
Abstract
Background:
Although motor disturbances parallel the course of dementia, worsening both quality of life and social costs, the pathogenesis remains still unclear.
Objective:
Through the combination of cerebrospinal fluid (CSF) biomarkers assessment and transcranial magnetic stimulation (TMS) protocols, here we provided a cross-sectional study to understand pathogenic mechanisms of Alzheimer’s disease (AD)-related early motor disturbances.
Methods:
The motor phenotype, as defined with Unified Parkinson’s Disease Rating Scale (UPDRS) part 2-3, Rating Scale for Gait Evaluation in Cognitive Deterioration (RSEGCD) and Tinetti scale, together with CSF profile of amyloid-β 42 (Aβ42), total-tau, and phosphorylated-tau were determined in 37 AD patients and compared to 18 patients with vascular dementia (VaD). A TMS protocol of short afferent inhibition (SAI) was further applied on a subset of AD patients. Clinical, biochemical, and neurophysiological data were then compared and correlated in order to find significant associations.
Results:
AD patients exhibited subtle locomotor impairment and slight extrapyramidal signs. Main motor features (UPDRS part 3, RSGECD, and Tinetti scale scores) correlate with Aβ42 levels but not with t-tau and p-tau. AD patients also presented SAI impairment directly related to UPDRS part 3 score and Aβ42 levels. Motor disturbances of VaD group did not differ statistically from AD and did not correlate with CSF biomarkers.
Conclusions:
The association of motor disturbances with low Aβ42 CSF levels and individual SAI suggests that amyloid-mediated degeneration of cholinergic system may account for early AD-related motor impairment, providing interesting insights either for frailty stratification of patients or personalized therapies.
INTRODUCTION
Motor disturbances and cognitive decline strictly intertwine in dementias. Indeed, motor impairment, especially gait slowing, may long precede cognitive symptoms, representing a sort of risk factor (“Motoric Risk Cognitive or MCR syndrome”) [1, 2]. Likewise, mobility limitations and falls complicate late stages of dementias, leading to negative health outcomes and high annual total costs of care for patients [3–6]. Therefore, the spectrum of dementia-related motor disturbances is wide and changes during the disease course, depending on both the etiology and the stage of disease [5, 8]. Emerging evidence now suggests that cognitive and motor dysfunctions may origin from the degeneration of the same brain networks [6], however the phenomenology and the pathogenic mechanisms of dementia-related motor disturbances remain greatly undefined yet.
Alzheimer’s disease (AD) is the first cause of dementia [9]. In last decades, biochemical and neurophysiological techniques allowed exploring in vivo the underlying neurodegenerative process. In fact, we know that cerebrospinal fluid (CSF) levels of both total and phosphorylated tau proteins (t-tau and p-tau) and 42-aminoacid isoform of amyloid-β peptide (Aβ42) reflect main AD neuropathological changings (neuronal degeneration, phosphorylation of tau protein with tangles formation and the accumulation of Aβ42 into plaques, respectively) [10–12]. Moreover, through a transcranial magnetic stimulation (TMS) protocol of short-latency afferent inhibition (SAI) it has been revealed an aberrant central cholinergic transmission, responsible for a peculiar disruption of sensory-motor integration [13–16].
In this study, we tried to characterize pathophysiology of AD motor disturbances through a multimodal approach. Specifically, we provided standardized clinical assessment of main motor features and subsequent correlation analysis with CSF biomarkers in AD patients compared to patients with vascular dementia (VaD). Then, we evaluated SAI in a subset of AD patients, looking for further informative interactions among clinical, biochemical, and neurophysiological parameters.
