
Abstract
Targeting the early oligomers formed by the amyloid-β (Aβ) peptide of 40 and 42 amino acids is considered one promising therapeutic approach for Alzheimer’s disease (AD). In vitro experiments and computer simulations are often used in synergy to reveal the modes of interactions of drugs. In this account, we present our contribution to understanding how small molecules bind to Aβ40/Aβ42 peptides, based either on extensive coarse-grained and all-atom simulations, or a variety of experimental techniques. We conclude by offering several perspectives on the future of this field to design more efficient drugs.
Keywords
INTRODUCTION
The amyloid-β (Aβ)42 intrinsically disordered protein, of sequence DAEFRHDSGYEVHHQKLV FFAEDVGSNKGAIIGLMVGGVVIA, produced from the amyloid-β protein precursor (AβPP) by β-secretase and γ-secretase, forms amyloid plaques by a nucleation-condensation polymerization process with nonspecific interactions. The population of the smallest pathological oligomers (dimers, trimers, hexamers, or dodecamers) is dependent on agitation, temperature, concentration, ionic strength, and sample preparation [1–6]. Aβ42 aggregates faster and is more toxic than Aβ40, and familial mutations make Aβ peptides either more toxic (H6R, D7N, A21G, E22G, E22Q, E22K, E22Δ, and D23N) or protective in patients (A2T and A2V in its heterozygous form) [6–20]. Designed mutations or chemical modifications at specific positions can turn on/off the aggregation and toxicity properties [21–27] by preventing amyloid formation and increasing neurotoxicity (phosphorylation of S26) [21, 22], accelerating amyloid formation (lactam bridge between D23 and K28) [6], producing less toxic fibrils (mutation L34T) [24] and oligomers (mutations L34T [25] and G33I and G33A [26]), producing more toxic oligomers (mutation K16N) [27], or modulating the toxicity and self-assembly (azobenzene photoswitch at G25-S26-N27 [23]).
Despite progress in the determination of Aβ fibril structures and polymorphism, characterized by 3-fold and 2-fold symmetries of Aβ40 fibril (depending on sample origin, AD-derived brain or synthetic) and different Aβ42 fibril structures (depending on the seeding and buffer conditions) [28–34], the structures of the monomers and small oligomers of Aβ40/Aβ42 alone or in the presence of drugs remain to be determined. Here, we report on our recent computational and experimental studies on this aspect. It is worth noting that at the moment, it is not possible to do accurate simulations at a molecular basis by including all complex biological entities, for instance, the neurovascular unit including astrocytes, neurons endothelial cells of blood-brain barrier, etc., as well as all known Aβ protein receptors and metabolites. The present simulations include only Aβ peptides and inhibitors in water under physiological pH, ionic strength, etc., and this is why we combined the computational studies with the studies in cell cultures.
COARSE-GRAINED AND ALL-ATOM SIMULATIONS OF DRUG/Aβ PEPTIDES
Atomistic molecular dynamics (MD) simulations in explicit solvent using the Anton computer were able to elucidate the detail of how 12 proteins of 10–80 amino acids fold into their native states within 1 ms [35] and how the cancer drug dasatinib finds its Src kinase target binding site within 15μs [36]. This speedup in solving the equations of motion is not sufficient, however, for understanding the early-formed non-fibrillar aggregates because of the number of degrees of freedom and the timescale to be explored (days at in vitro conditions) [37, 38]. As a result, it is necessary to study small-size oligomers and use advanced conformational sampling methods, such as replica exchange molecular dynamics (REMD) [39] or simulated tempering [40] coupled either to atomistic protein force fields (e.g., CHARMM22*, AMBER99sb-ildn, and OPLS-AA) [35, 43] with water models (e.g., TIP3P) or coarse-grained (CG) models [38, 44]. For CG models that eliminate many unimportant degrees of freedom and replace groups of atoms by a single bead, there are, however, two main issues: 1) how to derive effective potentials that maintain the all-atom physical behavior in a water environment [44]; and 2) how to account for the hydrodynamics effects if we use an implicit solvent [45, 46].
We recall that the six-bead CG OPEP (Optimized Potential for Efficient peptide structure Prediction) model (an all-atom backbone with CG side-chains) and force field we developed have been extensively used with success on many proteins [47–53] and protein complexes [54]. Also, we were the first to observe β-barrels [55] during self-assembly of amyloid peptides that were validated by X-ray micro crystallography and all-atom simulations [44, 56]. It is worth noting that atomistic force fields perform well on proteins with well-defined and stable 3D structures, but provide different equilibrium ensembles on intrinsically disordered proteins, so the best force field remains to be determined [57–61].
Many drug molecules have been screened against Aβ aggregation using computer simulations [62–67]. In this account, we focus on four systems, and in what follows, the N-terminal spans residues 1–16, the central hydrophobic core (CHC) spans residues 17–21, the loop region covers residues 22–29, and the C-terminal region covers residues 30–42.
1,4-naphthoquinon-2-yl-L-tryptophan (NQTrp)
NQTrp was found to reduce the toxicity of wild-type (WT) Aβ42 oligomers toward a cultured neuronal cell line and transgenic AD Drosophila model. The nuclear magnetic resonance (NMR) structure of NQTrp bound to Aβ12 - 28 monomer at a molar ratio 0.5:1 showed three dominant binding sites between NQTrp and the Aβ18 - 21 region, but no nuclear Overhauser effects were observed [68]. CG OPEP simulations of Aβ17 - 42 trimer in solution, followed by all-atom docking and MD simulations, showed that the curcumin, EGCG, 2002-H20, resveratrol, and NQTrp drugs have more favorable binding energies for the most populated predicted Aβ structures than for the fibril state, and NQTrp can have multiple binding modes even within a given pocket [69]. Moving to CHARMM22* REMD simulations of Aβ1 - 28 dimer with two NQTrp, and NMR experiments with different Aβ1 - 28 to NQTrp ratios, our results showed that NQTrp has no “binding-site” type interaction [70].
Epigallocatchine gallate (EGCG)
EGCG was reported to redirect the aggregation of the Aβ42 peptide into off-pathway oligomers [71]. We performed an all-atom REMD study on Aβ42 dimer with the OPLS-AA force field and TIP3P solvent in the presence and absence of EGCG molecules with a molar ratio 2:10 (Aβ: EGCG) as used experimentally [72]. Upon EGCG binding, the bend, turn, coil, and helix contents remain constant and only the β content is reduced from 8% to 4%, but the β-strand reduction is significant within residues 1–16 (varying from 10% to 1%), the CHC and residues 39–42 (varying from 20% to 5%). Interestingly with EGCG, the CHC/CHC, and C-terminal/C-terminal interactions observed in pure Aβ42 dimer are greatly reduced, leading to a significant increase of 8% of the cross-collision sections of Aβ42 dimer. The EGCG molecules are buried in the interface between the peptides and bind essentially to the hydrophobic residues of the CHC and C-terminal region by van der Waals interactions, and to the N-terminal D1, E3, R5, D7, and E11 residues by hydrogen-bonds, consistent with the picture derived from isothermal titration calorimetry [73]. Overall, this simulation on Aβ42 dimer with 10 EGCG predicts that 5% of free Aβ42 monomers can associate to larger toxic and non-toxic aggregates. Whether the association of two possible drug candidates or the use of larger drug molecules can prevent the formation of larger Aβ42 aggregates was explored in the next two cases.
SEN304-INH3
The SEN304 (d-[(chGly)-(Tyr)-(chGly)-(chGly) (mLeu)]-NH(2), with D chirality, ch for cyclo-hexyl and m for a N-methyl group) inhibitor [74] and the penta N-methylated peptide 3 (INH3) [75] inhibitor were tested separately on amyloid aggregation and toxicity using multiple experimental techniques. Our computational goal was to determine whether these designed molecules aimed at targeting the Aβ16 - 22 and Aβ32 - 37 regions, respectively, could act in synergy, stabilize the monomer of Aβ40, and prevent its aggregation. To this end, we present unpublished results of the REMD simulation of Aβ40 monomer with two SEN304 and two INH3 molecules. A total of 64 replicas ranging from 300 to 400 K was used, each replica for 400 ns, using the CHARMM22* and TIP3P water force fields, starting from a randomly chosen configuration and orientation of the five molecules. The procedure described in [70] was used to obtain all necessary force field parameters for SEN304 and INH3. The first 50 ns of each replica were excluded from analysis.
