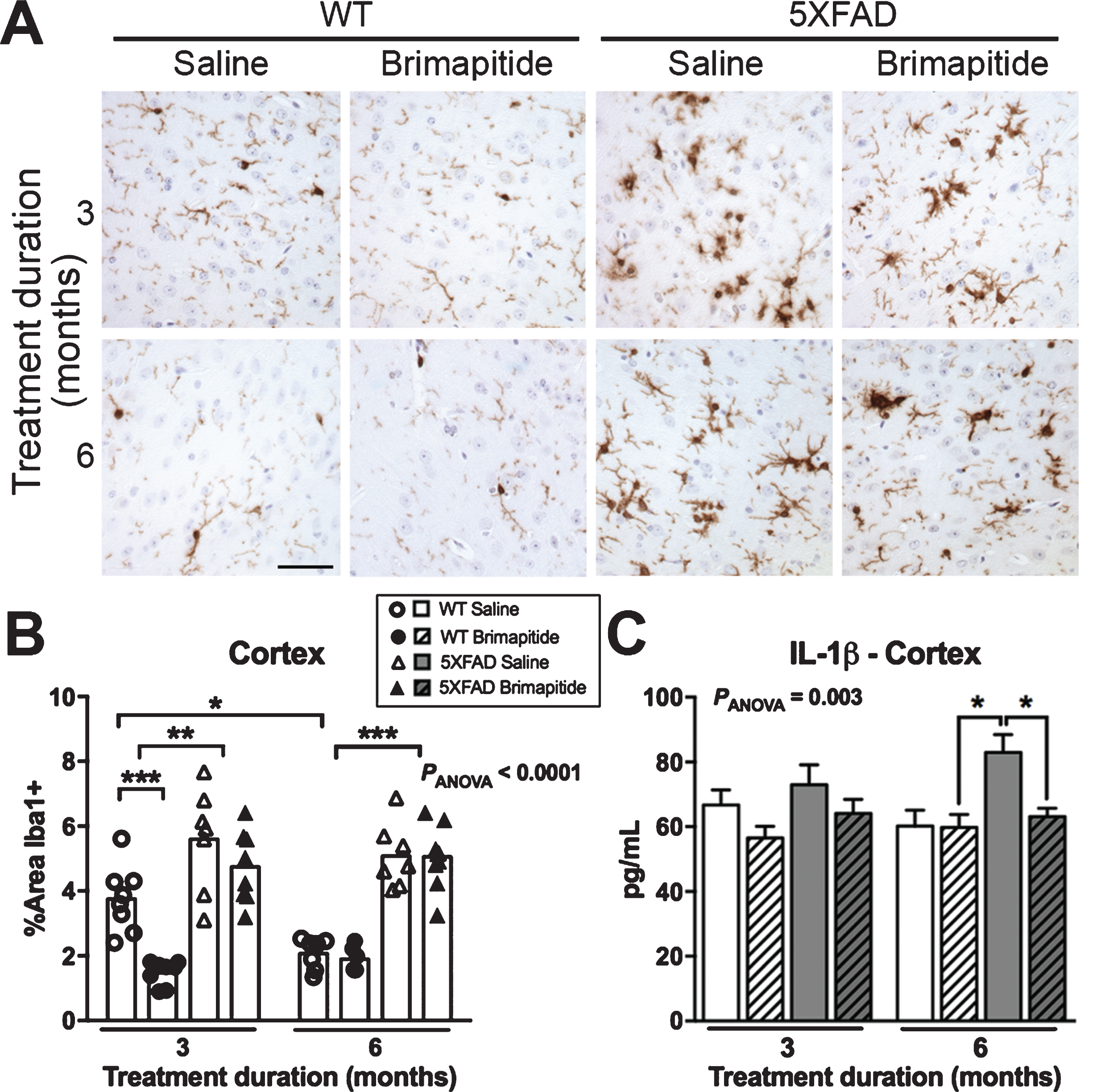
Inflammation in WT and 5XFAD mice. Mice were IV treated every 3 weeks, for 3 or 6 months, with 10 mg/kg Brimapitide or saline. A) Sagittal brain sections from WT littermates and 5XFAD mice incubated with an antibody against Iba1, visualized by DAB staining and micrographed in the cortex. B) ImageJ quantification of Iba1 labelling in the cortex of WT and 5XFAD mice. C) LUMINEX analysis: Level of IL-1β cytokine in pg/mL, in the cortex (RIPA homogenates) of WT and 5XFAD mice. Data are means±SEM. Number of analyzed animals: 6–9. * p < 0.05, ** p < 0.01, and *** p < 0.001. Scale bar = 50μ m.
