
Abstract
Background:
The Religious Orders Study and Rush Memory and Aging Project are both ongoing longitudinal clinical-pathologic cohort studies of aging and Alzheimer’s disease (AD).
Objectives:
To summarize progress over the past five years and its implications for understanding neurodegenerative diseases.
Methods:
Participants in both studies are older adults who enroll without dementia and agree to detailed longitudinal clinical evaluations and organ donation. The last review summarized findings through the end of 2011. Here we summarize progress and study findings over the past five years and discuss new directions for how these studies can inform on aging and AD in the future.
Results:
We summarize 1) findings on the relation of neurobiology to clinical AD; 2) neurobiologic pathways linking risk factors to clinical AD; 3) non-cognitive AD phenotypes including motor function and decision making; 4) the development of a novel drug discovery platform.
Conclusion:
Complexity at multiple levels needs to be understood and overcome to develop effective treatments and preventions for cognitive decline and AD dementia.
Keywords
Introduction
For more than a century, careful clinical characterization followed by examination of neural tissues after death has been an important approach for identifying the neuropathologic determinants of dementia [1, 2]. The vast majority of older adults studied in clinical-pathologic studies are recruited at tertiary care dementia centers [3, 4]. In the early 1990 s, community-based cohort studies of aging and dementia started obtaining autopsies. This is important as autopsies from community participants differ from autopsies of individuals evaluated at dementia centers [5]. The first community-based studies were the Nun Study, the Honolulu Asia Aging Study (HAAS), and the Hisayama Study [6–8]. Participants in the Nun Study were over age 75 at entry, and all agreed to organ donation; however, it was not explicitly designed as a study of risk factors for incident AD dementia. Both HAAS and the Hisayama Study were population-based studies of risk factors for AD dementia. Both added organ donation for the relatively small number that agreed. Later, other community-based studies in the USA and Europe started to obtain autopsies [9].
The Religious Orders Study (ROS) and Rush Memory and Aging Project (MAP) began in 1994 and 1997, respectively (together referred to as ROSMAP). They are both cohort studies of risk factors for cognitive decline and incident AD dementia, and other health outcomes. They share all essential attributes of analytic cohort studies, and both require an agreement for organ donation as a condition of study entry. The last review of these studies summarized findings through the end of 2011 [10, 11]. Here, we describe the current datasets, summarize progress and study findings with emphasis on results from 2012 through 2017, and contextualize the findings with the other advances in the field. As ROSMAP serves as a resource for the aging and dementia research community, we hope the review will orient potential users of the resource to the wealth of data and findings which can be leveraged for future studies.
Materials
Participants
ROS started in 1994 and enrolls nuns, priests, and brothers from across the US. MAP started in 1997 and enrolls lay persons from across northeastern Illinois. Evaluations are annual and all participants in both cohorts are organ donors. This includes brain, spinal cord, nerve, and muscle for those autopsied at Rush (Illinois, southeastern Wisconsin, and northwestern Indiana), and brain only for those autopsied elsewhere (California, central Illinois, central Indiana, Iowa, Kentucky, Louisiana, Maryland, Minnesota, Missouri, New York, Ohio, Pennsylvania, Tennessee, Texas, Washington DC, central and western Wisconsin). All MAP and a few hundred ROS donate blood annually. A large common core of data is shared by both studies allowing efficient merging of data. The two studies support additional sub-studies that address a wide range of other aspects of aging. Many sub-studies are restricted to MAP as nearly all participants are within driving distance of Rush and it is easier for staff to assess them more frequently. The parent studies and sub-studies were all approved by an Institutional Review Board of Rush University Medical Center and all participants signed an informed consent, Anatomical Gift Act, and a repository consent to share data and biospecimens.