METHODS
Subjects and clinical assessment
The study prospectively involved 64 patients, admitted for cognitive decline to the Neurology Unit of Tor Vergata General Hospital (Rome, Italy) between 2014 and 2017. All patients signed an informed consent and underwent demographic and medical recording, complete neurological examination, diagnostic lumbar puncture (LP), and apolipoprotein E (APOE) genotype determination. After diagnostic work-up, patients were divided into two main groups: AD (n = 37) and VaD (n = 18). Probable AD was diagnosed according the 2007 revised diagnostic criteria [17]. VaD patients fulfill available diagnostic criteria for vascular dementia [18]. Diagnostic accuracy was also improved by means CSF biomarkers analysis [19]. 9 patients were excluded from the study because taking neuroleptics or diagnosed with other defined form of dementia (frontotemporal dementia, Lewy body disease, and Creutzfeldt-Jakob disease). None of remaining patients was taking anti-dementia, anti-parkinsonian, or neuroleptic drugs. The study was conducted according with ethical principles of Helsinki declaration and Local committee recommendations. In both groups, motor abilities were assessed using the Unified Parkinson’s Disease Rating Scale (UPDRS) part 2 and part 3, evaluating motor experiences of daily living and severity of extrapyramidal signs respectively; the Tinetti scale for static and dynamic balance; the Rating Scale for Gait Evaluation in Cognitive Deterioration (RSGECD), a novel clinical rating scale for gait and mobility in patients with cognitive impairment [20]. The level of cognitive impairment was indexed with Mini-Mental State Examination (MMSE) score, adjusted for age and educational level, and the Montreal Cognitive Assessment (MoCA) score (full psychometric evaluation was performed for diagnostic purpose).
CSF sampling and analysis
Patients underwent LP for diagnostic purposes within 2 days from the clinical assessment, following standard procedures [21] in the morning, lying in lateral position with atraumatic needles. CSF collection, storage and processing were realized according procedures described elsewhere [22]. One CSF sample was used for chemical and microscopic analysis (>5 cells/μl would be excluded), another one for the determination of biomarkers levels (t-tau, p-tau, Aβ42) through commercially available sandwich enzyme-linked immunosorbent assays [22].
TMS protocol of SAI
TMS recordings were performed at the IRCCS Fondazione Santa Lucia (Rome–Italy) on a subset of 12 AD patients in comparison to 7 age/sex matched healthy controls (CTL, non-blood relatives of patients, without history or clinical sings of neurological diseases). Enrolled AD patients were consecutively selected among patients who received CSF analysis and clinical assessment, depending on the consent, the absence of contraindications to TMS and ability to accomplish the protocol. Magnetic stimulation was performed using a high power Magstim 200 magnetic stimulator (Magstim Co, Whitland, Dyfed, UK). The magnetic stimuli had a nearly monophasic pulse configuration, with a rise time of 0.1 ms, decaying back to zero over 0.8 ms. A figure of eight coil with external loop diameters of 9 cm was held over the left motor cortex at the optimum scalp position to elicit motor responses in the contralateral first dorsal interosseous (FDI) muscle. The optimal position was marked on the scalp with a felt pen to ensure identical placement of the coil throughout the experiment. The handle of the coil pointed backward and was perpendicular to the presumed direction of the central sulcus, about 45° to the midsagittal line. The direction of the induced current was from posterior to anterior and was optimal to activate the motor cortex trans-synaptically. Surface muscle responses were recorded via two 9 mm diameter Ag–AgCl electrodes with the active electrode over the motor point of the muscle and the reference on the metacarpophalangeal joint of the index finger. Muscle responses were amplified and filtered (bandwidth 3–3000 Hz) by D150 amplifiers (Digitimer, Welwyn Garden City, Hertfordshire, UK). Data were collected on a computer with a sampling rate of 10 kHz per channel and stored for later analysis using a CED 1401 A–D converter (Cambridge Electronic Design, Cambridge, UK). All selected subjects were able to understand and carry out the simple task required for this electrophysiological study— that is, to keep fully relaxed. The resting motor threshold (RMT) was defined as the lowest intensity that produced MEPs of >50μV in at least five out of 10 trials with the muscles relaxed. Determination of RMT was done in