The convergence of the simulations was assessed by several metrics (data not shown). Fig. 1A reports the secondary structure of the Aβ40 peptide along the sequence at 315 K. This temperature was selected because it is near physiological temperature. Our results show that the presence of the four drug molecules lead 25% of β-strand at residues E11 and V12, and a high α-helix probability spanning the CHC (with a maximum of 25%) and the residues 30–36 (with a maximum of 67% for residue I32). Figure 1B shows the distribution of free Aβ monomer as a function of the minimal distance between any heavy atoms of Aβ and any heavy atoms of the four drugs. Using a standard cut-off distance of 0.35 nm, we see, in contrast to the simulations of Aβ1 - 28 dimer with two NQTrp, that the population of free Aβ40 monomer is close to zero, indicating a tight binding between the receptor and the drugs. Figure 2 shows 20 clusters obtained from the backbone dihedral angle principal component analysis (dPCA) analysis [72] of the Aβ peptide. First, all 20 clusters differ in the conformations of the Aβ monomer. The states S1, S3, S12, and S20 are essentially random coil, states S2, S5, S7, S8, S9, and S10 display some α-helices at different positions, while states S4, S11, S14, S15, and S16 display mixed α-β configurations. Also, all clusters differ in the positions and orientations of the four drugs. To get a better understanding of the binding, Fig. 3 shows the contact probability map between the Aβ40 monomer and the drugs. Both drugs are very mobile and bind preferentially to the CHC and residues 30–36, but transient interactions are also observed with the residues V24 and N27 of the loop and C-terminal residues (F4, Y10, and V12). SEN304 was designed based on modifying the self-recognition element Aβ16 - 20 sequence [74]. While it does indeed bind to this region, it also binds to residues 30–36.
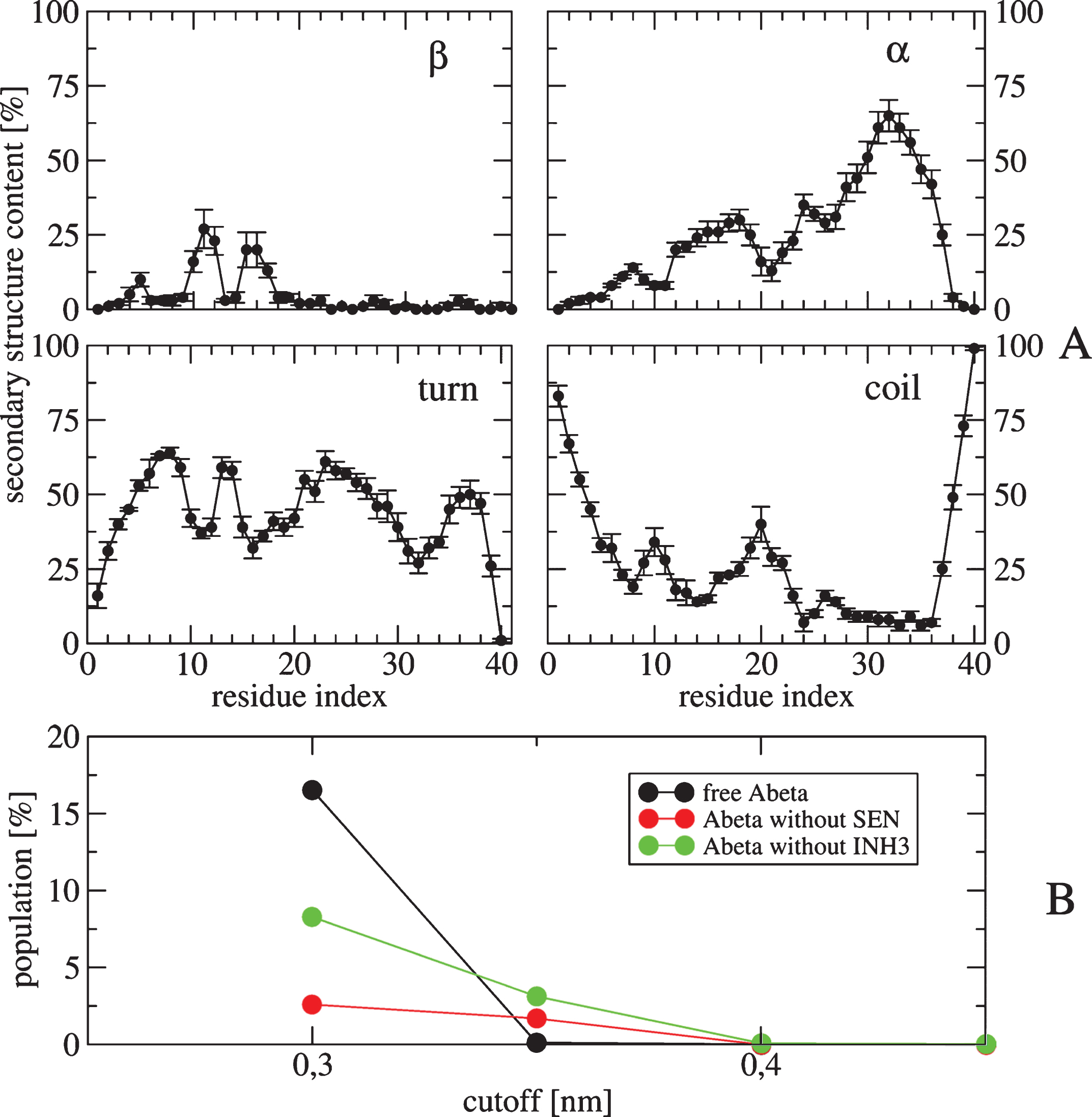
A) Secondary structure contents (in %) along residues of the Aβ40 obtained from 50–400 ns of the REMD trajectory at 315 K. The average values are: 4.6% for β-strand, 24.2% for helix, 43.8% for turn and 27.2% for coil. Error bars are also shown. B) Populations of the free monomer Aβ40 (black), Aβ40 in contact with two INH3 molecules but free from two SEN (red), Aβ40 in contact with two SEN drugs but free from two INH3 (green).