Through December 31, 2017, the studies enrolled 3,414 persons of whom 72.6% are female, 88.2% are non-Latino White, 6.3% are African American, 5.5% are Latino (including African American Latinos), and the remainder are other racial groups. Their mean age was 78.3 years and education 16.9 years, and blood was collected from 94.3% . There have been 1,232 cases of incident mild cognitive impairment (MCI) and 764 cases of incident dementia, and only 7.8% have withdrawn. There have been 1,717 deaths and 1,506 (87.7% ) brain autopsies and 834 spinal cord, nerve, and muscle autopsies. Of those autopsied, 67.2% are female, 94.8% are non-Latino White, and the remainder were members of other racial groups. Their mean age was 89.1 years and mean education is 16.9 years. Of those autopsied, 31.0% were without cognitive impairment, 23.0% had MCI, 41.4% had AD dementia with or without another condition, and the remainder had another cause of dementia.
The layers of data now available (or currently being generated) in one or both cohorts are illustrated in Fig. 1. These are documented in the Rush Alzheimer’s Disease Center Resource Sharing Hub (http://www.radc.rush.edu). The Hub also includes all information and links required to request data and biospecimens, including downloadable data use and material transfer agreements. The Hub automatically updates with ongoing data collection and is actively expanded when new data is available for sharing.
Potential risk factors

Multi-layered omics, neuropathologic, and clinical data in ROSMAP.
Both studies collect a wide range of exposure data that includes genomic, experiential, psychological, and medical risk factors [12–46]. This includes continuous daily recordings of physical activity with an omnidirectional accelerometer [47]. From these recordings quantitative metrics of physical activity, sleep and circadian rhythms are extracted [48]. We administer the Food Frequency Questionnaire [49]. Genome-wide data has been generated [50], and we recently generated whole genome sequencing.
Multi-level omics
Several additional layers of brain and blood molecular genomics were generated. Data generated from the dorsolateral prefrontal cortex, include DNA methylation, H3K9Ac, miRNA, and RNAseq. We are currently generating 5hC methylation, another histone mark, proteomics and metabolomics from the same region, plus RNAseq and DNA methylation from other brain regions. Proteomic and metabolomic data is also being generated on blood, DNA methylation from CD4+ lymphocytes, and RNAseq from monocytes. Finally, we are in the process of establishing 50 iPSC lines from participants.
Neuropathologic and neurobiologic traits
