
Abstract
Kainic acid (KA) was recently identified as an epileptogenic and neuroexcitotoxic agent that is responsible for inducing learning and memory deficits in various neurodegenerative diseases, such as Alzheimer’s disease (AD). However, the mechanism by which KA acts upon AD remains unclear. To this end, we presently investigated the roles of KA in processing amyloid-β protein precursor (AβPP) and amyloid-β protein (Aβ) loads during the course of AD development and progression. Specifically, KA treatment clearly caused the upregulation of tumor necrosis factor α (TNF-α) via activation of the PI3-K/AKT, ERK1/2, and p65 pathways in glial cells. TNF-α secreted from glial cells was then found to be responsible for stimulating the expression of BACE-1 and PS1/2, which resulted in the production and deposition of Aβ in neurons. Finally, the accumulation and aggregation of Aβ lead to the cognitive decline of APP23 mice. These results indicate that KA accelerates the progression of AD by inducing the crosstalk between glial cells and neurons.
INTRODUCTION
Kainic acid (KA), a glutamate-related chemical, was recently identified as being responsible for inducing learning and memory deficits in various neurodegenerative diseases [1]. After KA administration, rodents were shown to exhibit behavioral changes in their performance in a water maze, object exploration tasks, and the open-field test in association with selective damage to the hippocampus. Deficiencies in short-term and long-term spatial learning were also observed [2]. Apart from these syndromes, KA administration has been shown to result in selective neurodegeneration in rodents. For instance, focal injection of KA resulted in hippocampal damage that occurred primarily in the CA3 region [3]. Similarly, systemic injection of KA was shown to produce extensive neuronal death, primarily within the hippocampus hilus, CA3, and CA1 regions [4]. Although the reasons for neuronal death are varied between animal species and strains [5, 6], most investigations have described the role of KA in cell death due to its high neurotoxicity [7].
Although KA has shown its effects on learning and memory deficits in neurodegeneration, its specific role in Alzheimer’s disease (AD) has been largely overlooked. Indeed, KA is involved in regulating inflammation, neuronal excitability and neuronal cell death, which results in neurodegeneration [8–10]. Among these mechanisms, inflammation has been deemed a major factor in brain aging and age-related neurodegenerative diseases, such as AD [11]. In this regard, over the past two decades, several inflammatory mediators, such as TNF-α, IL-1β, and IL-6, that are produced by multiple endogenous sources including microglia, astrocytes, and brain endothelial cells, have been found to be upregulated in the AD brain [12, 13]. Long-term exposure to these inflammatory cytokines leads to cognitive impairment during the course of AD development [14]. Among these pro-inflammatory cytokines, TNF-α expression was reported to be enhanced in KA-stimulated microglial cells [15], which indicates this cytokine’s potential contribution to the mediating effects of KA on the pathogenesis of AD.
In light of the above observations, the current study investigated whether TNF-α has the ability to mediate the effects of KA on the abnormal cleavage of amyloid-β protein precursor (AβPP) and amyloid-β (Aβ) deposition that result in the cognitive decline of APP23 mice. Consequently, injection or intranasal administration of KA was found to increase the expression of tumor necrosis factor α (TNF-α) via activation of the PI3-K/AKT, ERK1/2, and p65 pathways in glial cells. TNF-α production was found to stimulate the expression of BACE-1 and PS1/2, which result in the production and deposition of Aβ in neurons. More importantly, the accumulation and aggregation of Aβ lead to cognitive decline in APP23 mice. These results indicate that KA accelerates the progression of AD by inducing the crosstalk between glial cells and neurons.
MATERIALS AND METHODS
Reagents
KA, TNF-α, and the inhibitors U0126, LY294002, and QNZ (4-N-[2-(4-Phenoxyphenyl)ethyl]-1,2-dihydroquinazoline-4,6-diamine) were obtained from Sigma-Aldrich Corp (St. Louis, MO, USA); Antibodies specific for β-actin, p-ERK1/2 (#4370), ERK1/2 (#4695), p-AKT (#4060), AKT (#9272), p-p65 (#3033), p65 (#8242), TNF-α (#3707), BACE-1 (#5606), PS1 (#5643), PS2 (#9979), Aβ (#14974) and HRP-labeled secondary antibody (#7074) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). DAPI was obtained from Beyotime Institute of Biotechnology (Haimen, JS, China). All reagents for the qRT-PCR and SDS-PAGE experiments were purchased from Bio-Rad Laboratories. All other reagents were from Invitrogen (Carlsbad, CA, USA) unless otherwise specified.
