
Abstract
Spanning over three decades of extensive drug discovery research, the efforts to develop a potent and selective GSK3 inhibitor as a therapeutic for the treatment of type 2 diabetes, Alzheimer’s disease (AD), bipolar disorders and cancer have been futile. Since its initial discovery in 1980 and subsequent decades of research, one cannot underscore the importance of the target and the promise of a game changing disease modifier. Several pharmaceutical companies, biotech companies, and academic institutions raged in a quest to unravel the biology and discover potent and selective GSK3 inhibitors, some of which went through clinical trials. However, the conundrum of what happened to the fate of the AstraZeneca’s GSK3 inhibitors and the undertaking to find a therapeutic that could control glycogen metabolism and aberrant tau hyperphosphorylation in the brain, and rescue synaptic dysfunction has largely been untold. AstraZeneca was in the forefront of GSK3 drug discovery research with six GSK3 drug candidates, one of which progressed up to Phase II clinical trials in the quest to untangle the tau hypothesis for AD. Analysis of key toxicity issues, serendipitous findings and efficacy, and biomarker considerations in relation to safety margins have limited the potential of small molecule therapeutics as a way forward. To guide future innovation of this important target, we reveal the roller coaster journey comprising of two decades of preclinical and clinical GSK3 drug discovery at AstraZeneca; the understanding of which could lead to improved GSK3 therapies for disease. These learnings in combination with advances in achieving kinase selectivity, different modes of action as well as the recent discovery of novel conjugated peptide technology targeting specific tissues have potentially provided a venue for scientific innovation and a new beginning for GSK3 drug discovery.
ALZHEIMER’S DISEASE AND GSK3
Alzheimer’s disease (AD) is one of the most common forms of dementia and appears to increase exponentially with age [1]. The etiology of AD has many facets and only a very minor component is attributed to familial AD of genetic origin [2]. AD is characterized by a progressive loss of episodic memory and cognitive and behavioral dysfunction. One of the most affected brain structures is the entorhinal cortex hippocampus circuitry which plays a key role in memory acquisition and consolidation [3]. Impairments of these brain structures in AD are believed to underlie the impairments in memory that characterize this chronic neurodegenerative disease.
Glycogen synthase kinase 3 (GSK3) has been regarded as a critical molecular link between the two major histopathological hallmarks of the disease, extracellular plaques which are composed of the protein amyloid-β (Aβ) and neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein [4, 5]. GSK3 is a highly conserved protein-serine/threonine kinase that was first isolated from skeletal muscle as one of several enzymes that phosphorylated the enzyme glycogen synthase [6]. In mammals, GSK3 is encoded by two highly related genes encoding GSK3α and GSK3β, respectively; however in the brain, GSK3β isoform acts as a key switch that controls numerous signaling pathways [7]. The dysregulation of this kinase has been linked to the development of AD and related dementias, cancer, type 2 diabetes, schizophrenia, depression, and bipolar disorder. Given its relevance in pathophysiological processes, GSK3β is widely considered a therapeutic target of interest [8, 9].
GSK3 activity is regulated by phosphorylation of the Tyr279/Tyr216 residue which is important for enzymatic activity [10, 11]. In contrast, inactivation of GSK3 can be achieved through phosphorylation of Ser21/Ser9 residues within the N-terminal domain on GSK3, respectively [12]. GSK3 is also regulated upon interaction of the Wnt ligand, its receptor Frizzled and co-receptor LRP5/6. This interaction releases GSK3 from a multi-protein complex formed by β-catenin, axin, and adenomatous polyposis coli [13], which prevents GSK3-mediated β-catenin degradation and induces β-catenin– dependent gene transcription.
GSK3 phosphorylates the microtubule associated protein tau resulting in its hyperphosphorylation and subsequently paired helical filamentous tau (PHF-tau) formation, a key component of NFTs [14, 15]. In post-mitotic neurons, tau associates with microtubules and stabilizes their polymerization. In AD, increased GSK3β activity has been identified in postmortem AD brains [16]. Evidence indicates that the phosphorylated state of tau is closely associated with AD pathology [17] GSK3 phosphorylation sites on tau are believed to be abnormally phosphorylated in AD. Furthermore, Aβ40,42 induces the formation of tau fibrils resembling PHF-tau in culture [18], and is also thought to increase GSK3 activity. PHF-tau is deposited as an insoluble misfolded aggregate protein in the somatodendritic compartment in postmortem AD brain tissue [19], and it is highly resistant to the action of phosphatases and proteases as it is often truncated at the C-terminal domain. These studies suggest that the inappropriate activation of GSK3 in the AD brain could play a role in the pathophysiology of PHF-tau formation.