A wide range of neuropathologic traits are generated. These include quantitative measures of AD pathology by histochemistry and immunohistochemistry, and Braak Stage, NIA-Reagan, and NIA-AA pathologic criteria for AD [51–55]. Other measures include macro- and microscopic infarcts, athero- and arteriolarsclerosis, amyloid angiopathy, Lewy bodies, TDP-43, hippocampal sclerosis, and (on subsets) activated microglia and white matter pallor. Arteriolar sclerosis as well as AD pathology and Lewy bodies are also recorded in the spinal cord [56–61]. This is complemented by measures of resilience including presynaptic proteins and neuron density [62]. We also are generating data on targeted proteomics.
Structural and functional neuroimaging
Antemortem 3D MPRAGE, diffusion weighted imaging, 2D fast spin echo, 2D FLAIR, QSM, and resting state functional MRI is done on a subset of participants [63]. We also perform ex vivo imaging in many cases both fresh and fixed [64, 65].
Quantitative clinical phenotypes
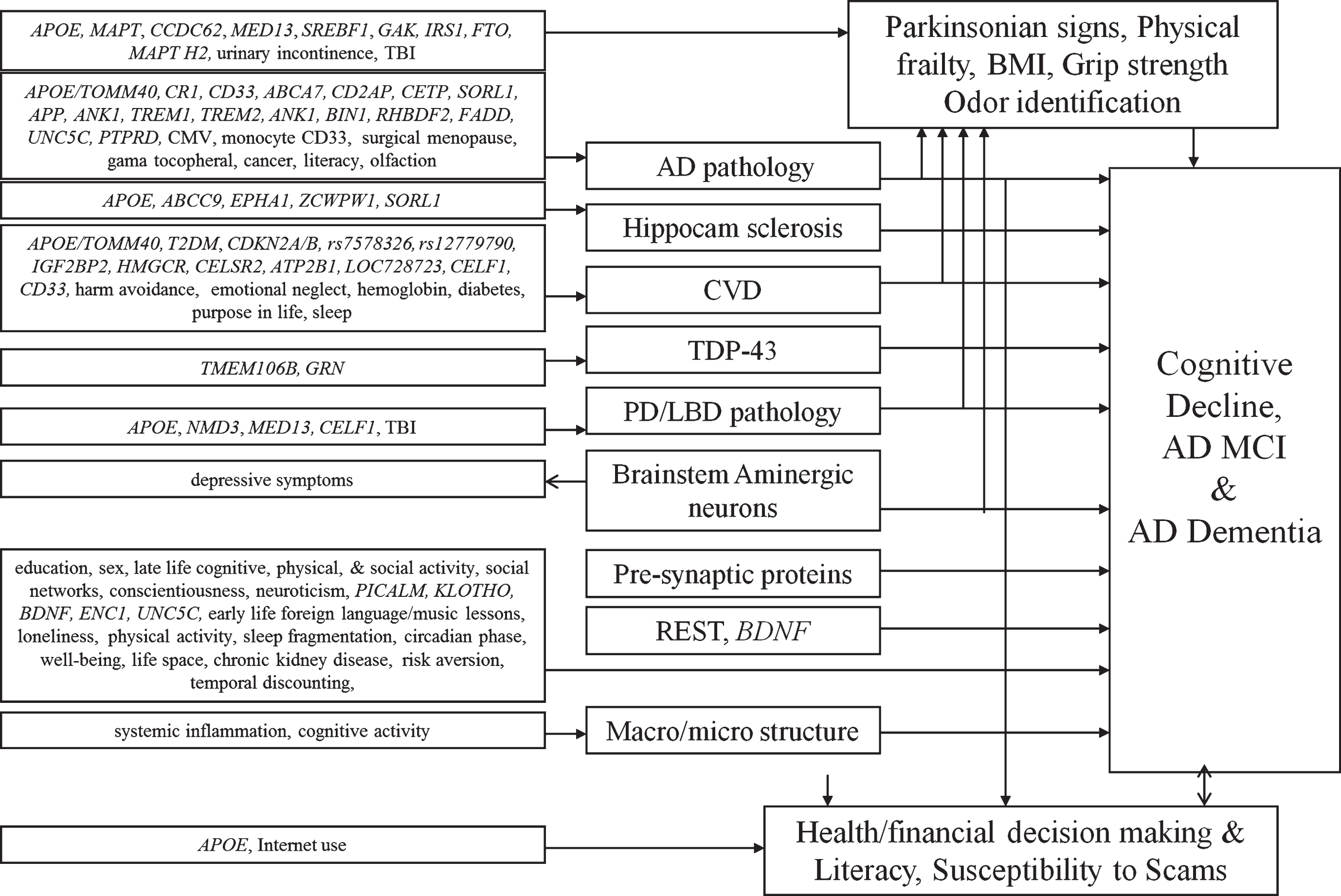
Neurobiologic pathways linking risk factors to AD clinical phenotypes.
Twenty-one cognitive performance tests with 19 in common, 17 of which are summarized as measures of global cognition, and scores of episodic memory, semantic memory, working memory, perceptual speed, and visuospatial ability [15, 66–68]. Parkinsonian signs summarized as a continuous measure of parkinsonism, and domains of gait, bradykinesia, rigidity, and resting and/or postural tremor, and a categorical measure of parkinsonism [69–72]. Other motor performance tests include quantitative measures of upper and lower limb performance [42, 73–77]. Since 2012, annual gait testing now includes a body sensor with a triaxial accelerometer with three gyroscopes [78–82].
We also have studies of behavioral economics, decision making, and related behaviors in MAP. This includes measures of risk aversion and temporal discounting, health and financial decision making, health and financial literacy, and susceptibility to scams and fraud victimization, and related psychological measures (e.g., purpose in life) [40, 83–86].
Syndromic clinical phenotypes
Clinical diagnosis of dementia, especially AD dementia, and MCI are documented [87, 88]. We also make diagnoses of stroke and vascular cognitive impairment, Parkinson’s disease (PD), and depression [89–91]. Other diagnoses are made by history and examination of medications. Diagnoses are rendered annually and a final diagnosis prior to death is generated after review of all data blinded to neuropathology. For some participants, we have linkages to Medicare data.