Cell culture and treatment
Mouse D1A and neuroblastoma (n)2a cells were grown (37°C and 5% CO2) on 6-cm tissue culture dishes (106 cells per dish) in appropriate media. The cells were treated with KA (5μM) or TNF-α (1 ng/ml) in either the absence or presence of inhibitors, including U0126 (10μM), LY294002 (10μM) or QNZ (1μM). After 24 h, the total RNA or protein were extracted by Trizol or lysis buffer.
Western blot
The tissues were homogenized in lysis buffer consisting of 15 mM HEPES (pH 7.4), 0.5 mM EDTA, 0.5 mM EGTA and the protease inhibitor cocktail. Protein content was determined by BCA kits (Thermo-Fisher Scientific, Rockford, IL, USA). The equal protein aliquots, assessed by immunoblot, were loaded onto SDS-PAGE. The gels were then transferred onto a polyvinylidene fluoride membrane. The membranes were probed with p-ERK1/2, ERK1/2, p-AKT, AKT, p-p65, p65, TNF-α, BACE-1, PS1, PS2 and HRP-labeled secondary antibody were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). β-actin was used as a loading control. All western hybridizations were performed at least in triplicate using a different cell preparation each time.
Real-time PCR
The Total RNA was extracted using Trizol (Invitrogen, Shenyang, China). The concentration of RNA was quantified by nanodrop devices (Thermo Fisher Scientific, Beijing, China). Complimentary DNA templates were prepared from 1 mg of the total RNA primed with oligo dT primers using reverse transcriptional kits according to the manufacturer’s instructions (Promega Corporation, Beijing, China). The cDNA was amplified using 35 cycles of real-time PCR assays with the iQ5 real-time PCR detection system (Bio-Rad) and appropriate primers. The primers used for the real-time PCR were as follows: for mouse TNF-α (NM_013693) F-atggcctccctctcatcagt, R-aaggtacaacccatcggctg; for BACE-1 (NM_011792) F-gaccactcgctatacacggg, R-gaggctgccttgatggactt; for PS1 (NM_008943) F-caagagctgctgtccaggaa, R-gctggcaacgctttcttgaa; for PS2 (NM_011183) F-atgtcctccctgatgctcct, R-atgaacaccagggccatgag; and for GAPDH (NM_001289726) F-ccacagtccatgccatcact, R-ctcagatgcctgcttcacca.
Immunohistochemistry (IHC)
The brains were perfusion-fixed with 4% paraformaldehyde solution and postfixed for 48 h, followed by cryoprotection in 30% sucrose for 72 h. The tissue slides were prepared in 10μm thicknesses by cryostats. For staining, the brain slides were incubated with TNF-α antibody overnight at 4°C and with secondary antibody for 1 h at room temperature. IHC images were obtained by Leica DM750 microsystems. Images were processed using Image J software [16].
Intracerebroventricular injection (i.c.v.)
WT or APP23 mice were subjected to cryoanesthesia until their activity slowed. A needle was inserted perpendicularly to the skull surface, two-fifths of the distance along the line between the eye and lambda, with a depth of 2 mm. After bilateral injection of the ventricles, the pups were rewarmed slowly by placing them on a warming pad until they were warm and active.
TNF-α and Aβ immunoassay
The samples were extracted from the brains of the C57/BL6 or APP23 mice according to the manufacturer’s instructions (PeproTech, Suzhou, China). The concentrations of TNF-α or Aβ were then determined by a specific commercial ELISA kit (PeproTech, Suzhou, China). The tests were carried out according to the instructions from the manufacturer. The total protein used for the ELISA was used as a loading control, and the results are expressed as pg of TNF-α or Aβ per mg of total protein.
Morris Maze test
The rats were subjected to water maze trials immediately after treatment with KA (n = 10–13/group). The bath was divided into four quadrants and a hidden escape platform (10 cm in diameter) was submerged in the center of one quadrant. The mice were trained to learn and find the hidden platform based on several cues external to the maze. Three trials were conducted on each day with 5 min intervals for the 6 consecutive days. The mean time spent to escape onto the platform was recorded. In addition, the test session consisted of a single probe trial where the platform was removed from the tank, and each mouse was allowed to swim for 60 s in the maze. Performance was monitored with an AnyMaze video-tracking system (Stoelting Co., Wood Dale, IL, EUA).
Nest construction
The mice were housed separately in cages before the test. Eight pieces of paper were put into the cages to create conditions for nesting. The nest scores were analyzed by the following standard according to a 4-point system [17]: 1, no biting/tearing, with random dispersion of the paper; 2, no biting/tearing of the paper, with gathering in a corner/side of the cage; 3, moderate biting/tearing of the paper, with gathering in a corner/side of the cage; and 4, extensive biting/tearing of the paper, with gathering in a corner/side of the cage.