step width of 1% of maximal stimulator output. Short latency inhibition was studied using the technique that has been recently described [13]. Conditioning stimuli were single pulses (200μs) of electrical stimulation applied through bipolar electrodes to the right median nerve at the wrist (cathode proximal). The intensity of the conditioning stimulus was set at just over motor threshold for evoking a visible twitch of the thenar muscles. The intensity of the test cortical magnetic stimulus was adjusted to evoke a muscle response in relaxed right FDI with amplitude of approximately 1 mV peak to peak. The conditioning stimulus to the peripheral nerve preceded the magnetic test stimulus by different interstimulus intervals (ISIs). ISIs were determined relative to the latency of the N20 component of the somatosensory evoked potential induced by stimulation of the right median nerve. The active electrode for recording the N20 potential was attached 3 cm posterior to C3 (10–20 system) and the reference was 3 cm posterior to C4. Five hundred responses were averaged to identify the latency of the N20 peak. ISIs from N20–4 ms to N20 +8 ms were investigated in 4 ms steps (ISI 16, 20, 24, 28 ms). Ten stimuli were delivered at each ISI. The subject was given audiovisual feedback at high gain to assist in maintaining complete relaxation. The inter-trial interval was set at 5 s (±10%), for a total duration of approximately 5 min. Measurements were made on each individual trial. The mean peak-to peak amplitude of the conditioned MEP at each ISI was expressed as a percentage of the mean peak-to-peak amplitude size of the unconditioned test pulse in that block.
Statistical analysis
After having tested data distribution with Shapiro-Wilk test, Mann-Whitney-U-test or one-way ANOVA were used, as appropriate, to evaluate differences in clinical scores and CSF biomarkers concentrations between the two group. Differences depending on the APOE status were instead measured by means the one-way ANOVA within groups. Spearman’s test was applied separately in each group to observe correlations among clinical data and CSF biomarkers; in the presence of a significant correlation, linear regression was further performed. For SAI protocol, mean values of AD patients were compared with those of CTL through repeated measures ANOVA with GROUP (AD versus CTL) and ISI (16, 20, 24, 28 ms) as within-subject factors. The Greenhouse-Geisser correction was used for non spherical data. Mauchley’s test examined for sphericity. When a significant main effect was reached, paired t tests with Bonferroni’s correction were used to characterize the different effects of the specific time points or ISIs. On the cohort of AD patients subject to TMS, Pearson’s test among SAI values (ISI 16, 20, 24, 28 ms), motor and biochemical data and successive linear regression were performed in order to observe correlations of sensori-motor integration with motor abilities and CSF biomarkers. Statistical significance was set at p < 0.05. Statistical analysis was performed using IBM–SPSS Statistic 22.
RESULTS
Demographic, clinical, and biochemical data
Main demographical, clinical, and biochemical data of both groups are summarized in table 1 (results are expressed as mean±standard deviation). Mann-Whitney-U-test showed no significant differences in age and gender distribution, disease duration, and cognitive abilities (mild cognitive impairment at MMSE and MoCA). In addition, motor features were similar in both groups. Specifically, UPDRS part 3 and part 2 indicated the mild presence of extrapyramidal signs with preserved motor daily activities [23]; Tinetti and RSEGCD scores demonstrated respectively an initial impairment of balance [24] and gait. CSF biomarkers analysis provided expected results, with AD presenting lower Aβ42 and higher t-tau and p-tau levels (table 1).
Demographical, clinical and biochemical data of AD and VaD groups. M, male; F, female; m, months; y, years
In AD group, Spearman’s test revealed significant correlations of UPDRS part 3, RSEGCD and Tinetti scale scores with Aβ42 levels (respectively: R = –0.419, p = 0.012; R = –0.479, p = 0.004; R = 0.368, p = 0.03) but not with t-tau and p-tau. Further analysis with linear regression confirmed the associations, estimating significant predictive value of CSF Aβ42 levels on UPDRS part 3, RSEGCD and Tinetti scale (respectively: T = –7.043, p = 0.006; T = –10.93, p = 0.003; T = 15.21, p = 0.02) (Fig. 1). Conversely, no significant correlations emerged between clinical data and CSF biomarkers in VaD group. As expected, in both groups MMSE correlated with MoCA and t-tau with p-tau (data not shown).