Results
Several themes have dominated our work over the past six years. One is the relation of neuropathologic and resilience indices to cognitive decline, MCI, and AD dementia. Second are the neurobiologic pathways linking risk factors to cognitive decline, MCI and AD dementia. A summary of these associations are illustrated in Fig. 2. Third is a comparable portfolio centering on motor structure and function including parkinsonism, and a fourth on behavioral- neuro-economics and decision making. Finally, we describe our emerging novel drug discovery pipeline.
Due to the large number of annual assessments over so many years, we calculated the attributable risk of death due to incident AD dementia [92]. Time from incident AD dementia to death was less than 4 years with a hazard ratio of more than 4. Upweighted to the US population resulted in an estimated 500,000 deaths attributable to incident AD dementia in 2010 putting it on par with cancer and heart disease.
Relation of neuropathology and resilience indices to cognitive decline, MCI, and AD dementia
We demonstrated that mixed pathologies were the most common cause of AD dementia with AD pathology including amyloid angiopathy, several indices of cerebrovascular disease, including macro- and micro-infarcts, atherosclerosis, arteriolarsclerosis, Lewy body disease, TDP-43, and hippocampal sclerosis all having additive effects on the odds of AD dementia [93–102]. Some pathologies, such as atrophy and white matter changes, were assessed with in vivo or ex vivo imaging and were also separately related to dementia [103–110].
A dozen years ago we first reported that pathologic AD was present in about a third of older persons without dementia or MCI and that it was related to episodic memory [55]. In a follow-up we showed that neocortical amyloid-β, mesial temporal PHF-tau tangles, and macroscopic infarctions were all related to episodic memory and amyloid-β related to working memory in persons without cognitive impairment [111]. We recently extended this work showing that TDP and hippocampal sclerosis are associated with cognitive impairment in persons without pathologic AD, similar to earlier findings with AD pathology, Lewy bodies, and infarcts [101].
An important feature of the study design, the repeated measures of cognition, permits one to examine the relation of pathologies to the trajectory of cognitive decline over up to a quarter century prior to death. Using several approaches including latent variable models, change point models, Markov chain models, and sigmoidal models we find that the effects of neuropathology on cognitive decline emerge many years prior to death [112–123]. These data further illustrate the continuum of the clinical AD phenotype that seamlessly evolves from normality, to minimal then mild cognitive impairment, and eventually to dementia. Further, they show that effects of pathology, including hippocampal volume, measured at death are related to cognitive changes many years prior to death, including in persons who died without dementia [120, 124].
Using a Markov chain model, we illustrated the effects of multiple pathologies on the “horserace” between dementia and death [121]. Without any pathology there is nearly a 20% likelihood of cognitive impairment prior to death. By contrast, with AD, infarcts, and Lewy body disease, the risk triples to nearly 60% . We recently reported nearly 250 different unique combinations of pathologies accounting for cognitive decline in just over 1000 persons [122]. The most common, pathologic AD alone was less than 10% . Nearly 100 people had a combination that was not present in any other person. Further, the magnitude of the effect of each pathology on cognitive decline varied widely depending on the specific combination present.
Interestingly, we found that when we link common neuropathologies to cognitive decline, we explain less than half of the person specific differences in slopes [124, 125]. This likely results from several factors. First, neuropathologies are neither measured perfectly nor completely, and downstream effects of measured pathologies are only captured on a subset of participants [105–108]. In addition, we are not measuring all of the brain pathologies known to be associated with the named diseases. For example, we only have soluble pathologies on a small subset of participants [126–131]. Further, it is likely that new associated pathologies will be discovered in the future.
However, another important factor is neural reserve or resilience. We define resilience as a continuous (latent) variable defined as cognitive decline not explained by extant pathologies, i.e., residual cognitive decline [132]. When viewed in this way, every person has some resilience. However, one can be more or less resilient relative to the average person and therefore have a slower or faster rate of residual cognitive decline. We found several genomic and neurobiologic indices of resilience were associated with a slower rate of decline including presynaptic proteins, neuron density, and BDNF expression [122, 132–137]. We are also finding genes associated with a faster rate of cognitive decline [138].