Animals and treatment
C57BL/6 and APP23 mice were obtained from The Jackson Laboratory (Stock #030504). C57BL/6 and APP23 mice weighing ∼20 g and of 4 months of age at the start of the experiments were used in the current study. They were housed in a controlled environment (22–25°C, relative humidity, 12-h light/dark cycle with free access to food and water). Following at least a 2-week acclimatization period to the facility, the mice were weight-matched and then randomly assigned to one of the following three groups: WT, WT treated with KA, APP23 and APP23 treated with KA. For intranasal administration, APP23 mice at the age of 3-month-old was caught and administered with KA (5μg/20μl/d) by Hamilton syringe. All animals were observed daily for clinical signs of disease. All the animal procedures were approved by the Institutional Animal Care and Use Committee of Jilin University, which was in compliance with the Guidelines for the Care and Use of Laboratory Animals of the U.S. National Health Institute.
Statistical analysis
Data are presented as mean±S.E. The statistical significance between group comparisons for behavioral data was determined by one-way analysis of variance (ANOVA), followed by post hoc Tukey’s multiple comparison test.
RESULTS
KA stimulates expression of TNF-α in 3-month-old APP23 mice.
To assess the potential roles of KA in AD, the WT and APP23 mice were challenged with 3μg/5μl KA. Using intracerebroventricular injection (i.c.v), the expression of TNF-α was determined by immunohistochemistry (IHC) after 24 h. When compared with the vehicle-injected (i.c.v) controls, the positive staining was obviously enhanced (Fig. 1A). To further validate this observation, real-time PCR and ELISA were carried out. The results demonstrated that the mRNA and protein expression of TNF-α were increased compared to the vehicle-injected controls, which was set to 100% (Fig. 1B, C and Supplementary 1A). These observations clearly revealed the ability of KA to upregulate the expression of TNF-α in the brains of APP23 mice.
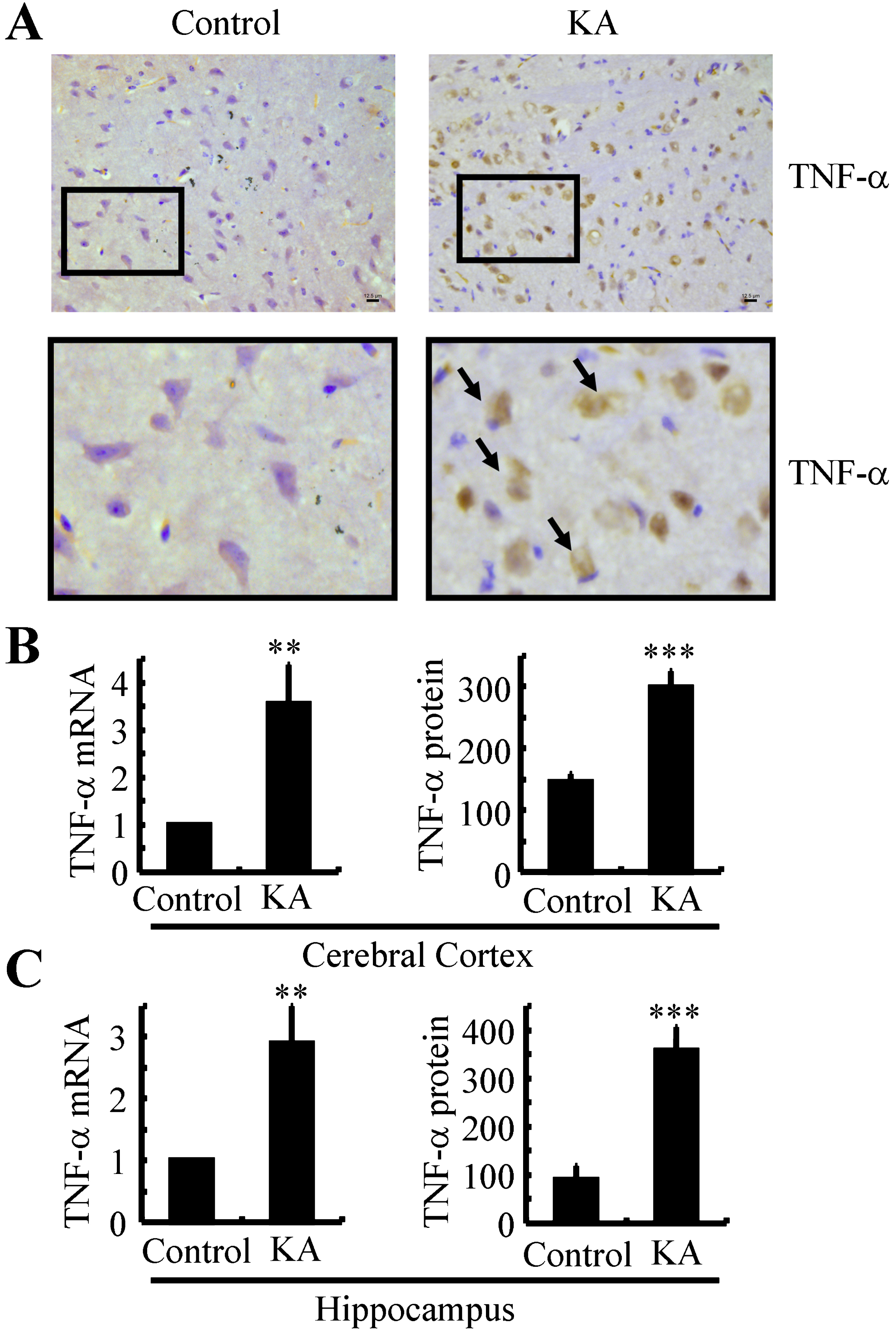
TNF-α was upregulated in KA-treated APP23 mice. A) APP23 mice were injected (i.c.v) at the age of 6 months with KA (3μg/5μl) for 24 h. The brains of the APP23 mice were collected and sectioned by cryostats. The slices were immunohistochemically stained with anti-TNF-α specific antibody. Arrowheads are pointing to the positive staining of TNF-α. Bar = 12.5μm. B, C) The mRNA and protein expression of TNF-α in the cerebral cortex and hippocampus of 6-month-old APP23 and C57BL/6 mice were determined by real-time PCR and ELISA, respectively. GAPDH and total protein served as internal controls. **p < 0.01, ***p < 0.001 with respect to the vehicle-treated controls.