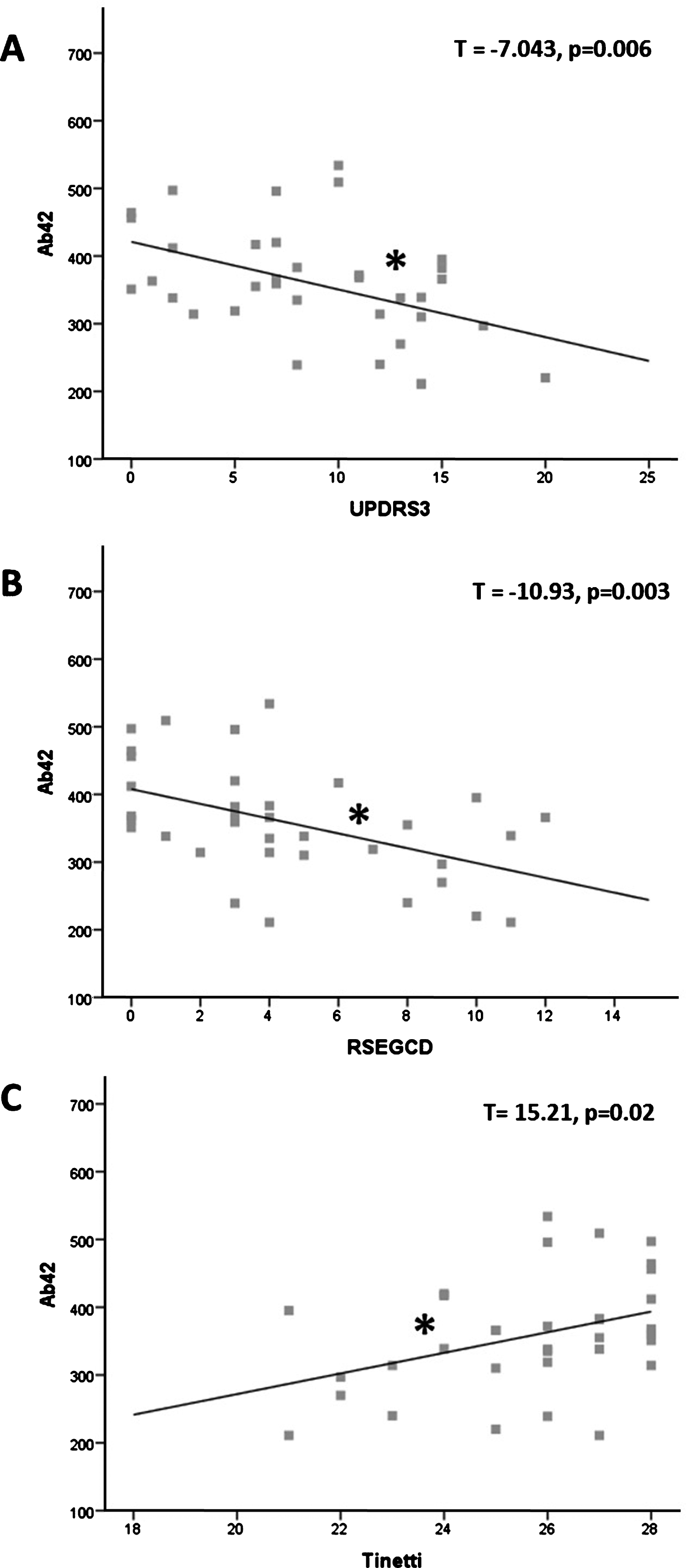
Correlations between Aβ42 and motor scores. A) Inverse association between Aβ42 levels and UPDRS pars 3 score. B) Inverse association between Aβ42 levels and RSEGCD score. C) Direct association between Aβ42 levels and Tinetti scale score. Aβ42 is expressed in pg/ml; asterisks indicate statistical significance.