Neurobiologic pathways linking risk factor to cognitive decline, MCI, and AD dementia
We first examine change in cognition and show that cognitive decline in African Americans and Latinos in our cohorts was similar to whites [139–141]. We also conducted a series of change point models in persons who developed AD dementia and showed that change in cognition began years prior to onset of AD dementia, and among persons who developed MCI, change in cognition began long before diagnosis [142]. Next, we summarize four sets of risk factor associations.
Genomic risk factors
We examined the effects of the TOMM40 haplotypes to cognitive decline, incident AD, and neuropathology [143–146]. Due to its strong linkage disequilibrium with APOE, we restricted one analysis of Caucasians to persons with APOEɛ3/3 genotype and found that both ’523-L and ’523-S/S S/S poly-T genotype were related to faster cognitive decline, especially episodic memory, a finding similar to APOE4 [146]. In another study we examined racial differences and found that among Caucasians nearly all APOE4 carriers had ’523-L whereas less than half of the African Americans had this haplotype [144]. In African Americans, the ɛ4-’523-L haplotype had stronger effect on risk of AD dementia than other APOE4-’523 haplotypes. This contrasts with the effects of APOE4 among African Americans which is much weaker than in Caucasians [147]. Interestingly, the effect of the ’523-L poly-T genotype was attenuated and no-longer significant controlling for AD and other neuropathologies, again similar to what we found for APOE4 [148–151]. By contrast, ’523-S/S S/S association with unchanged in analyses with neuropathologies suggesting that the two haplotypes work via different pathologic mechanisms.
We also examined in more detail are several single nucleotide polymorphisms (SNP) that emerged from prior genome wide association studies (GWAS) [152]. We found that CR1, SORL1, and CD33 were all associated with cognitive decline, AD pathology and amyloid angiopathy [153–159]. CD33 also modulated TREM2 in monocytes [156]. When examining all genomic variants from prior GWAS, we found some were associated with AD pathology but as a result of mixed pathologies and resilience, others were associated with co-morbid pathologies (i.e., ZCWPW, SORL1, and APOE with hippocampal sclerosis, CELF1 with Lewy bodies and microinfarcts, and ABCA7 with macroinfarcts), and some were not associated with any pathology [160]. We used DNA methylation to delve further into the known genomic variants and found associations between DNA methylation in several AD genes [161–163].
We found other genomic variants associated with cognitive decline and AD, and some associated with other pathologies or with no pathologies [164–172]. We also used GWAS to identify genomic variants associated with resilience and found two genes, ENC1 and UNCSC, that also showed evidence with DNA methylation and expression [162]. Interestingly, we also found that UNCSC was associated with amyloid angiopathy [173]. We did not find evidence of an association of the fragile X permutation expansion with cognition [174]. We also conducted GWAS for neuropathologic traits [175, 176]. Finally, we identified a variant in TMEM106B and expression of GRN associated with TDP-43 [177].
Experiential risk factors
Using change-point models, we found that education was associated with better cognition, a slower rate of cognitive decline and a delayed change point but a more rapid rate of decline after the change point [142]. Further, life-time cognitive activities as well as foreign language and music instruction were associated with a slower rate of cognitive decline including among Latinos [178, 179]. To address the potential for reverse causality, we used a cross-lagged model to show that cognitive activities initially predicts cognitive decline but as cognition becomes poor, cognition predicts decline in late-life activity [180]. Further, late-life cognitive activity was not related to common neuropathologies [181]. However, it was related to brain microstructure by neuroimaging, which partially mediated the association of cognitive activity with level of cognition [182]. We also found that total daily physical activity measured by actigraphy was associated with risk of AD dementia [183] and negative social interactions were associated with incident cognitive impairment [184].
We also found that both the DASH and Mediterranean diets were associated with a slower rate of cognitive decline [185]. We created the MIND diet which combines elements of the other two diets and found a stronger association with cognitive decline and AD dementia risk [186, 187]. Green leafy vegetables and seafood were individually associated with cognitive decline, the latter driven by consumption of foods high in long-chain omega-3 fatty acids [188–190]. Interestingly, in matched plasma and brain samples, we found lower levels of oleic acid isomers and omega-3 and omega-6 fatty acids as well as oleic acid in AD plasma [191]. By contrast, we only found lower docosahexaenoic acid (DHA) in brain. Interestingly, we found that fish consumption was associated with measures of AD pathology [192]. This is one of very few non-genomic factors that we found directly associated with measures of AD pathology. The finding was restricted to those with APOE4, but that could result from greater power. Higher α-linolenic acid (18:3 n-3) was associated with fewer cerebral macroinfarctions. Finally, we found that γ-tocopherol concentrations were associated with less AD pathology [193].