KA activates the expression of TNF-α via the PI3-K/AKT and ERK1/2 pathways in glial cells
As TNF-α was reported to be expressed in glial cells [13], D1A cells were selected for the following experiments as the in vitro model of glial cells. In the present study, TNF-α was detected in the conditioned media and in the D1A cells after KA (5μM) insult for 24 h. Our results showed that the KA-treated D1A cells displayed higher expression and production of TNF-α compared with the untreated glial cells (Fig. 2A). These observations demonstrated the potential contributions of glial cells in TNF-α production in KA-injected APP23 mice.

KA stimulates the expression and production of TNF-α in a PI3-K- and ERK1/2-dependent mechanism in glial cells. A, C, D) D1A cells were incubated with KA (5μM) in either the absence or presence of a PI3-K inhibitor, LY294002 (10μM), or an ERK1/2 inhibitor, U0126 (10μM), for 24 h. The mRNA and protein were extracted before determining the expression of TNF-α (A, C) and the phosphorylation of AKT and ERK1/2 (D). GAPDH and β-actin served as internal controls. B) KA (3μg/5μl) was injected into APP23 mice over 24 h. The phosphorylation of AKT and ERK1/2 was measured by western blots. β-actin served as the internal control. *p < 0.05, **p < 0.01, ***p < 0.001 with respect to the vehicle-treated controls. # #p < 0.01, # # #p < 0.001 when compared to the KA-treated groups.
To better understand the mechanism behind the increased TNF-α in APP23 mice after KA treatment, we employed western blots to examine phospho-AKT and phospho-ERK1/2 in the brains within all the groups of mice (APP23 mice before and 24 h after KA insult). Twenty-four hours after the injection of KA (3μg/5μl) to the APP23 mice, the phosphorylation of AKT and ERK1/2 were significantly upregulated compared to that of the WT mice (Fig. 2B). To further verify the key roles of the PI3-K/AKT and ERK1/2 pathways in regulating the expression of TNF-α, D1A cells were treated with KA (5μM) in either the absence or presence of inhibitors, including a PI3-K/AKT inhibitor, LY294002 (10μM), and an ERK1/2 inhibitor, U0126 (10μM). The results revealed that preblockade with LY294002 or U0126 significantly attenuated the expression of TNF-α in the KA-treated D1A cells (Fig. 2 C). In addition, the efficacies of LY294002 and U0126 in facilitating the phosphorylation of AKT and ERK1/2, respectively, were confirmed (Fig. 2D). Based on these findings, the PI3-K/AKT and ERK1/2 pathways appear to play key roles in mediating the effects of KA on inducing the expression of TNF-α in glial cells.