GSK3α was reported to regulate Aβ production by positively modulating the γ-secretase complex [20], although this area of research is still being debated [21]. Inhibition of GSK3 activity with nonspecific GSK3 inhibitors, such as lithium chloride and valproic acid in in vitro and in animal models of AD, has been shown to decrease Aβ production. More recently it was shown that specific inhibition of GSK3β, reduced BACE1-mediated cleavage of AβPP through a NF-κB signaling-mediated mechanism and consequently Aβ production by decreasing BACE1 gene transcription and expression [22]. This is an important finding since the expression level and activity of BACE1 is reported to be elevated in AD patients [23]. Furthermore, inhibition of GSK3 signaling reduced Aβ deposition and neuritic plaque formation, and rescued memory deficits in a double transgenic AD mouse model [43]. Given the role of GSK3 in PHF-tau and the subsequent link to Aβ production, it appears that GSK3 could act as a common molecular link between amyloid plaque pathology and NFT pathology in AD. This is an area of intense research and such hypotheses will still require substantial validation, both pre-clinically and in the clinic.
Synaptic loss is one of the best correlates of cognitive deficits in AD [24]. It has been proposed that the mechanism allowing information storage in the brain involves changes in synaptic plasticity, including long-term potentiation (LTP) and long-term depression (LTD). LTP inhibits GSK3β activity, and it is required for LTD, which indicates that the two phenomena are interrelated and that LTP regulates LTD [25]. In addition, within the synaptic compartment, tau and Presenilin 1 (a component of the γ-secretase complex that generates Aβ), may be additional targets for GSK3β. Specific overexpression of GSK3β in neurons causes a drastic decrease in postsynaptic density number and volume in hippocampal granule neurons [26], a phenomenon that could induce cognitive impairment and altered LTP production [27, 28]. Since Aβ is thought to induce synaptic toxicity [29], and GSK3β activation is required for the pathological effect of Aβ on synaptic plasticity, it is tempting to speculate that GSK3β inhibitors could protect synapses from the deleterious effects of Aβ [30].
THE EARLY GSK3 DRUG DISCOVERY YEARS
During an era where genetics pointed to amyloid targets as the future for drug discovery research in the attempt to cure AD, several pharmaceutical companies including Zeneca, Astra, Mitsubishi Tanabe, Bristol Myers Squibb, and others were debating whether testing the alternate abnormal tau hyperphosphorylation hypothesis was worth an investment. Several academic researchers had proposed that tau, a microtubule associated protein present in axons, was hyperphosphorylated in AD brains and this aberrant hyperphosphorylated tau did not bind effectively to microtubules leading to destabilization. Consequently, axonal transport could no longer proceed efficiently resulting in synaptic and cognitive dysfunction [31–33]. Based on cellular, transgenic mouse data and expression and phosphorylation profiles in AD brain, GSK3 was implicated as a major kinase in the aberrant hyperphosphorylation of tau leading to NFTs in AD [34, 35]. Accordingly, inhibition of pre-tangle pathology via GSK3 inhibition would be expected to slow down the progression of NFT formation and neurodegeneration in AD. In addition, given the evidence that GSK3 inhibitors might be able to suppress the production of glucose by the liver, as well as enhance its conversion into glycogen, GSK3 was also an important target for type 2 diabetes.