Psychological risk factors
We previously showed that depressive symptoms were associated with risk of AD dementia and did not change as AD dementia developed suggesting that the association is not reverse causality [13, 35]. Recently, we controlled for neuropathology and showed that it did not influence the association, nor were depressive symptoms a consequence of typical pathologies that cause dementia [194, 195]. Interestingly, lower density of dopamine neurons in the ventral tegmental area was associated with more depressive symptoms [196]. We previously found that rate of cognitive decline increases several fold about four years prior to death, a concept referred to as terminal decline [197, 198]. We found that conscientiousness was related to a slower rate of terminal decline and that this trait attenuated the association of Lewy bodies with terminal decline [199].
We found that cognitive decline was associated with several aspects of reduced well-being, or eudaimonic happiness [200]. One aspect of well-being, purpose in life, was associated with risk of AD dementia and modified the relation, of pathology to cognitive decline [201]. It was also associated with reduced odds of cerebral infarctions [202], as well as with reduced hospitalization [203]. By contrast, childhood emotional neglect and harm avoidance were both associated with increased odds of cerebral infarction [204, 205]. Further, neuroticism modified the association of vision with cognition [206]. Finally, loneliness was associated with AD risk and cognitive decline, but not with neuropathologies [30]; and we identified numerous genes in the amygdala and the dorsolateral prefrontal cortex related to loneliness [207, 208].
Medical factors
We first examined cerebrovascular disease factors. Lower body mass index (BMI) was related to cognitive decline in both African Americans and whites [209]. Also, lower hemoglobin was related to macroscopic infarcts [210]. We did not find associations of antiphospholipid antibodies to any measure of cerebrovascular disease or of genetic variants associated with homocysteine to be associated with any pathology [211, 212]. Diabetes was associated with subcortical macroscopic infarcts [213]. Interestingly, we found that insulin resistance in brain was related to measures of AD pathology [214]. We found that initiation of anticholinergic medicine had a negative impact on the slope of cognitive decline [215]. Also, antibodies to cytomegalovirus were related to cognitive decline and AD dementia in both African Americans and whites and was associated with measures of AD pathology [216, 217]. Better odor identification on a smell test was positively associated with cognition, and worse scores were associated with loneliness and depressive symptoms [218]. When we examine the anterior olfactory nucleus, we find co-localization of amyloid-β, PHF-1, and cCaspase-6, and the level of PHF-1 and caspase-6 were positively correlated [219]. In two other papers we found that surgical menopause was related to cognitive decline and neurofibrillary tangles, and history of cancer was associated with a lower likelihood of AD dementia and PHF-tau tangles [220, 221]. Finally, in an in vivo imaging study restricted to persons without dementia, we found that c-reactive protein and tumor necrosis factor-alpha were associated with cognition and brain microstructure [222]
Using data generated with the accelerometer, we developed a metric of rest-activity fragmentation as a proxy for sleep fragmentation [48, 223]. This measure was related to cognition and incident AD dementia, as well as lower cortical gray matter volume in the inferior frontal gyrus pars orbitalis and lateral orbitofrontal cortex [224, 225]. It modified the relation of APOE4 to measures of AD pathology and was directly associated with measures of cerebrovascular disease [226, 227]. The same data was used to generate circadian rhythms. We found inter-daily variability associated with the metabolic syndrome [228]. Further, we investigated the influence of several clock genes, using genomic, epigenomic, and transcriptomic data on circadian and seasonal rhythms [229–233]. Separately, we found that sleep fragmentation and circadian rhythm disruption were related to neuron counts in the ventrolateral preoptic/intermediate nucleus of the hypothalamus, and the suprachiasmatic nucleus [229].