TNF-α is responsible for increasing the expression of BACE-1, PS1/2 and the production of Aβ by activating NF-κB in neurons
Next, we addressed the effects of constitutive TNF-α on glial cell-evoked synaptic modulation. To this end, n2a cells were treated with TNF-α (1 ng/ml) for 24 h. After incubation with TNF-α in cultures, the neurons showed significantly higher BACE-1 and PS1/2 expression than did the neurons from the untreated n2a cells (Fig. 3A, B). Interestingly, TNF-α treatment stimulated the phosphorylation of the NF-κB p65 subunit in the n2a cells (Fig. 3C). To further verify the key roles of NF-κB in regulating the expression of BACE-1 and PS1/2, n2a cells were treated with TNF-α in either the absence or presence of inhibitors, including the NF-κB inhibitor, QNZ (1μM). The results revealed that preblockade with QNZ significantly attenuated the expression of BACE-1 and PS1/2 in TNF-α-treated n2a cells (Fig. 3D). Similarly, QNZ blocked the effects of TNF-α on the production of Aβ in n2a cells (Fig. 3E). Therefore, NF-κB plays a key role in mediating the effects of TNF-α on inducing the expression of BACE-1 and PS1/2, as well as on the production of Aβ in neurons.

TNF-α stimulates the expression of BACE-1 and PS1/2 via NF-κB pathways in neurons. N2a cells were treated with TNF-α (1 ng/ml) in either the absence or presence of an NF-κB inhibitor, QNZ (1μM), for 24 h before extracting the total mRNA and protein. The mRNA and protein expression of BACE-1 (A, D) and PS1/2 (B, D) were determined by real-time PCR and western blots. GAPDH and β-actin served as internal controls. C) The phosphorylation of the NF-κB, p65 subunit was determined by western blots, and β-actin served as the internal control. E) The production of Aβ was measured by immunoassay kits. The total protein was used as the internal control. In select experiments, n2a cells were treated with KA (5μM) for 24 h. The mRNA and protein expression of BACE-1 and PS1/2 were determined by real-time PCR and western blots. GAPDH and β-actin served as internal controls. *p < 0.05, **p < 0.01, ***p < 0.001 with respect to the vehicle-treated controls. #p < 0.05, # #p < 0.01 when compared to the TNF-α-treated groups.
As TNF-α is primarily expressed in glial cells [18]. The above results have mimicked the phenomenon that TNF-α originated from glial cells has ability to stimulate the expression of BACE-1 and PS1/2 in neurons. We will continue to elucidate whether KA has ability to induce the expression of BACE-1 and PS1/2 without the involvement of glial cells. To this end, we treated n2a cells with KA (5μM) for 24 h. By western blots and qRT-PCR, we found that KA treatment clearly induced the expression of BACE-1 and PS1/2 (Fig. 3F). However, we further found that KA treatment clearly induced the synthesis of TNF-α in n2a cells (Supplementary Figure 2). In view of this result, TNF-α is still an essential factor in mediating the effects of KA on inducing the expression of BACE-1 and PS1/2 without the involvement of glial cells. In another word, these observations demonstrate that KA stimulates the expression of BACE-1 and PS1/2 in a TNF-α-dependent manner.
KA stimulates the production and deposition of Aβ by inducing the expression of BACE-1 and PS1/2 in APP23 mice
As AD is pathologically characterized by the production and deposition of Aβ, we further determined the effects of KA on Aβ production in vivo. For animal models of AD, APP23 mice showed 5 folds increase in plaque-associated human Aβ1 - 42 peptides compared to that of younger ones [19]. In addition, the APP23 mice showed loss of spatial memory, which is coincided with the first appearance of hippocampus and cortex [19]. Thereby, 3-month-old APP23 mice were intranasally treated with KA (5μg/20μl/d) for 3 months before collecting the brains. Using real-time PCR and western blots, the mRNA and protein expression of TNF-α, BACE-1, and PS1/2 were measured. The data demonstrated that KA upregulated the expression of TNF-α, BACE-1 and PS1/2 in APP23 mice (Fig. 4A, B). Of note, the roles of KA in upregulating the expression of BACE-1 and PS1/2 are not limited to APP23 mice. We also found that intranasal treatment of WT mice with KA (5μg/20μl) for 24 h increased the expression of BACE-1 and PS1/2 in WT mice (Supplementary Figure 1). As Aβ is cleaved from AβPP by β- and γ-secretases, we further measured the production of Aβ in KA-treated APP23 mice. Using an ELISA assay, the production of Aβ was elevated in the KA-treated APP23 mice (Fig. 4C). In addition, we found that the formation of APs was clearly enhanced by the KA treatment (Fig. 4D). Thus, KA stimulates the production and deposition of Aβ by inducing the expression of BACE-1 and PS1/2 in APP23 mice.