While several companies successfully identified small molecule GSK3 inhibitors [36], in the following article, we focus specifically on the quest to identify suitable orally available GSK3 inhibitors as a disease modifying therapy for AD from AstraZeneca. Embarking on a drug discovery project targeting GSK3 in 1997, two independent high throughput screening (HTS) campaigns; one at Zeneca and the other at Astra resulted in the screening of approximately 2 million small molecule chemical compounds. Following the merger of AstraZeneca in 1999, the two geographically separated projects were combined into one project in Sweden in July, 2000. From the HTS assay, six chemical series were selected. These were the oxindolequinazolines, anilinoquinazolines, thiazoles, pyrimidines, thiazolidinediones, and pyrazines. The pyrazines and oxindolequinazolines were pursued based on druggability, intellectual property and the potential to expand and optimize structural activity relationships [37], and subsequently AZD2858 (Fig. 1a) from the pyrazine series progressed through two species 28-day GLP toxicology studies, and AZD1080 (Fig. 1b) from the oxindolequinazoline chemical series was progressed through Phase I clinical trials for AD [38]. Some of the other selective GSK3 inhibitors [39] were abandoned due to poor physico-chemical and drug-like properties.
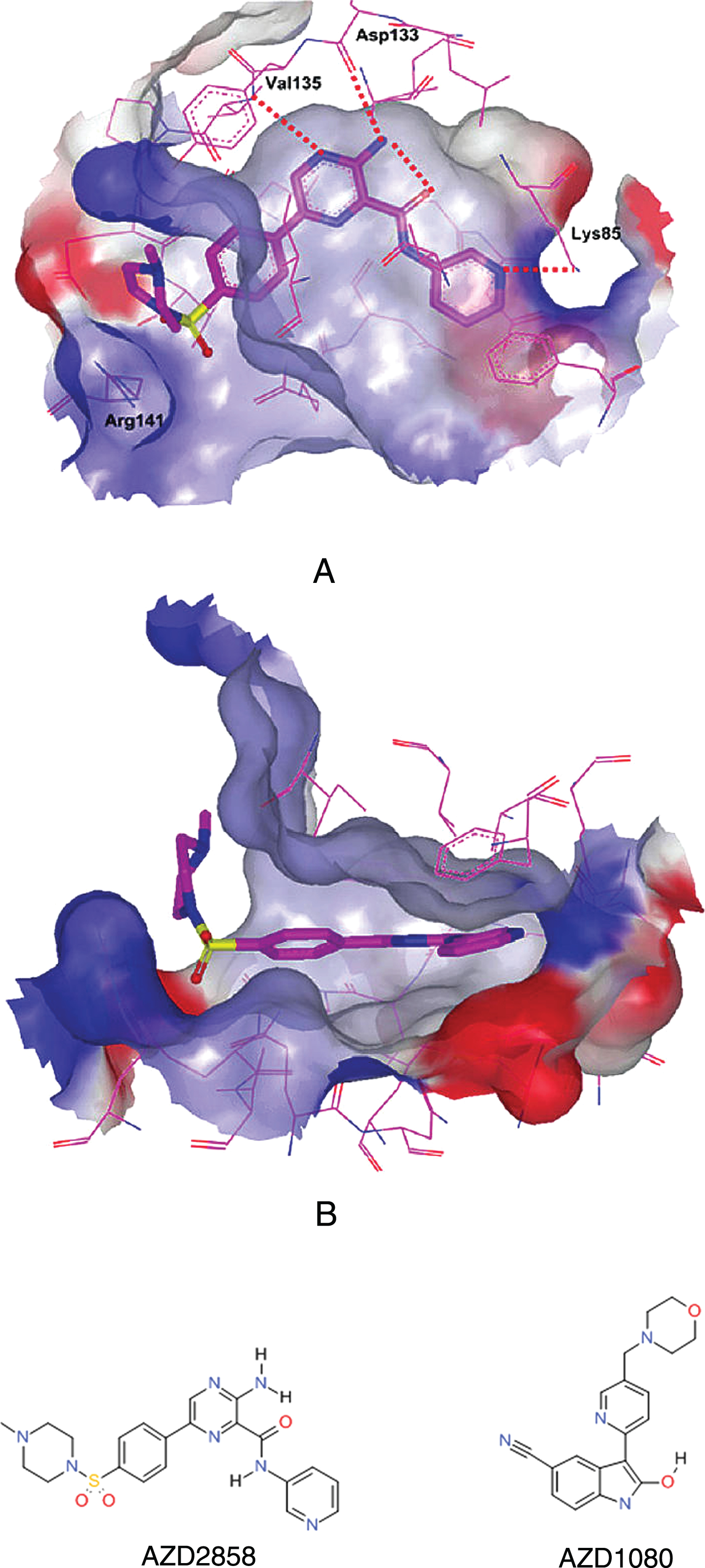
X-ray crystal structure of GSK3 inhibitor AZD2858 (1a) that progressed through 28 day GLP toxicology studies (A: top view; B: side view) in the ATP pocket of GSK3β and the Phase I clinical candidate AZD1080 (1b).