Risk factors, neuropathology, and motor structure and function
Parkinsonism was progressive and associated with adverse health outcomes [183, 230–233]. Changes in motor structure and function were strongly correlated with changes in cognition and both related to the same neuropathologies. Parkinsonism was associated with risk of death, MCI, and AD dementia, and common brain pathologies were related to parkinsonian signs, and progression of physical frailty, respiratory function, and cognitive decline [234–239]. Further, neurons in the locus coeruleus were related to parkinsonian signs and to cognition [134, 240]. Thus, it was not surprising that many risk factors for cognitive decline and AD dementia are also risk factors for motor outcomes including physical and social activity, social isolation, neuroticism, harm avoidance, extraversion, and antihypertensive medications [232, 241–244]. By contrast, traumatic brain injury was related to progression of parkinsonism and PD pathology but not change in cognition or AD pathology [245]. Sleep, was also associated with motor outcomes in both African Americans and Caucasians, and with PD pathology [246–248]. We also conducted a candidate SNP analysis examining PD risk alleles with a variety of motor clinical and pathologic phenotypes [249]. We are just beginning to explore the spinal cord examining the distribution of α-synuclein, atherosclerosis, white matter pallor, and their association with brain pathology and motor function [259–261]. Recently, we added a body sensor, a triaxial accelerometer with 3 gyroscopes, which participant’s wear on a belt, which continuously records 3 acceleration and 3 angular velocity signals during annual gait testing. We examined the metrics derived from these recording during several gait and balance tests and their relation to IADL, parkinsonism, and physical activity [78–82]. Finally, using resting state fMRI we interrogated connectivity in relation to chronic musculoskeletal pain [250]. In a separate study, we found that physical activity modified the relation of white matter hyperintensities with motor function [251].
Behavioral- and neuro-economics and decision making
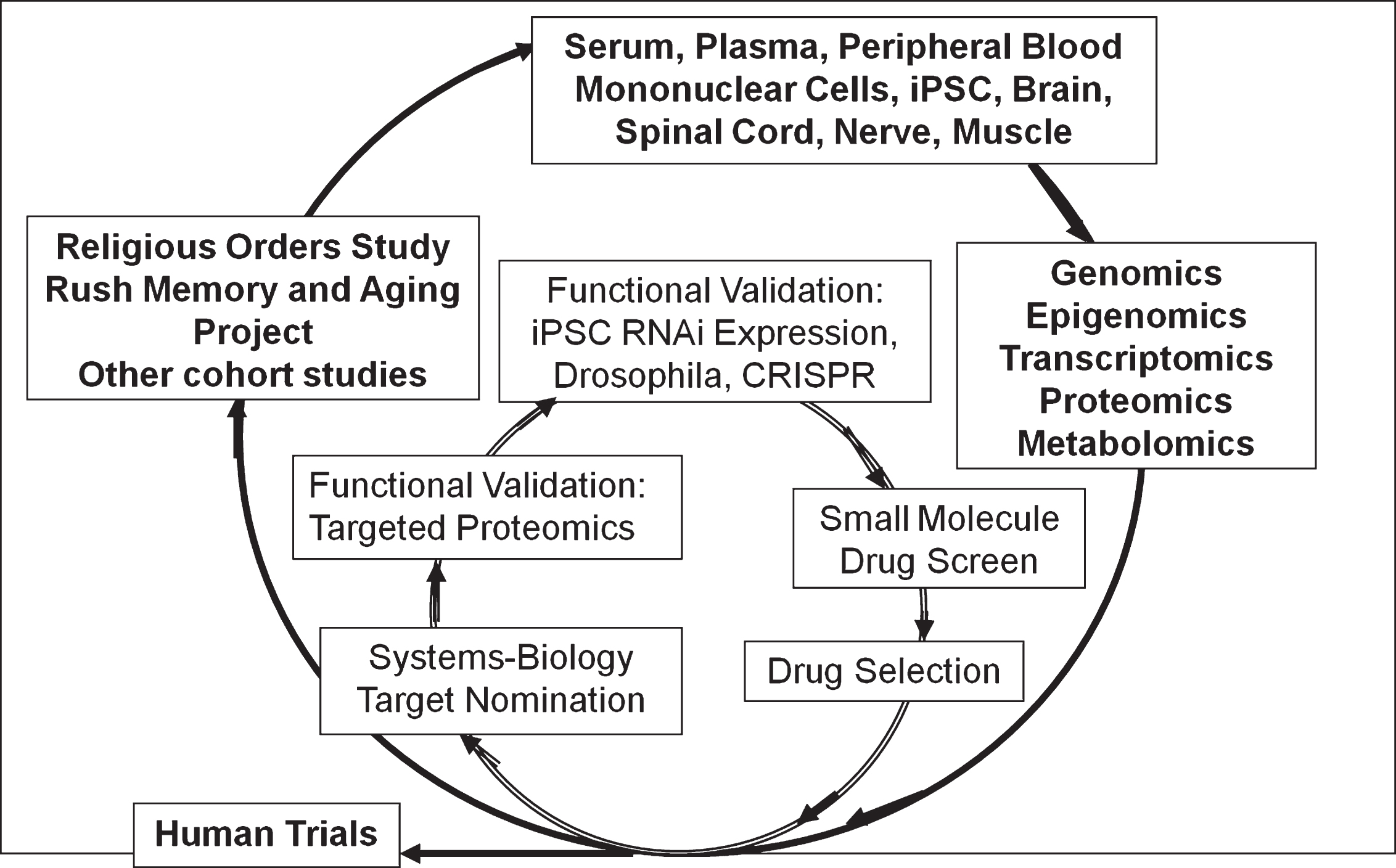
Cohort studies generating ante- and postmortem biospecimens which are used to generate multi-layered omics data. These data feed a systems biology computation pipeline for therapeutic target nomination. There are two stages of functional validation, one with targeted proteomics using brain tissue from the same cases and the other with a variety of high throughput ex vivo models. High value targets will then move to small molecule drug screen and eventual drug selection.