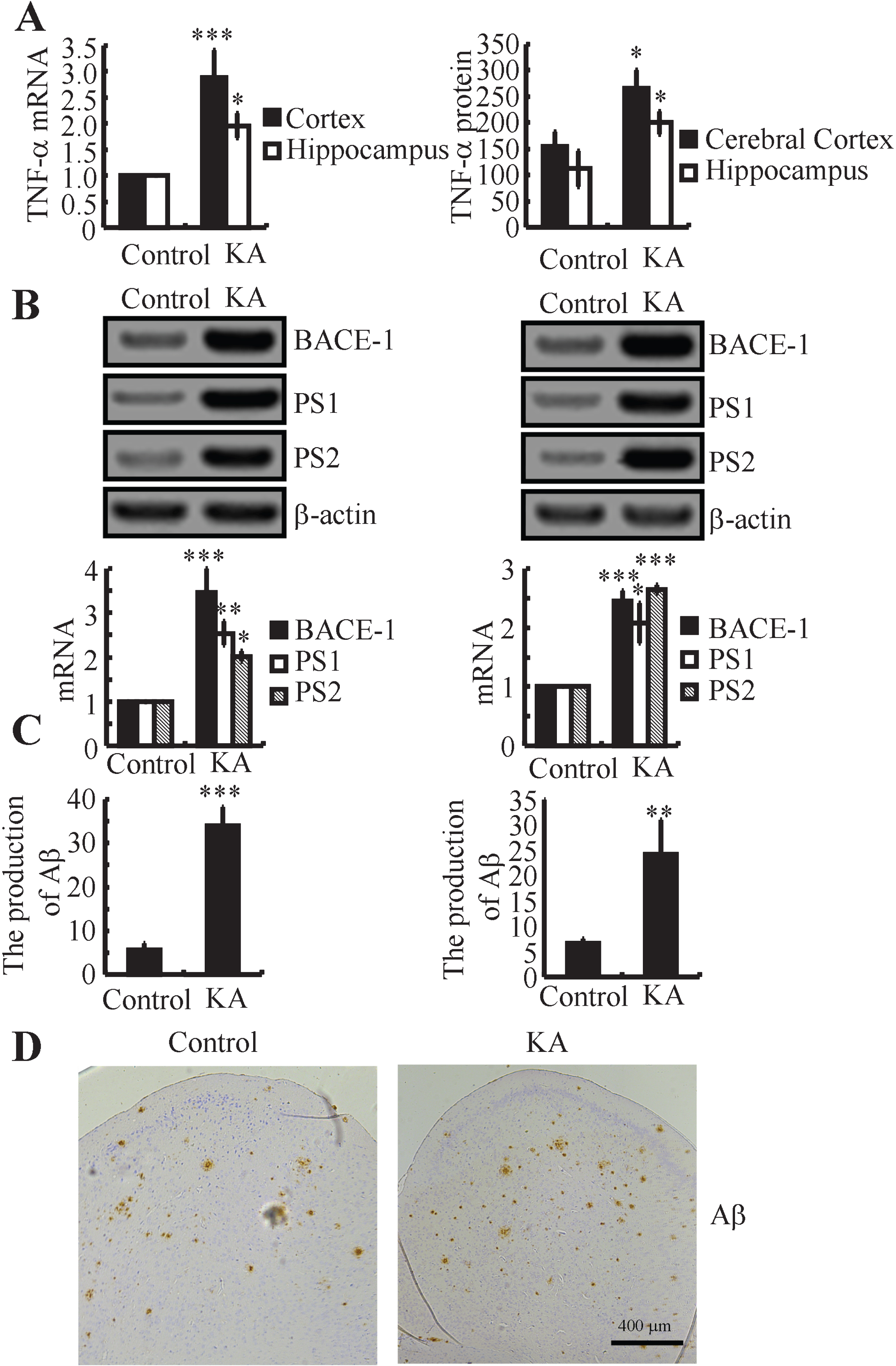
KA treatment induced the production and deposition of Aβ in a BACE-1- and PS1/2-dependent mechanism in APP23 mice. A) APP23 mice at the age of 3-month-old were intranasally treated with KA (5μg/20 g) for 24 h. The mRNA and protein expression of TNF-α was determined by real-time PCR and ELISA. GAPDH and total protein served as internal controls. B) APP23 mice at the age of 3-month-old were intranasally treated with KA (5μg/20 g/d) for 3 months. The mRNA and protein expression of BACE-1 and PS1/2 were measured by western blots. β-actin served as the internal control. C) The production of Aβ was measured by immunoassay kits. The total protein was used as the internal control. (D) The cerebral cortexes of the APP23 mice were determined by immunohistochemistry. *p < 0.05, **p < 0.01, ***p < 0.001 with respect to the vehicle-treated controls.