DISCUSSION
In addition to confirm that Brimapitide, depending on the treatment duration, can efficiently reduce brain JNK activation, improve cognition and modulates brain Aβ plaque load in AD transgenic animals [11, 19] our report demonstrates for the first time in 5XFAD mice, a model marked by clear-cut neuronal degeneration, Brimapitide at 10 mg/kg can attenuate cellular apoptotic markers, and decrease brain IL1-β levels after 6 months of treatment.
We have found a positive effect on Aβ plaque burden, but not Aβ42 concentration, after a 3-month Brimapitide treatment in 5XFAD mice. Our result is comparable to the decrease of Aβ42 peptides and plaques seen in aged-matched FAD:JNK3+/+mice [11]. After 6 months of Brimapitide treatment, no significant change was seen and it is known that 6-month-old 5XFAD mice have almost reached the plateau of Aβ42 level [24]. This could also explain why we found no difference in AβPP production or phosphorylation with age. In 5XFAD mice, pAβPP levels were augmented compared to age-matched WT mice. Thr668 phosphorylation site leads to an increase of the amyloidogenic processing by BACE1. These results can be compared with the data obtained in the AD mouse model APPswe/PS1dE9 heterozygous for JNK1 (APPswe/PS1dE9/jnk1+ /–) [25]. In these mice they observed a decrease of BACE1 expression, without changes in Aβ burden. General inhibition of JNK with Brimapitide seemed to be more efficient. At 3 and 6 months of treatment Brimapitide induced a tendency to decrease BACE1 activity, which could partly be explained by the modifications of AβPP phosphorylation. We confirmed in vivo the effect of JNK on pAβPP already demonstrated in cell cultures [17].
Our results showed that JNK inhibition induced by Brimapitide partially prevents the working and associative memory declines detected in 5XFAD mice after 6 months of treatment. A comparable finding has been previously reported in TgCRND8 mice but with higher doses at 22 mg/kg [19]. As we did not observe any effect after a 6-month treatment with Brimapitide on Aβ burden, the cognitive action of Brimapitide could be explained by the reduction of cell stresses including apoptosis and neuroinflammation. Although, only minimal effects were seen, as NeuN labeling was unchanged, and IL-1β was the only cytokine being modified after Brimapitide treatment. In addition, Brimapitide has previously been shown to have an effect on Aβ42 oligomers formation [19] and a reduction in oligomers levels could also account for the improvement in cognitive ability in the absence of effects on plaque load. It could also be due to a possible direct action of JNK inhibition on memory mechanisms including synaptic plasticity in 5XFAD mice [26]. These assumptions remain out of the scope of this study, and merit further analysis.
We demonstrated that Brimapitide treatment for 6 months at 10 mg/kg decreased brain JNK and c-Jun activation levels. We found an age-related increase of activated JNK in controls and 5XFAD mice. This finding has been already observed in the brain of normal animals [27] and in the AD mice model Tg2576/PS1 [28] and could be explained in AD mice by amyloid deposition. Our findings revealed that c-Jun activation level (JNK activity) was only increased in 5XFAD mice compared to WT mice and a small effect on this kinase could have multiplied actions on its target activity [29]. We could only measure a significant decrease of JNK phosphorylation after 6 months of Brimapitide treatment in WT and 5XFAD mice but this could be due to our time point for brain analysis. Indeed, we sacrificed the mice 3 weeks after the last injection, at the time mice were normally re-injected with the peptide. JNK may be very quickly re-phosphorylated before the mouse sacrifice. This would explain why we were still able to measure c-Jun inhibition after 3 and 6 months of treatment (only statistically significant after 6 months for WT mice), as JNK, just re-activated could not yet act significantly on c-Jun activity. Target engagement of Brimapitide on JNK activation and kinase activity has been previously demonstrated in vitro and in vivo [15, 16].