The first clinical candidate AZD2858, a highly potent (4.8 nM) and selective orally bioactive brain penetrant GSK3 inhibitor, emerged in 2003 with excellent drug-like properties [37]. AZD2858 demonstrated dose-dependent inhibition of tau hyperphosphorylation in rodent brain hippocampus and inhibition of gliosis, a marker of neurodegeneration in a transgenic model overexpressing GSK3. Furthermore, AZD2858 inhibited PHF-tau formation in cellular models.
THE GLASS: HALF FULL OR HALF EMPTY?
Since GSK3 is a master switch regulating cell fate specification, ranging from proliferation and differentiation to regulation of glucose homeostasis, we anticipated some challenges in preclinical toxicology studies. In the regulatory IND enabling toxicology studies for AZD2858, we unexpectedly found that AZD2858 caused a rapid and robust increase in bone formation, including thickening trabeculae and osteoblast proliferation, in rats and dogs. The effect was only partially reversible four weeks after discontinuation of AZD2858 administration. These findings led to the hypothesis that inhibition of GSK3 affects the Wnt signaling pathway, thereby causing proliferation in bone tissue [40]. Consistent with this hypothesis, two key downstream targets of the Wnt signaling pathway, β-catenin and cyclin D1, were shown to be increased in the femur, tibia, and femoro-tibial joint in rats treated with a high dose of AZD2858. These serendipitous findings raised the possibility that AZD2858 could be potentially used for the treatment of bone disorders such as osteoporosis, fractures, and bone loss during myeloid leukemia. Further investigative studies clearly demonstrated that Wnt activation by inhibiting GSK3 caused β-catenin stabilization in mesenchymal stem cells and stimulated commitment towards osteoblasts and osteogenic mineralization in vitro. Furthermore, GSK3 inhibition by AZD2858 led to time- and dose-dependent increases in bone formation biomarkers, as well as reductions in resorption biomarkers, indicating increased bone anabolism and a reduced bone resorption. Surprisingly, the resulting bone formation appeared normal and was resilient, as analyzed by histomorphometry and biomechanical testing [41, 42]. In alignment with this, GSK3 inhibition was able to drive direct bone repair in an unstable fracture milieu in rats. Unfortunately, the severity of the findings in the preclinical toxicology studies were not conducive to further development of this compound for chronic treatment for an AD indication. There were numerous other target organs identified (including the bile duct, see below) and the compound was also shown to be genotoxic (clastogenic) which ultimately prevented further progression to the clinic. In parallel, AstraZeneca embarked a new project focused solely on trying to identify GSK3 inhibitors that could mimic the beneficial effects of AZD2858 in osteoporosis and bone disorders, potentially with local applications during dental or orthopedic surgery, similar to how bone morphogenic proteins are used.
A SECOND ATTEMPT
The toxicity findings raised the possibility that we needed to minimally engage with the GSK3 target in vivo and inhibit its activity by approximately 15–20% upon which a stoichiometric balance in tau phosphorylation would favor microtubule stabilization in neurons, but not drastically inhibit basal cellular functions of GSK3 elsewhere. This resulted in a quest to identify a GSK3 inhibitor from a distinct chemical series that was slightly less potent albeit highly brain penetrant. In 2004, a second candidate drug, AZD1080 was identified which selectively inhibited GSK3 (Ki 30 nM), and had a two-fold higher brain to plasma ratio than the previous clinical candidate. AZD1080, had a more suitable pharmacokinetic profile, and was from a separate chemical series (oxindolypyridine), and it was able to mitigate the toxicological effects on the musculoskeletal system that had been identified with AZD2858. AZD1080 specifically binds to and inhibits GSK3β within the ATP pocket of the catalytic domain (Fig. 1b). The crystal structure showed that the inhibitor binds through three hydrogen bonds, to the backbone atoms of Val-135 (both the amide N and the carbonyl O), a residue located in the hinge/linker region alongside of the ATP-binding pocket of the enzyme (Figure 1b).