The one-way ANOVA analysis excluded associations between motor impairment and APOE status in both groups.
SAI assessment
TMS procedures were well tolerated in all subjects. The N20 latency and amplitude were within normal limits in all AD patients and CTL and did not differ between the two groups (20.6±2.9 versus 20.8±3.1). RMT to TMS (mean±standard deviation) was lower in AD patients compared to CTL (AD = 39.8±0.84%; CTL = 43.9±1.28%; p = 0.009). Baseline mean MEP amplitude did not differ between AD patients and CTL across all protocols (AD = 1.09±0.58 mV; CTL = 1.12±0.49 mV). Repeated measure ANOVA tests performed on SAI measures revealed that SAI effects were reduced in AD patients compared to the CTL, showing a significant main effect of GROUP (F(1,17) = 12.606; p = 0.002) and ISI (F(3, 51) = 8.286; p < 0.001), but not a significant GROUP* ISI (F(3,51) = 0.312; p = ns) interaction (Fig. 2A). Pearson’s test and linear regression analyses both revealed that, in AD patient, the individual SAI at ISI 20 is inversely related with Aβ42 levels (respectively R = –0.652, p < 0.05; T = –0.160, p < 0.05) and directly with UPDRS3 (respectively R = 0.731, p = 0.016; T = 2.16, p = 0.03) suggesting that weakening of sensorimotor integration reflects both motor impairment and amyloidopathy (Fig. 2B).
DISCUSSION
In this study, we performed a multimodal analysis of motor disturbances occurring at early stages of AD providing evidence for plausible underlying mechanisms. According with available literature [5, 25], our cohort of AD patients presented subtle locomotor impairment, slight extrapyramidal signs and preserved motor daily activities. Such a mild motor phenotype resulted mostly similar to that of VaD patients. Indeed, no significant differences emerged among motor clinical scores between the two conditions.
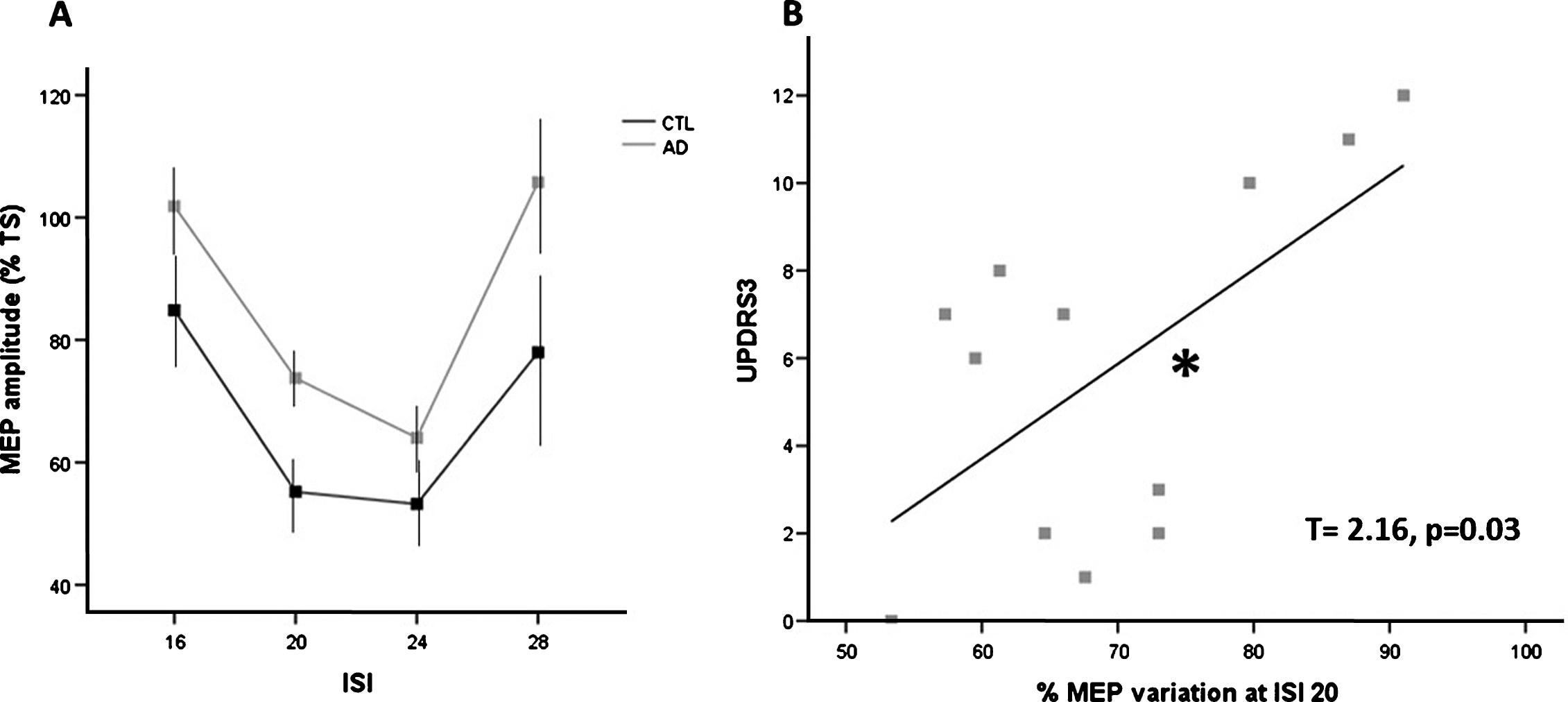