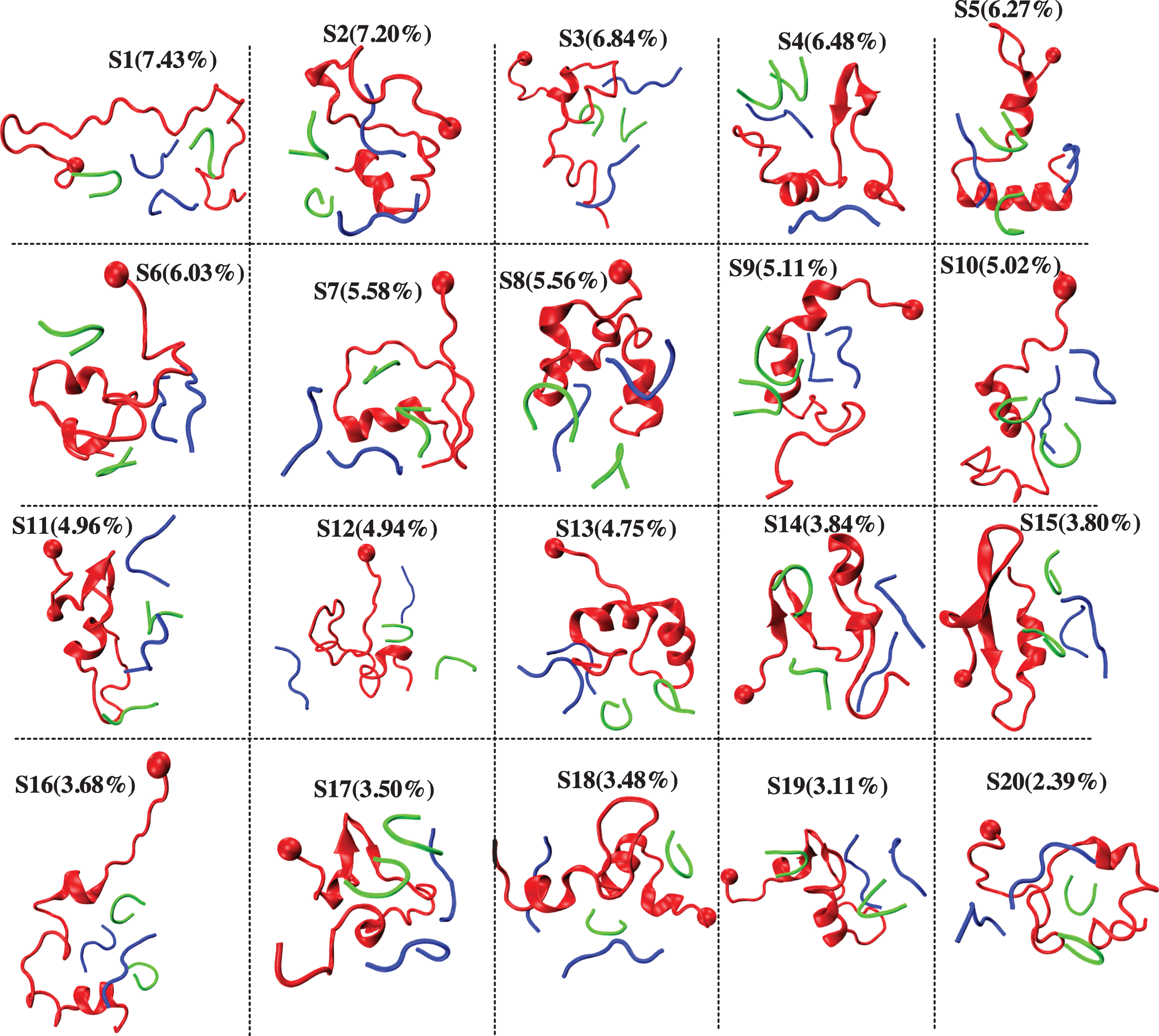
Representative structures and populations of 20 conformational states of the Aβ40 monomer (red) in the presence of two SEN (green) and two INH3 molecules (blue). The ball indicates the first residue of Aβ40.
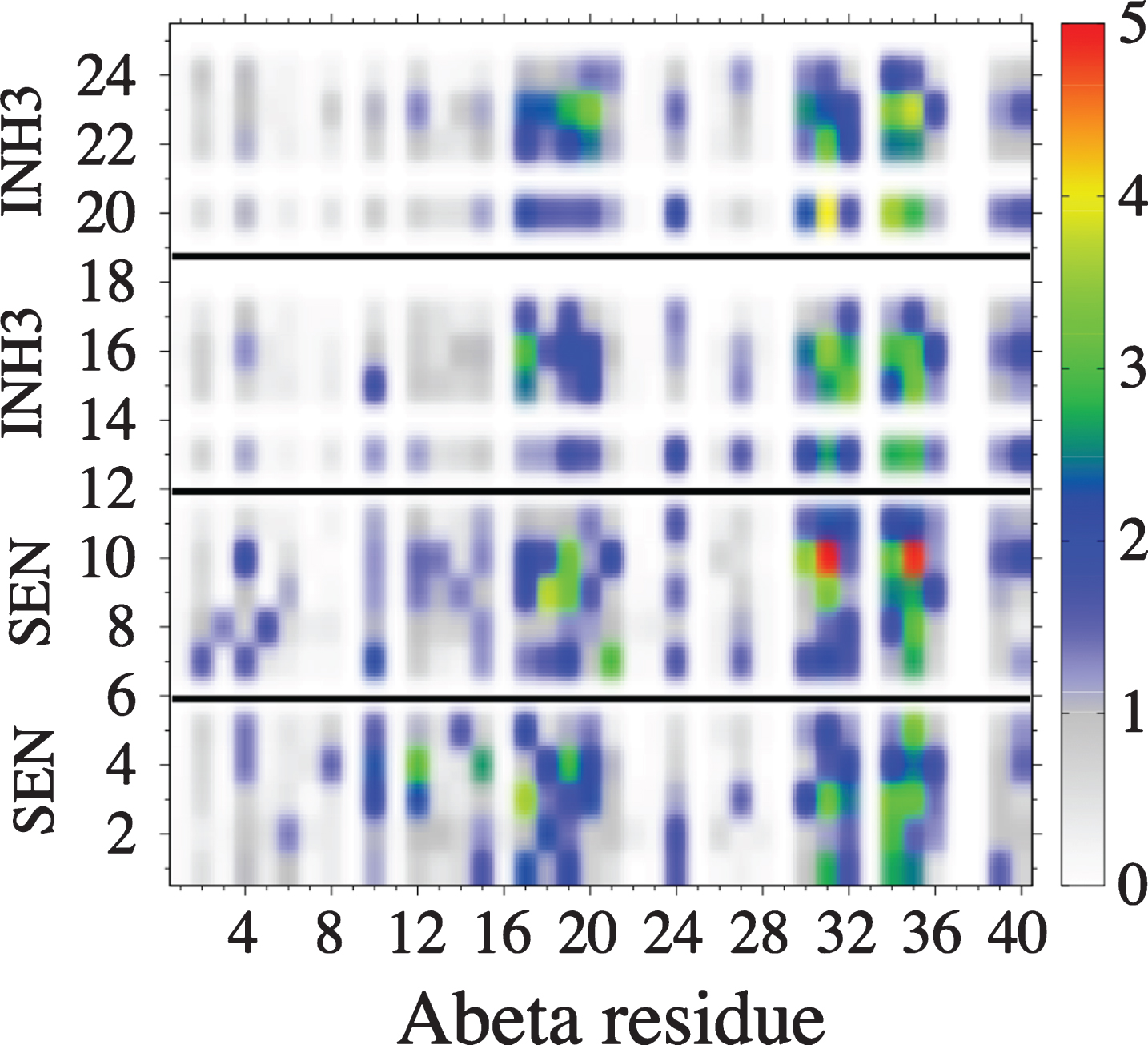
Probability (in %) of forming contacts between residues of the monomer Aβ40 (horizontal axis) and those of four drugs (vertical axis). Residue indices of drugs are (1–5: first SEN), (7–11: second SEN), (13–18: first INH3), and (20–25: second INH3). A contact is considered if the shortest distance between heavy side-chain atoms of the Aβ40 and those of drugs is smaller than 0.45 nm.
Chignolin
The last system, for which we present unpublished atomistic REMD results, is Aβ40 with four chignolin peptides. Our selection of chignolin (GYDPETGTWG) was motivated by two experimental results. First, the phage-display selected protein ZAβ3 of 58 amino acids inhibits Aβ40 fibrillation at stoichiometric concentrations, with the bound Aβ40 conformation featuring a β-hairpin comprising residues 17–36 [76]. Also, the complex interface displays a four-stranded β-sheet consisting of the Aβ17 - 36 region and the residues 15–19 of the two ZAβ3 proteins. This β-hairpin covering 17–36 has been proposed as an intermediate conformation on the pathways to amyloid fibrils [6, 77–79]. Second, our idea was to find a peptide inhibitor that would stabilize the β-hairpin in Aβ by favoring a four-stranded β-sheet in the complex, form a β-hairpin like structure alone in aqueous solution, and be not cytotoxic. Looking at the literature, we find that the 10-residue chignolin is monomeric in aqueous solution, forms a β-hairpin like conformation by NMR, and is one of the most stable peptides [80]. Atomistic simulations have shown that many force fields can predict the correct fold and thermal stability of chignolin in explicit solvent [81–83].