One of the main findings of our study is that Brimapitide can decrease neuronal apoptotic stress in 5XFAD mice using a reduced concentration as compared to previous studies. As expected [20, 23], we observed an increase of cell loss and caspase-3 apoptotic pathway in 5XFAD mice. The levels of activated form and activity of caspase 3, and the levels of pBcl2 [Ser87] were enhanced in 5XFAD mice, especially in 9-month-old mice. It is known that JNK induces the phosphorylation of Bcl2 on Ser87 decreasing its anti-apoptotic role [30] and our data shows that Brimapitide (10 mg/kg) was efficient to reduce caspase 3 protein and activity levels as well as pBcl2Ser87 concentrations after 6 months of treatment. In WT mice treated for 6 months with Brimapitide, a decrease of the anti-apoptotic form of Bcl2 was detected that remains to be investigated. This could be due to an adverse reaction of JNK inhibition in a healthy brain, and could be an issue for clinical trials. After 6 months of treatment, the inhibition of JNK in WT mice could lead to the phosphorylation of Bcl2 on Ser87, via ERK, another MAPK able to phosphorylate Bcl2 [31]. Unfortunately, we did not observe any expected increase in NeuN labeling load following Brimapitide treatment in 5XFAD mice. Brimapitide treatment seems to reduce apoptotic pathways but not the loss of NeuN positive cells. This result may be linked to the fact that apoptosis could be also non neuronal or also to the possibility that NeuN negative neurons may be more sensitive to Brimapitide-induced neuroprotection. Future Fluorojade studies will be conducted. This histological staining labels degenerating neurons, and may be more suitable for our study.
It has been shown before that JNK interferes with inflammation [5] and our data demonstrated a reduction of brain IL-1β levels induced by Brimapitide treatment. Earlier reports have described the mRNA upregulation of inflammatory markers in 6-month-old 5XFAD mice [32], as well as increased protein levels in 4-month-old mice [33]. Microglia load is augmented in 5XFAD mice compared to same-age WT littermates as seen in a previous study [20] but Brimapitide has not effect on this marker after 6 months of treatment.
This study which shows new properties of Brimapitide on brain neuronal stress markers has some limitations. Only one dose of Brimapitide was tested in animals and it is necessary to plan lower concentrations in order to determine the appropriate minimum efficacious doses to envisage possible human studies. Although apoptotic markers were reduced by Brimapitide, no modification of cell loss was noted in treated animals with the method employed. Stereological analysis and Fluorojade staining for example would be needed in further studies. The reasons for the counter effect of long-term Brimapitide treatment on Bcl2 anti-apoptotic function in WT mice remains unknown and needs to be further analyzed in the objective of testing the efficacy of this peptide in human clinical trials.
In conclusion, our report adds to new effects of Brimapitide in a severe model of AD. So far, Brimapitide is efficacious in two AD transgenic models to modulate the cognitive decline and seems to induce neuroprotection and was shown to be quite safe in a human phase 1A study [14]. The next phase will be to determine experimentally a dose ranging pharmacological study necessary to implement a neuroprotection approach with Brimapitide.
Footnotes
ACKNOWLEDGMENTS
The authors thank Patrice Castagnet, Jean-Marc Combette, Catherine Deloche, Pascal Millet and Julien Dumurgier for technical help and valuable comments on the manuscript. Brimapitide was provided by Xigen S.A. Data are available on share repository (DOI: 10.6084/m9.figshare.5047054). This work was supported by Inserm and Xigen S.A.