SAI and motor impairment. A) SAI at different ISI in AD and CTL groups. SAI was significantly reduced in AD group (p = 0.002, further results in the text). MEP amplitude is expressed as percentage variation of a Test Stimulus (TS, MEP evoked by single pulse magnetic stimulation alone). Error bars indicate standard error. B) Linear regression graph showing direct relationship between UPDRS pars 3 score and deficient SAI (percentage of MEP variation at ISI 20). Asterisk indicates statistical significance.
In the AD group, motor impairment was directly related to brain amyloidopathy, since UPDRS part 3, RSEGCD and Tinetti scale scores significantly correlated with Aβ42 CSF levels but not with t-tau and p-tau. Moreover, the TMS protocol, besides confirming the disruption of cholinergic transmission [14–16] and the correlation with brain amyloidopathy in AD patients [26], demonstrated a novel direct relation between the SAI impairment at ISI20 and UPDRS part 3, which preliminarily suggests an interesting link between cholinergic dysfunction and motor disturbances in AD.
Our results thus indicate that mild motor disturbances of AD early stages may arise from greater brain amyloidopathy rather than tauopathy. These findings, if one side collide with the well-established knowledge that tau pathology causes neurotransmitters systems disarray and clinical progression of AD [27–29], strengthen a previous observation on the parallel between gait speed decline and [18F]florbetapir PET uptake increase in putamen and other cortical regions [30], highlighting the role of amyloidopathy in the degeneration of motor networks. Actually, plaques deposition in nucleus striatum typically occurs at late stages of AD, causing overt extrapyramidal syndrome [31–33]. In this case, the AD population was instead at early phases of the disease and exhibited only mild locomotor impairment, thus suggesting the involvement of different motor network systems, apart from basal ganglia. Accordingly, the evaluation of sensorimotor integration through SAI protocol, although restricted to a smaller sample of patients, demonstrated that both amyloidopathy and motor disturbances were coupled with cholinergic dysfunction, indicating the probable intervention of cholinergic transmission in the pathophysiology of AD-related motor disorders.