REMD of the complex was performed with 64 replicas spanning 300–480 K, each replica for 400 ns, using CHARMM22* and TIP3P model. All replicas start from a designed structure shown in Fig. 4, where the initial conformation of Aβ40 is extracted from a predicted β-hairpin spanning residues 17–36 obtained from our previous Aβ40 dimer simulations [79], the initial configuration of chignolin is the NMR structure [80] and the four chignolin molecules are randomly orientated in a water box of approximately 200 nm3, resulting in a Aβ concentration of 8.3 mM. Figure 4 shows the free energy landscape (FEL) of the Aβ peptide in the complex at 315 K, using the trajectories 50–400 ns and dPCA analysis (with the first two V1 and V2 components). We have checked that the results are converged and are independent of the time windows using for analysis 50–225, 225–400, or 50–400 ns. The FEL displays eight minima (S1– S8) with populations varying between 20.4% (S1) and 4.5% (S8). In all states, the β-hairpin architecture spanning residues 17–36 is formed, which is otherwise lost in the absence of chignolin (data not shown). Additionally, in states S1, S3, S4, S6, and S7 the C-terminal region makes contacts with the N-terminal region, resulting in 3-stranded β-sheets. All chignolin peptides retain the β-hairpin conformation most of the time, as shown in the secondary structure composition at 315 K in Fig. 5. Consequently, the complex is stabilized by interactions between the β-strands of chignolin with those of the N-terminal region (residues 4–6), CHC region, and C-terminal region (residues 32–40) of Aβ peptide, as shown by the intermolecular contact map (Fig. 6).
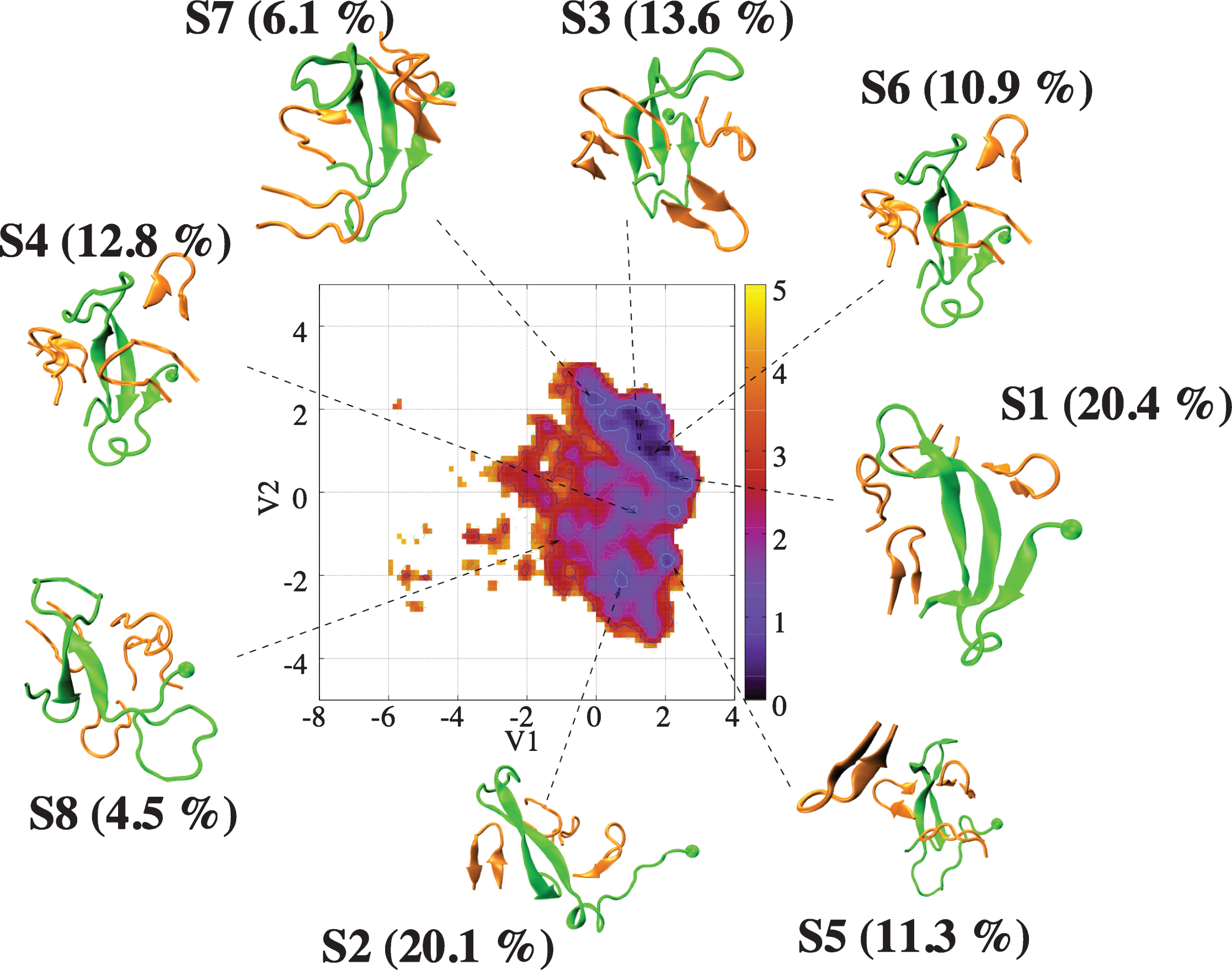
Free energy landscape (in kcal/mol) of Aβ40 (green) in the presence of four chignolin peptides (orange) as a function of the first two principal components (V1 and V2) obtained from PCA on the backbone dihedral angles of Aβ40. Shown are the centers of the free energy minima.
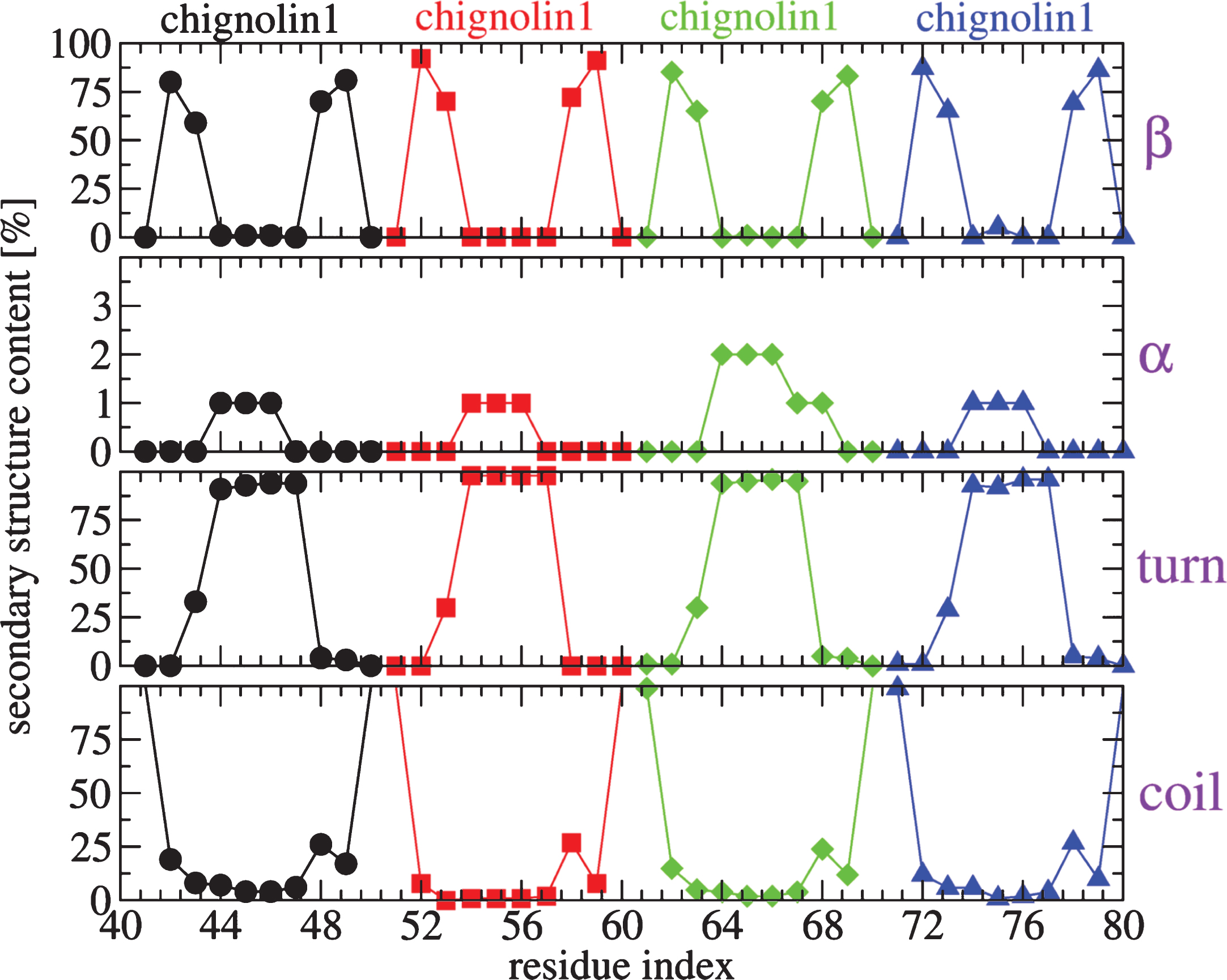
Secondary structure contents (in %) along residues of the four chignolins obtained from 50–400 ns of the REMD trajectory at 315 K.