The importance of AZD1080 as a specific GSK3 inhibitor was validated by its capacity to interfere with tau hyperphosphorylation in vitro and in vivo. In cells and in rodent brain following in vivo administration, AZD1080 is extremely efficacious at inhibiting tau phosphorylation, thereby addressing one of the fundamental tau hyperphosphorylation hypothesis in a preclinical setting. AZD1080 inhibited tau phosphorylation in cells over-expressing tau protein in a dose-dependent manner. AZD1080 is a brain permeable small molecule compound which has favorable oral bioavailability and pharmacokinetic (PK) profile. The PK-pharmacodynamic (PD) analysis suggests that peak exposure in the hippocampus is within 1 h while the effect on tau phosphorylation inhibition peaks at 6 h and the effect remains up to 24 h. This suggested that a shorter frequency of dosing regimen may be required in the clinic [38]. The advantage of such a finding is that larger safety margins could potentially be derived. The reason for this prolonged effect is unclear but could be attributed to the tight regulation of phosphorylation and dephosphorylation events on the tau protein.
GSK3 inhibitors have also been reported to influence cognitive processes under certain conditions, specifically in impaired systems. AZD1080 prevented the disruption of LTP induction caused by acute treatment with the NMDA receptor antagonist, MK-801 while AZD1080 applied to brain tissue slices obtained from non-compromised animals had no significant effect on LTP. These studies suggest that AZD1080 reverses synaptic plasticity and functional deficits in a dysfynctional neuronal system and the efficacious effect is likely because of modification of pathways downstream of GSK3 [38].
In the Phase I clinical trial, AZD1080 exposures that result in peripheral inhibition of GS activity in rodent PBMC correlated well with that observed in the Phase I multiple ascending dose study. These results demonstrated that for the first time a selective GSK3 inhibitor such as AZD1080 had the ability to inhibit the GSK3 enzyme in humans [38]. AZD1080 was well tolerated and demonstrated peripheral target engagement in Phase I clinical studies in healthy volunteers. Unfortunately, the histopathological changes observed in the gall bladder in the dog after chronic dosing, progressed to chronic cholecystitis without any exposure margins to what was believed to be clinically relevant doses, and the severity of these findings eventually forced us to abandon Phase II clinical trials.
For the subsequent programs, we tried yet another chemical series where we again identified biliary hyperplasia in rat as dose limiting toxicity, even though optimization efforts targeted increased fractional excretion through renal pathways. With the expectation that the margins to the biliary findings would erode, we explored two additional backup clinical candidates from separate chemical series, and initiated problem solving activities to understand drivers for the biliary toxicity. The physicochemical properties of these compounds in combination with the unique pharmacodynamics effects of GSK3 inhibition ultimately proved to be an unsurmountable hurdle with respect to preclinical toxicity.
DRUG TARGETING TO RELEVANT TISSUE: A NEW BEGINNING?
The ubiquitous expression of GSK3 in many tissues and organs has led to significant toxicological challenges following oral dosing and systemic exposure to the GSK3 inhibitors, ultimately leading to the failure of many GSK3 inhibitors either in the clinic or prior to their progression. However, GSK3 inhibitors are still currently pursued in clinical development. An example is Tideglusib (AMO-02), an inhibitor of GSK3β currently in clinical trials at AMO Pharma for the treatment of myotonic dystrophy. Nevertheless, as inhibition of GSK3 is such a powerful biological mechanism with the potential to positively impact many diseases, the challenge of how GSK3 inhibitors can be progressed as a potential therapeutic remains enigmatic.