Cholinergic system is a major contributor of motor behavior. In particular, locomotion is tuned by two main cholinergic projection systems, one from the pedunculopontine nucleus in brainstem and the other from the nucleus basalis of Meynert in basal forebrain [34]. The first one extensively innervates basal ganglia and thalamus, directly controlling gait and posture [35, 36], while both pathways reach brain cortex, participating to attention and executive functions [34, 37]. Specifically, the cholinergic system plays a key role in the top-down control of attentional orienting and stimulus discrimination [38], such that its dysfunction could potentially lead to increased fall propensity and reduced attentional capacity in both aging and neurodegenerative diseases. Accordingly, Pelosin and colleagues demonstrated an elegant parallelism of SAI deficiency with motor disturbances, showing progressive worsening of SAI along old adult non-fallers, old adult fallers and Parkinson’s disease patients [37]. However, the significant associations between gait speed, SAI and either postural or gait disorders in patients with Parkinson’s disease was further observed also in another compelling study from Rochester and coworkers [34]. Our findings thus, at least in part, overlap these latter ones; additionally, since we observed the association between amyloidopathy and the dysfunction of cholinergic system, we also accord with pathological data, showing an high vulnerability of cholinergic neurons to amyloid-mediated degeneration [39]. Previous fluorine-18 fluorodeoxyglucose (18F-FDG) PET studies demonstrated how AD-related CSF changes are associated to patterned cortical hypometabolism, mainly at frontotemporal level [40]. Unfortunately, here we do not have functional-anatomical data corroborating both biochemical and neurophysiological results; however, we could equally hypothesize that early AD motor disturbances may underlie an impairment of cholinergic transmission probably due to a great accumulation of amyloid-beta in cholinergic neurons.
Regarding patients with VaD, we did not find significant correlations between motor scores and CSF biomarkers. Thus, VaD-related motor impairment is not strictly dependent on amyloid cascade events or on tau pathology and extensive neurodegeneration. Otherwise, the absence of neurophysiological investigations in this group of patients limits further pathophysiological hypothesis. However, we could suppose that motor impairment origins from the decline of neurotransmitters system, specifically of the dopaminergic signaling, as suggested by the higher load of extrapyramidal symptoms (although not significant compared to AD). Indeed, either cortical and striatal dopamine transporter or dopamine D1 and D2-like receptors densities decline with aging, being responsible for cognitive, behavioral and motor dysfunctions [41]. Therefore, a dysfunction of the fronto-striatal fibers secondary to cerebrovascular lesions burden, remains the more likely underlying mechanism of motor impairment in this group [42].
Definitely our findings are still preliminary and present several limitations. It should be first considered that clinical rating scales used in the study might not have sufficient accuracy in detecting faint symptoms, which would be better assessed with kinematic analysis. Moreover, the absence of a control group including cognitive-intact age/gender-matched subjects limited understanding the contribution of age-related motor decline. TMS results, although confirming previous findings [26], should be replicated on larger samples; likewise, the assessment of gait in “dual task” conditions, specifically evaluating the control of attention orienting, could have further supported the weight of cholinergic dysfunction in AD-related motor impairment [37]. Possibly, future investigations may also include functional neuroimaging and PET studies, which are indeed crucial to deepen pathophysiological mechanisms.
Concluding, here we depicted the early motor phenotype of AD, which results clinically indistinguishable from VaD and encompasses mild locomotor impairment and slight extrapyramidal signs. Of interest, we found that the disruption of physiological cholinergic transmission, probably mediated by an amyloid-mediated mechanism, might represent a potential cause of AD-related motor impairment. Cholinergic dysfunction is thus critical for both cognitive and motor disturbances of AD, contributing to negative clinical course and making AD patients frailer. In this light, low Aβ42 CSF levels and/or deficient SAI configure two reliable biomarkers of frailty [43] which may lead the development of personalized therapeutic approach, limiting the high-cost and the disabilities of AD.