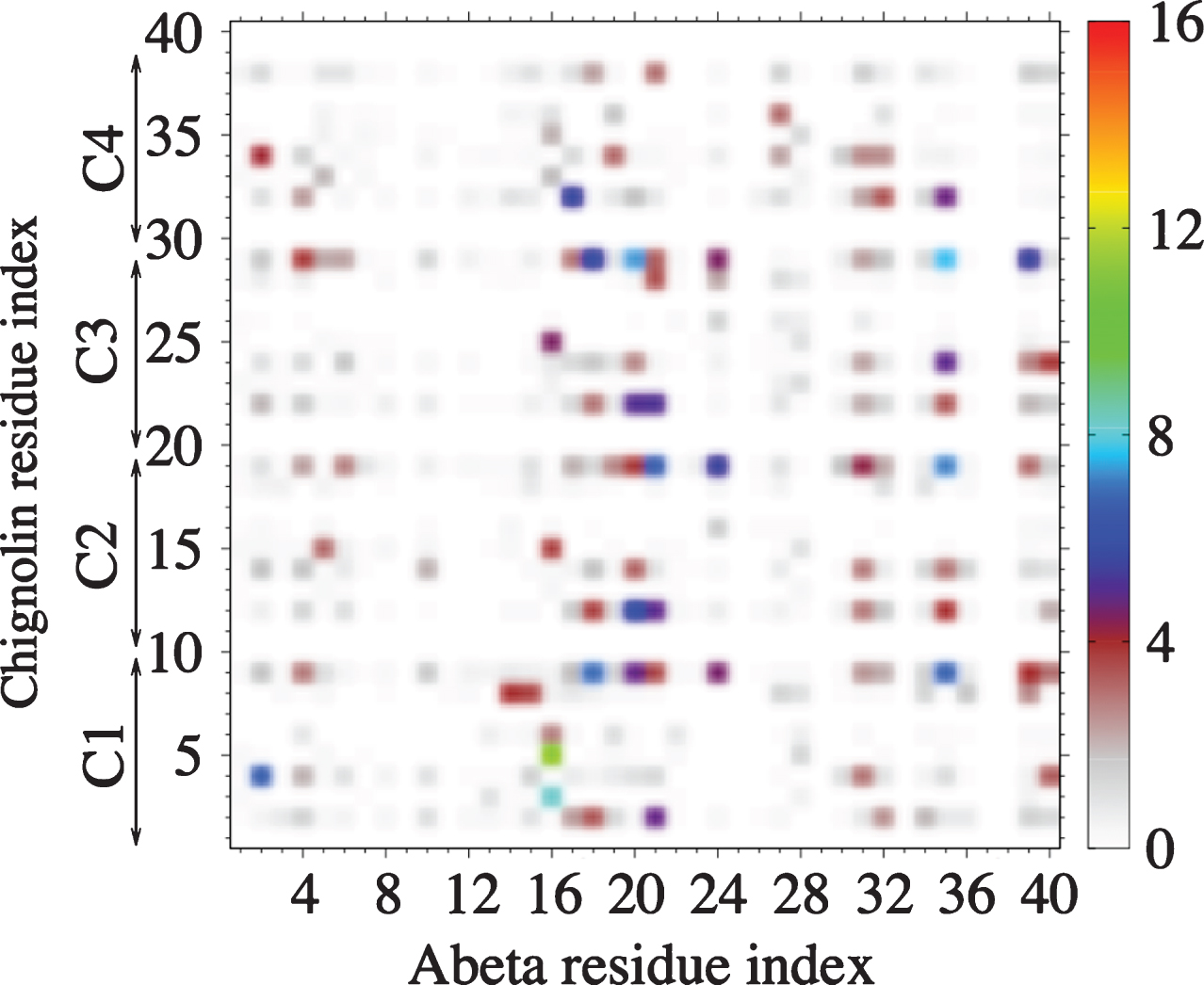
Intermolecular side-chain–side-chain contact probabilities (in %) of Aβ40 with four chignolins (denoted as to C1-C4) at 315 K. A contact is considered if the shortest distance between two heavy side-chain atoms is smaller than 0.45 nm.
DRUG TESTING USING CELL-BASED ASSAYS
To complement the possible therapeutic effects of the drugs studied by computational methods, we used SH-SY5Y cells acutely treated with Aβ42, and SH-SY5Y695 cells overexpressing Aβ [84], as in vitro models to mimic the neurotoxic effects of Aβ in AD. The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) cell viability assay is extensively used in studies measuring Aβ toxicity. Healthy cells reduce MTT, but this metabolic process is decreased when SH-SY5Y cells are treated with Aβ42. We used this assay to evaluate whether cells treated with 1μM EGCG, SEN304, INH3, chignolin, and NQTrp alleviated Aβ42 toxicity. EGCG and SEN304 significantly reduced Aβ42 toxicity, (p < 0.0001) compared to cells treated with 1μM Aβ42, while INH3, chignolin and NQTrp had no significant effect (Fig. 7). These results correlate very well with the capacity of these compounds to inhibit fibril formation of Aβ40 as assessed by Thioflavin T (ThT) fluorescence and atomic force microscopy (AFM), SEN304 and EGCG inhibit fibril formation in a dose dependent manner, whereas chignolin, INH3 and NQTrp show no significant inhibition (see Supplementary Figures 1–3). Note we previously reported that only very high concentration of NQTrp might partially rescue cells from Aβ, indicating that the reported anti-AD activity of NQTrp in in vivo models has to involve another mechanisms [85].
SEN304 and EGGC were further investigated by the cell viability assay at a range of concentrations (Fig. 7). Both EGCG and SEN304 reduced Aβ42 toxicity in a dose-dependent manner. EGCG seems to be the most potent (EC50 = 0.8μM) as it restores MTT reduction to 100% in cells treated with only 2.5μM drug; SEN304 (EC50 = 2.5μM) also restored MTT reduction to 100% when cells were treated with 10μM of this drug, in agreement with previous work [74]. Both drugs were tested without Aβ42 (dotted lines Fig. 7B) to ensure that the compounds are not toxic.