KA accelerates the cognitive decline of APP23 mice
Since KA stimulates the production of Aβ in APP23 mice, questions are easily raised as to whether KA affects the learning ability of APP23 mice. To determine the roles of KA in the learning ability of APP23 mice, the Morris water maze test was carried out. As expected, the KA-treated APP23 mice showed significantly impaired memory behavior compared with the control subjects (n = 10–13/group) (Fig. 5A-C). Specifically, KA treatment significantly increased the time and distance required for the APP23 mice to find the platform after training (Fig. 5A-C). Additionally, these results were also confirmed by probe tests 24 h after the last training trial. The KA-treated APP23 mice showed no preference toward the target quadrant, thus indicating significant memory impairment (Fig. 5D). Apart from the Morris water maze test, nest construction scores were measured to determine the loss of learning ability in the KA-treated APP23 mice. The results demonstrated that the KA treatment clearly decreased the learning ability of APP23 mice (Fig. 5E). In light of these observations, our findings demonstrated that KA treatment in the central nervous system could functionally impair memory behaviors of AD mice with a mechanism involving modulation of the accumulation of Aβ via TNF-α-dependent mechanisms.

KA impaired the learning ability of APP23 mice. APP23 mice at the age of 3 months were intranasally treated with KA (5μg/20μl/d) for 3 months. The learning abilities of the different groups of mice were determined by the Morris maze test or a nest construction assay (A-E). The number of mice used for the Morris maze tests or nest construction was 10–13 for each group. *p < 0.05 compared to the vehicle-treated mice.
DISCUSSION
The current findings clearly show that the systemic treatment with KA accelerated the cognitive decline of AD via inducing the production and deposition of Aβ via a TNF-α-dependent mechanism (Fig. 6). In detail, KA treatment stimulated the expression of TNF-α via the PI3-K/AKT and ERK1/2 pathways in glial cells. Reciprocally, TNF-α can exert its effects on neurons to increase the production and deposition of Aβ in a BACE-1- and PS1/2-dependent mechanism via activating the NF-κB p65 subunit. Aβ deposition will, finally, accelerate the cognitive decline of APP23 mice.

Proposed mechanisms of KA in accelerating the progression of AD. The intranasal administration of KA increased the expression of TNF-α via the PI3-K/AKT and ERK1/2 pathways in glial cells, which, in turn, stimulated the expression of BACE-1 and PS1/2 in an NF-κB-dependent mechanism in neurons. Highly induced BACE-1 and PS1/2 prompted the abnormal cleavage of AβPP, which lead to the production and deposition of Aβ in APP23 mice. The production and deposition of Aβ obviously impaired the learning ability of AD.
As a glutamate-related chemical, KA is involved in modulating neuroinflammation. Akiyama et al. [20] reported that KA injection (i.c.v) has the ability to activate microglial cells to gather around the injured pyramidal neurons. Consistent with this observation, Hoon et al. reported that KA treatment enhanced neuroinflammation by activating microglial cells [21]. Based on this mechanism, NF-κB seemed to be involved in neuroinflammation [22]. More closely, Oprica et al. reported that KA administration enhanced the expression of IL-1β in the brain [23]. In line with these observations, we extended the prior works to TNF-α regulation via KA treatment.
Apart from the above observations, we further found that the PI3-K/AKT and ERK1/2 pathways are involved in mediating the effects of KA on stimulating expression of TNF-α in APP23 mice. In agreement with our results, Henshall et al. reported that KA treatment induces the phosphorylation of AKT at the site of Ser473 in the CA3 region of C57BL/6 mice [24]. Apart from the PI3-K and AKT pathways, KA has the ability to activate ERK1/2 via phosphorylation in the rat hippocampus [25, 26]. In addition, wortmannin, a PI3-K specific inhibitor treatment, clearly decreased the protein expression of TNF-α in mast cells [27]. Apart from PI3-K, ERK1/2 also contributes to TNF-α production in rat Kupffer cells [28] and RAW 264.7 murine macrophages [29]. Along these lines, our data demonstrated that KA stimulates the expression of TNF-α via the PI3-K/AKT and ERK1/2 pathways, though we still could not exclude other pathways that involved in regulating the expression of TNF-α, such as JNK and p38 pathways [21].