One strategy to circumvent the issue of achieving therapeutic concentrations while limiting off-target effects is to specifically target the GSK3 inhibitor to the organ or even cells of interest. This can be achieved by, for example, conjugating a GSK3 inhibitor to a moiety which has a preference to bind to the target organ or cells, or encapsulating the GSK3 inhibitor in a drug delivery system which preferentially binds to the required organ or cells. This has recently been shown by conjugating a GSK3 inhibitor to an aspartic acid octapeptide which is known to adsorb to hydroxyapatite, the mineral portion of the bone, hence giving a molecular conjugate which targets bone fractures. In addition, the conjugated molecule assembles into micelles, which extends its circulation time while maintaining its fracture-targeting abilities. Another recent example has been reported43 using a bone-targeted nanoparticle (NP) for delivery of a GSK3 inhibitor [43]. The NPs were functionalized with a peptide with high affinity for tartrate-resistant acid phosphatase (TRAP) to achieve enhanced delivery to fractured bone. The TRAP binding peptide nano particles (TBP-NPs) showed improved accumulation at fractured bone as well as uptake in regenerative cell types such as mesenchymal stem cells and osteoblasts in vivo in mice. Thus, the approach of loading GSK3 inhibitor in TBP-NPs has the potential to enhance bone regeneration with improved therapeutic window using systemic delivery which is not currently possible.
Similar applications are being pursued in the field of metabolic diseases. Inhibition of GSK3, including specific GSK3β-cell knockouts, have been reported to increase proliferation of β-cells, leading to an increase in β-cell mass and improved glycemic control. One possibility is to direct a GSK3 inhibitor to the pancreatic β-cells via linking to an antibody, peptide, or a separate small molecule which targets a receptor or uptake mechanism, which is expressed specifically on β-cells (see Fig. 2). This strategy will allow the exploitation of the powerful and disease modifying action of GSK3 inhibition via a cell specific mechanism. Similar approaches have recently been explored to limit the systemic exposure of kinase inhibitors and thus improve their therapeutic index and/or restrict their activities to the cell type of interest. This should enable their application outside the oncology field. Novartis researchers have for example identified dual GSK3-DYRK1 inhibitors as inducers of pancreatic β-cell proliferation [44]. To assess the potential of this series to expand beta cell mass while maintaining beta cell function and to limit the risk of nonspecific proliferation of other cell type, they subsequently designed a delivery system by linking the dual kinase inhibitors to (+)-dihydrotetrabenazine derivatives [45]. (+)-Dihydrotetrabenazine is a ligand of vesicular monoamine transporter 2 and its derivative have been used as beta cell imaging agent. Although this conjugation strategy proved to positively impact biodistribution toward pancreatic accumulation, it negatively impacted the proliferative potency of the resulting conjugates thus requiring further optimization and underlining the requirement of highly potent cargos. Another recent example is the design of an antibody drug conjugate to deliver dasatinib, a very potent but unselective Bcr-Ab1 tyrosine kinase inhibitor and Src family kinase inhibitor, to human T lymphocytes by targeting the CXCR4 receptor [46]. These conjugates were shown to deliver dasatinib selectively to human T cells in vitro and to possess excellent in vitro immunosuppressive activity. These recent examples highlight the potential of using molecular conjugates to restrict the activity of potent kinase inhibitors to a specific target cell type and to improve their therapeutic index. An additional challenge when applying these these molecular Trojan horse approaches to GSK3 inhibition for an AD purpose, would be the need for the conjugates to cross the blood-brain barrier (BBB) to achieve exposure. This is, however, an intense area of research, and screening for brain penetrant antibodies or optimization of fusion proteins for BBB penetration have been reported. Moreover, for less complex small molecule drug and peptide drug conjugates, the use of an additional component enabling BBB could be envisaged in the form of, for example, a Transferrin receptor or GLUT1 transporter ligand.

Diagram showing drug targeting approach via homing moiety to the specific region (e.g., neurons, beta pancreatic cell), linker chemistry (green) and a GSK3 inhibitor (AZD2858)
In addition, understanding and achieving kinase selectivity, for example, via allosteric inhibition or targeting unique kinase conformations, and both discovering and introducing polypharmacology into a molecule (for example, through the re-emergence of phenotypic screening), has advanced significantly in recent years. We believe that combining appropriate kinase selectivity, polypharmacology, and targeting of the inhibitor to the cell type of choice will provide novel treatment options for GSK3 inhibitors in AD, metabolic disease and beyond. The beneficial effects of GSK3 inhibition has been demonstrated in a wide range of in vitro, cellular, and in proof-of-concept preclinical studies [4, 48]. However, this data needs to be translated both from an efficacy and safety standpoint to the clinic [49].
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-9934).