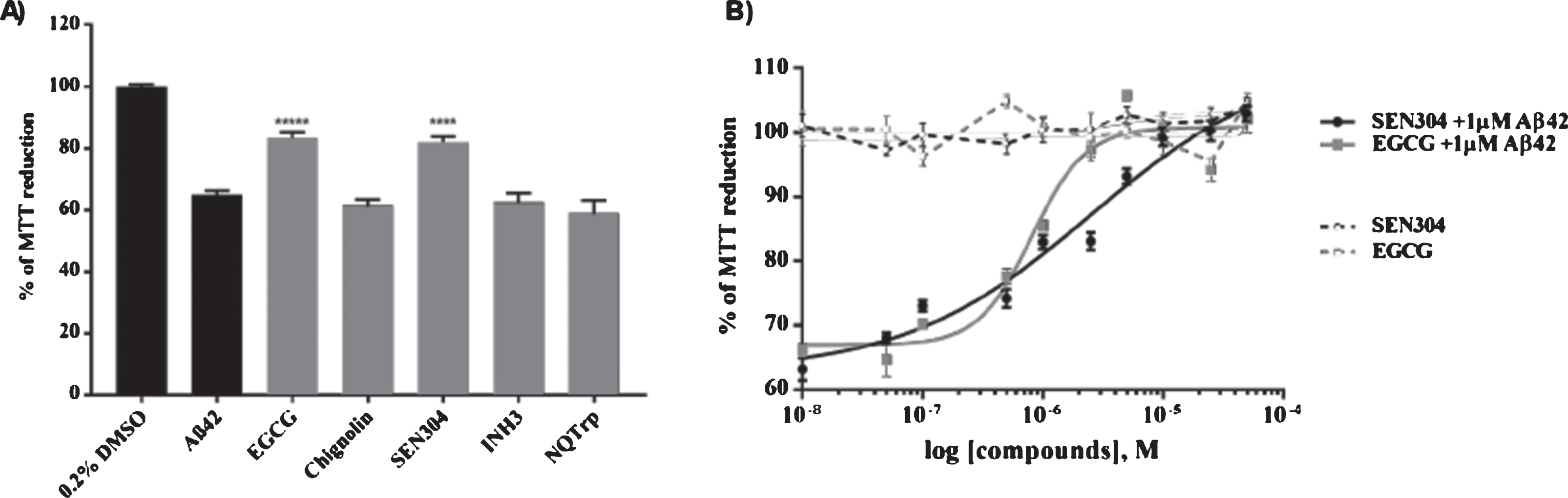
A) MTT screening of drugs. SH-SY5Y cells were treated with a mixture of the indicated drug and Aβ42 with both at 1μM in triplicate. After 24 h incubation at 37°C, an MTT assay was performed to measure the cell metabolism (% of MTT reduction) and each compound’s ability to attenuate toxicity caused by Aβ42. Error bars represent standard error of mean (SEM). ****p < 0.0001. B) MTT dose-response curves of SEN304 and EGCG. SEN304 and EGCG were added to SH-SY5Y cells at different concentrations ranging from 50μM to 10 nM along with 1μM Aβ42 (solid lines). Compounds were also incubated at the same concentrations without Aβ42 (dotted lines). Data from the drug response curves of all the compounds were fitted using a 4PL dose response model to give their EC50 values. Error bars represent SEM, n = 3.
Next, we used the Meso Scale Discovery system (MSD) to assess whether 5μM SEN304 or EGCG affected AβPP processing and secretion in SH-SY5Y695 cells. SEN304 decreased the amount of Aβ40, Aβ42, and sAβPPβ below 80% compared to the non-treated cells. It also increased the signal of sAβPPα to ∼115%. EGCG had no effect on Aβ40 and Aβ42, slightly increased sAβPPα and had no effect on sAβPPβ (Fig. 8).
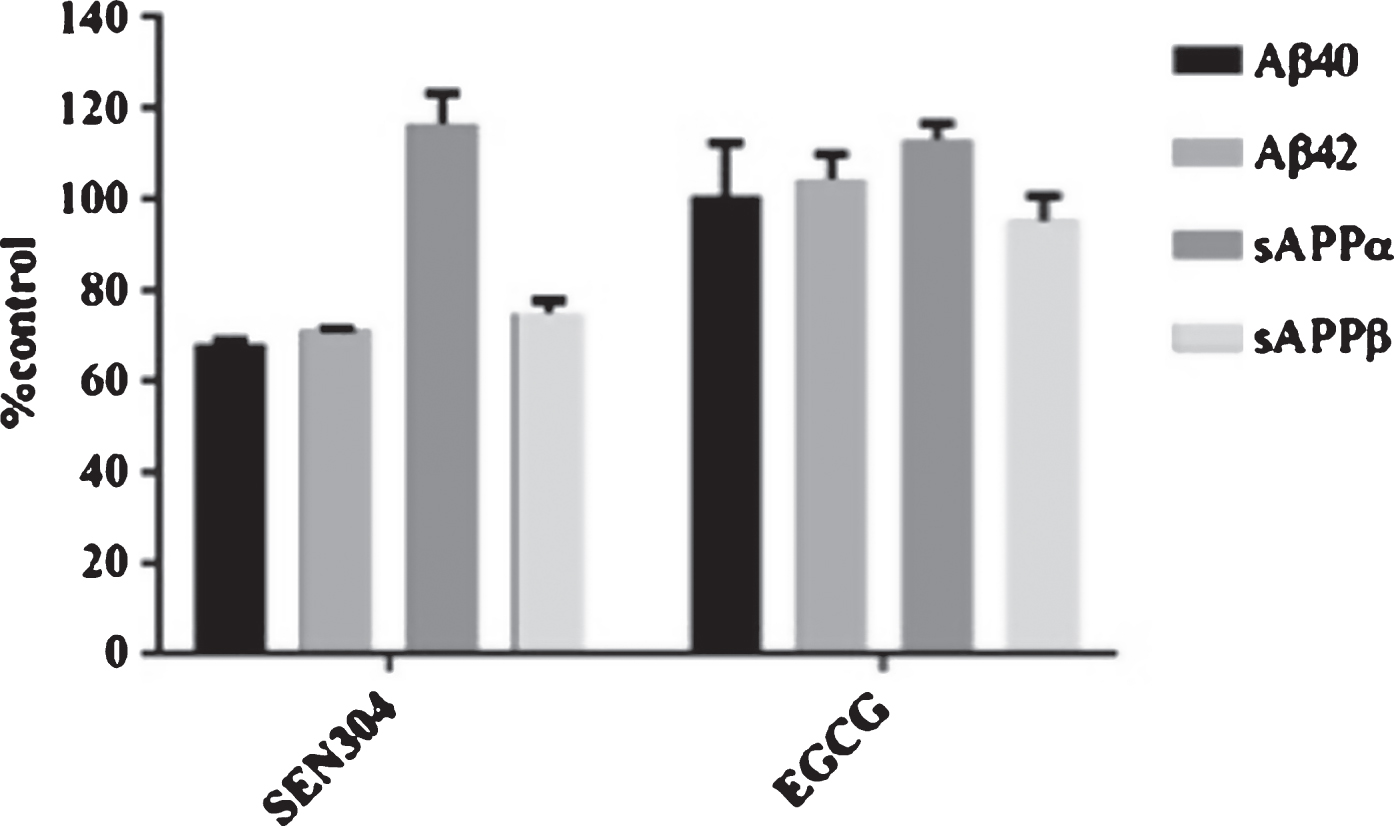
SH-SY5Y APP695 cells were incubated with 5μM of EGCG or SEN304 for 24 h. After incubation, 25μl of media from the cells was placed in the MSD immunoassay V-Plex Aβ peptide panel for measuring secreted Aβ38, Aβ40, and Aβ42 concentrations. Another 25μl was added to the sAβPPα/sAβPPβ kit to measure secreted sAβPPα and sAβPPβ concentrations. Data are represented as the mean of each parameter evaluated, n = 2 and the error bars represent standard deviations.