As TNF-α is not a passive molecule, it was further found to induce the production and deposition of Aβ via stimulating the expression of BACE-1 and PS1/2 in neurons. In agreement with our observations, TNF-α has been reported to be responsible for the cleavage of AβPP [30–33]. For instance, TNF-α treatment induces the expression of BACE-1 in APPsw Tg mice [33]. In addition, TNF-α stimulates γ-cleavage of AβPP in HEK293 cells [31]. For this reason, TNF-α is thought to have the ability to induce the expression or phosphorylation of γ-secretases, including presenilin (PS1), PS2 and nicastrin (NCT) in HEK293 or human neuronal cells [30, 32].
Since the concept has been universally accepted that the production of Aβ was abnormally cleaved by β-secretase (BACE-1) and γ-secretase [34], it is possible to speculate that TNF-α treatment is critical for the production of Aβ, especially over the course of AD development. In agreement with this hypothesis, we observed that KA has ability to upregulate the production of Aβ in APP23 mice. Apart from these observations, BACE-1 and PS1/2 have been regarded as a biomarker of AD. For instance, BACE-1 was reported to be responsible for generating Aβ in neurons [35]. In addition, PS was reported to combine with APH-1 and PEN2, to generate an active form of γ-secretase complex that cleaves AβPP and deposits Aβ [36]. Along these lines, our data demonstrated that TNF-α is the key molecule for mediating the effects of KA on inducing the production of Aβ1–42 in neurons.
As the critical roles of TNF-α in KA-induced Aβ, it is urgent to know the localization of this secreted cytokine. For TNF-α, it has been reported that TNF-α is not only expressed in microglia cells, but also in astrocytes, especially under the conditions of AD [37]. Interestingly, we extended prior works to find positive staining of TNF-α in neurons. As the highly expression of TNF-α receptors on neuros [38, 39], we still could not exclude the possibility that the immunoreactivities of TNF-α in neurons are originated and translocated from glial cells. For example, TNF-α produced from microglia cells was found to be critical for LPS-induced neuron degeneration [40]. Apart from neurodegeneration, TNF-α is also responsible for regulating the communications between other cell types and neurons [41, 42]. We thereby conclude that KA-stimulated the expression of BACE-1 and PS1/2 via a glial cells-dependent manner for its ability to produce TNF-α.
To keep the discussion focused, we will continue to discuss the roles of KA in AD. As AD is characterized by the Aβ aggregation and cognitive decline, we further found that KA treatment accelerated the deposition of Aβ and the loss of learning ability in 6-months-old APP23 mice. Although we could not find direct evidence to support our data, KA has shown its effects on impairing memory. For instance, KA has ability to induce damage of hippocampus neurons of mice [43], which might be because of its ability to stimulate mitochondrial oxidation [44]. For these reasons, KA has been used to establish neurodegenerative models [45]. By these experimental models, KA was verified to be effective on inducing abnormal distribution of phosphorylated neurofilaments in neurodegenerative diseases [46]. In addition, it has been verified to impair the learning ability and memory [47]. More closely, KA showed its efficacy on inducing AD [48]. More interestingly, the inflammatory gene expression and glial derived neurotrophic factor are involved in KA-induced cognitive decline [49, 50]. Along these lines, our data fill the gaps between KA and AD
However, KA is originally found to be an effective chemical for establishing epilepsy model [51]. We thereby suspected if there is relationship between epilepsy and AD. Indeed, many patients with AD are at increased risk for developing seizures and epilepsy [52]. In animal models, there is emerging evidence that AD can lead to pathological excitability in neuronal circuits, which is involved in temporal lobe epilepsy [53]. More interestingly, Volicer et al. [54] found that the 5 patients (7%) of their AD cohort who developed seizures following hospitalization had a more significant decline in language function compared to AD patients without seizures. These findings clearly indicated the possibility to treat AD by the drugs of epilepsy. However, data on treatment of seizures and epilepsy in AD are extremely limited. Rao et al. [55] noted that 79% of the patients in their cohort with epilepsy had an “excellent” response to treatment (>95% reduction in seizure frequency).
When considered together, these data supported our conclusion to reveal the roles of KA in regulating the expression of TNF-α, which is responsible for the production and deposition of Aβ via inducing the expression of BACE-1 and PS1/2 during the course of AD development. By finding the efficacy of KA on AD, we may provide novel therapeutic targets to combat AD via epilepsy.
Footnotes
ACKNOWLEDGMENTS
This study was supported by grants from The First Hospital of Jilin University (JDYY52014019), Norman Bethune Program of Jilin University (NO. 2015419,2015421), Health and Family Planning Commission of Jilin Province of China (NO. 2014Q028) and from the National Natural Science Foundation (No.81471216; No.81671186; No.81671177; No.31600820) as well as from the Swedish Research Council (K2013-66X-22337-01-3).