We studied whether the drugs could also decrease oxidative stress caused by Aβ42, as some studies suggest that an increase in oxidative stress could be one of the factors preceding AD and that it could also promote Aβ production [86, 87]. First, we performed a DCFH assay which allowed us to assess whether SEN304 or EGCG decreased the increase of reactive oxidative species (ROS) in cells treated with 5μM Aβ (Fig. 9A). DCFH measures different types of ROS species, including H2O2, hydroxyl radicals (•OH), and nitrile radicals (•NO 2) [88]. 5μM EGCG completely abolished oxidative stress caused by 5μM Aβ42, whereas SEN304 had no effect. To complement the measurements of oxidative stress we measured the ratio of glutathione (GSH)/ glutathione disulphide (GSSG). Glutathione is mostly found in its reduced form GSH, but when cells are exposed to oxidative stress, GSH is oxidized to GSSG. Thus, the ratio GSH/GSSG is a good measure of oxidative stress. EGCG partially restored GSH/GSSG ratio compared to cells treated with 5μM Aβ42 (Fig. 9B).
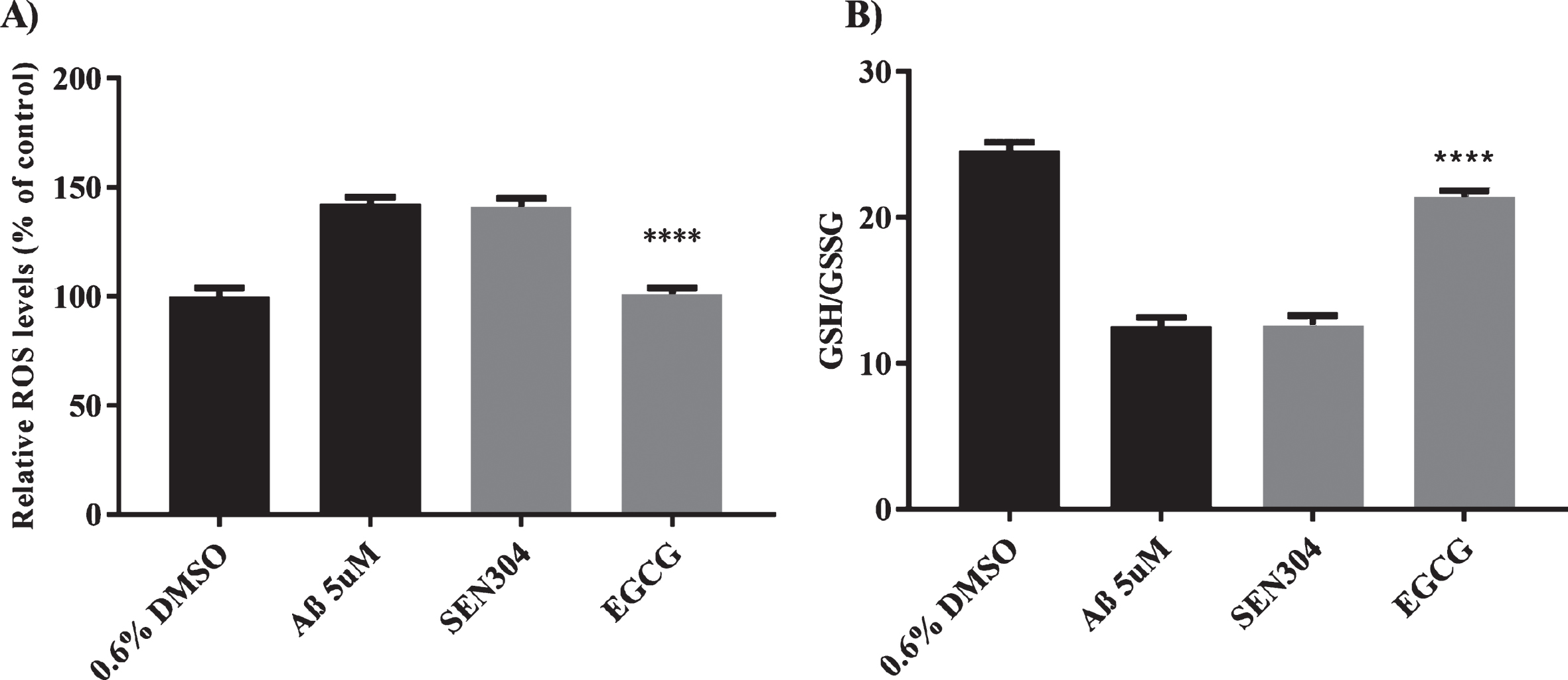
A) ROS levels measured using DCFH-DA. A) SH-SH5Y cells were incubated with 5μM of each of the primary screening hits and 5μM of Aβ42 5μM. Controls were treated with 0.6% DMSO or 5μM Aβ42. After 24 h incubation at 37°C, DCFH-DA assay was performed to measure ROS levels. B) GSH/GSSG ratio was measured using GSH/GSSG- GloTM. After 24 h incubation at 37°C GSH/GSSG- GloTM assay was performed to measure GSH/GSSG ratio. n = 3 and the error bas represent (SEM). Data was analyzed using ANOVA (one-sided) Dunnett’s post hoc test. ****p < 0.0001.
CONCLUSIONS AND PERSPECTIVES
We have reported in silico, biophysical, and cell assays for four drug molecules: NQTrp, SEN304, EGCG, and INH3, and one plausible inhibitor, the chignolin peptide. Our simulations and experimental results on NQTrp indicate that this compound is not appropriate for blocking Aβ aggregation and toxicity [70].
EGCG is the main catechin (antioxidant flavonoid) found in green tea. Several in vitro and in vivo studies have pointed to EGCG as a potential treatment for AD. For instance, EGCG inhibits Aβ toxicity in PC12 and neuroblastoma mice cells when measured with MTT [89, 90]. One possible mechanism of action for EGCG is as an aggregation inhibitor, by redirecting Aβ aggregation to off-pathway oligomers that are not as toxic as on-pathway oligomers [71]. In primary neurons from Swedish mutant APP mice, it was found that EGCG activates AβPPα processing [91]. Our data shows that EGCG increased MTT reduction in cells treated with Aβ42, but did not activate sAβPPα, or decrease Aβ40 or Aβ42 in the MSD immunoassay.
EGCG has been previously studied as a possible treatment for AD. Our data agrees with previous studies that suggest that EGCG inhibits Aβ fibril formation and Aβ toxicity when measured with MTT in a concentration dependent-manner [92]. Moreover, other groups have studied the increase of ROS in cells treated with Aβ fragments and have also found that EGCG significantly decreased the ROS signal [93]. Our data also shows that EGCG increased the ratio of GSH/GSSG in cells treated with Aβ. The effect of EGCG decreasing oxidative stress could be due to its action as ROS scavenger, but studies also suggest that it could be because it promotes the production of glutathione [94].
SEN304 is an optimized peptide based on the site recognition sequence (SRS) KLVFF corresponding to residues 16–20 of Aβ [95]. This sequence was identified as key for Aβ-Aβ interactions [96]. For this reason, this SRS was used as a template to design Aβ aggregation inhibitors, including SEN304. The peptide works by promoting a rapid aggregation of monomers in an alternative aggregation mode that produces larger, but less toxic aggregates [74, 97]. Our results agree with previous observation that SEN304 attenuates Aβ42 toxicity in SH-SY5Y cells assessed by MTT [74]. Surprisingly for an aggregation inhibitor, SEN304 also decreases production and secretion of Aβ40, Aβ42, and sAβPPβ and increases sAβPPα. There are no previous studies, to our knowledge, that investigated alternative mechanisms of action for SEN304, such as affecting AβPP processing. However, the 6E10 antibody, the captured antibody for the MSD Aβ42 panel, was previously used by Amijee et al. as the monoclonal antibody for a single-site ELISA to assess the effect of SEN304 on Aβ oligomer formation [74]. They found that SEN304 reduced the Aβ signal in the ELISA assay because SEN304 binds to Aβ monomers and oligomers. Considering this, and the fact that both SEN304 and 6E10 bind to the same Aβ region, it is possible that the decrease in Aβ40 and Aβ42 in the MSD assay is because SEN304 is already bound to the SRS KLVFF blocking the binding of 6E10. However, this does not explain why SEN304 decreases sAβPPβ and increases sAβPPα, as this would require affecting secretase activity. SEN304 may therefore have an additional mechanism of action besides the one it was initially designed for.
We have combined INH3 and SEN304 inhibitors in simulations. The absence of a clear pattern between Aβ/drug interactions indicates that the two molecules compete with each other, despite being designed to bind to different regions of Aβ. Hence, we would not expect any favorable synergy in retarding Aβ40 oligomerization compared to the effect of each drug taken separately. We also tested for the first time whether the chignolin peptide could be suitable as an inhibitor. Simulations report that Aβ retains the β-hairpin in the presence of chignolin, while experimental studies indicate that this molecule is not able to stabilize the β-hairpin in the monomer, and prevent Aβ aggregation and toxicity. This highlights the difficulty in designing new drugs in a milieu that simplifies cells. In a recent viewpoint, we have provided some reasons explaining why research on Aβ fails to give new drugs for Alzheimer’s disease [98]. These include, but are not limited to, differences between in silico, in vitro, and in vivo concentrations, the use of Aβ40 or Aβ42 peptides while AD brain plaques consist of many Aβ peptide sequences [99] and a stoichiometry that varies with the severity of the disease [100], and the neglect of the pas de deux between Aβ and the tau protein disease [101]. As recently stated by two recent articles [98, 102], it is also time to stop AD before it starts by primary prevention human trials aimed at investigating drugs designed to treat AD before brain pathology.
METHODS
Materials
The inhibitors were obtained as follows: EGCG (Sigma), SEN304 (purchased from Peptide Protein Research Ltd), NQTrp (synthesized as described in [85]), INH3 (from rPeptide) and chignolin (purchased from Genecust). Aβ42 was purchased from rPeptide. The GSH/GSSG-GloTM kit was purchased from Promega. V-Plex plus Aβ peptide Panel 1 (6E10) kit and sAβPPα/sAβPPβ kit were purchased from MesoScale Discovery. SHSY5Y cells were acquired from the European Collection of Authenticated cell cultures (ECACC). SH-SY5Y695 cells were kindly donated by Prof. Nigel Hooper.
Aβ42 preparation
Aβ42 lyophilized powder was dissolved in hexafluroisopropanol (HFIP) at a concentration of ∼1 mg/ml and vortexed in three cycles of 30 s to mix. After adding HFIP, the peptide was incubated at room temperature for 1 h to dissolve it completely and then aliquoted into 20 Eppendorf tubes of 50μl (50μg) each. Aliquots were lyophilized by streaming gaseous N2 to evaporate HFI, leaving the peptide coated onto the wall of the tube. The resulting lyophilized peptide aliquots were stored at –20°C until required. Anhydrous DMSO was added to the lyophilized aliquots of Aβ42 to obtain a concentration of 1 M. As DMSO is toxic for SH-SY5Y cells when it is present in concentrations above 1%, this stock was diluted in non-supplemented Opti-MEM without phenol medium to obtain a final concentration of 1μM Aβ42, 0.1% DMSO or 5μM Aβ42 0.5% DMSO, when added to the cells.
Cell culture
SH-SY5Y and SH-SY5Y695 human neuroblastoma cells were maintained in MEM Earle’s medium/ Ham’s F12 (1:1) supplemented with 10% fetal bovine serum (FBS), L-Glutamine (L-Q), 1% penicillin-streptomycin and 1% non-essential amino acids (n-aa). The cells were cultured in tissue flasks and incubated at 37°C, 5% CO2 atmosphere . When cells reached ∼80% confluency, they were either harvested for cell viability assays or passaged into new flasks.
Drug preparations
For MTT, an Aβ42-drug mixture was prepared by adding Aβ42 in non-supplemented Opti-MEM to achieve a concentration of 1μM Aβ42, 1μM drug, and 0.2% DMSO. For both DCFH and GSH/GSSG assays, drugs were tested at a concentration of 5μM with 5μM Aβ42 and 0.6% DMSO as controls in triplicates. For MSD assays, 1 mM stocks of drugs were diluted in Opti-MEM to achieve a final concentration of 5μM drug and 0.5% DMSO. For dose response curves, 10 concentrations of drugs, ranging from 50 to 0.01μM, were tested by incubating the cells with 1μM Aβ42. The drugs in different concentrations without Aβ42 were used as controls. All tested groups were incubated in triplicate.
MTT assay
100μl of SH-SY5Y cells was seeded in 96-well plates at 3×104 cells/well in MEM Earle’s medium/ Ham’s F12 (1:1) supplemented with 10% FBS, 1% L-Q, and 1% n-aa penicillin-streptomycin. The plates were incubated overnight at 37°C with 5% CO2 to allow cell adherence. After the incubation time, media was carefully removed from each well and 100μl of the Aβ42-drug mixture was added to wells in triplicate using reverse pipetting. The plate was returned for incubation for 24 h at 37°C with 5% CO2. After incubation, the MTT assay was performed. Firstly, 50μl of media was removed and 10μl of sterile MTT (2.5 mg/ml) was added to each well. The cells were incubated for 3 h at 37°C with 5% CO2. Then 100μl of acid-isopropanol (stock solution 100 ml of isopropanol and 398μl of HCl 37%) was added. To allow solubilization of the formazan crystals, the bottom of the wells was scraped with the micropipette tip and mixed thoroughly. The plates were covered with foil and placed in a plate-shaker for 15 min. The absorbance of the plates was measured using a Tecan Infinite M200 Pro microplate reader at 570 nm.
Percentage of MTT reduction (cell viability) was calculated as:
where X is the absorbance value of each well, A is the mean absorbance of the blank (buffer only), and B is the mean absorbance of the non-treated cells.
MSD assay
SH-SY5Y APP695 cells were seeded at a 5×104 cells/well in a 96-well plate. Cells were incubated overnight at 37°C, 5% CO2, to allow cell adherence. Media was replaced with Opti-MEM non-supplemented media containing drugs at 5μM with 0.5% DMSO. Treated cells were returned to the incubator for another 24 h. Aβ peptides (Aβ38, Aβ40, and Aβ42) and the soluble AβPP fragments (sAβPPα/β) were measured from the cell-media using the V-Plex Aβ panel (6E10) kit and the AβPPα/sAβPPβ multiplex kit, from Meso Scale discovery (MSD), respectively.
DCFH assay
100μl of SH-SY5Y cells at 3×105 cells/ml per well were seeded in MEM Earle’s medium/ Ham’s F12 (1:1) supplemented with 10% FBS, 1% L-Q, and 1% n-aa penicillin-streptomycin in a 96-black plate. The cells were incubated overnight at 37°C with 5% CO2 to let the cells attach to the bottom of the black 96-well plate. The cells were incubated for another 24 h with Aβ42-drug mixtures. A mother stock of DCFH at 100 M in DMSO was dissolved in PBS to achieve a concentration of 100μM DCFH and 0.1% DMSO. The media was replaced from all wells with the diluted DCFH solution and the plate was returned to the incubator for 30 min. Afterwards, each well was washed with 200μl of PBS to eliminate fluorescence coming from the media and ensure the measured fluorescence was coming from the cells only. The fluorescence was read using a Tecan Infinite M200 at an excitation of 480 nm and 530 nm emission. Data was normalized using the following formula:
GSH/GSSG
100μl of SH-SY5Y cells at 1×105 cells/ml were seeded in a 96-white well plate using MEM Earle’s medium/Ham’s F12 (1:1) supplemented with 10% FBS, 1% L-Q, and 1% n-aa penicillin-streptomycin and 1% L-Glutamine, and incubated overnight at 37°C with 5% CO2. The media was the replaced with 100μl of non-supplemented Opti-MEM containing its respective concentration of drug or control and incubated for another 24 h. For this assay, there were two sets in triplicate for each of the treatments: one set was used to measure total glutathione and one set to measure oxidized glutathione. Glutathione was measured using a GSH/GSSG-GloTM assay from Promega. The assay was performed as per manufacturer’s instructions. The plate was read in a Promega Glo-Max-Multi Detection system. The data was normalized to GSH/GSSG ratio using the following formula