We first examined health and financial decision making. We found both associated with risk of death, incident MCI and AD, and cognitive decline among persons without dementia. Further, several factors help maintain decision making, including literacy and access to resources (e.g., internet use) [252–257]. Next, we examined health and financial literacy. These also are associated with cognition, MCI, functional status, mental health, health promoting behaviors, and APOE4 [258–260]. Literacy is both a consequence of cognitive decline and a predictor of future cognitive decline and incident AD dementia [261–264]. Interestingly, AD pathology was associated with literacy controlling for cognition [264]. Among persons without dementia, we found that higher diffusion anisotropy was associated with better financial literacy, especially tracts connecting right hemisphere temporal-parietal brain regions [265]. Financial literacy was also associated with greater functional connectivity between the posterior cingulate cortex and the right ventromedial prefrontal cortex, the left postcentral gyrus, and the right precuneus, and negatively associated with functional connectivity with left caudate [266]. Greater temporal discounting was associated with increased risk of death, cognition, and cognitive decline [267–269]. Discounting also was positively associated with functional connectivity to the right middle temporal regions and ventromedial prefrontal cortex, and negatively associated with parahippocampal and right cerebellar regions [270]. Risk aversion was associated with decision making and cognitive decline [269, 271]. Using a seed in the anterior cingulate, we found that risk averse persons had greater connectivity to clusters within multiple brain regions (e.g., insula, inferior and orbital, frontal, parahippocampal), and those low in risk aversion had greater connectivity to numerous clusters (e.g., inferior temporal, superior, middle, and medial frontal regions) [272]. We also reported that susceptibility to scams was negatively associated with cognition, well-being, and literacy, MCI, and cognitive decline [85, 274]. There was also an inverse association between overall grey matter and susceptibility to scams [275]. Finally, cognitive decline and over-confidence in one’s financial knowledge was associated with fraud victimization [85].
Novel drug and biomarker discovery pipeline
The multilayer omics data are now being used to support a novel drug and biomarker discovery pipeline as part of the Accelerating Medicines Partnership-AD (Fig. 3) [276, 277]. We are still at the early stage of generating omics data, performing the quality control, and developing a basic understanding of relationships with AD quantitative endophenotypes with epigenomic and transcriptomic data [278–286]. We are just beginning to examine relations of omic between brain and blood and brain and neuroimaging as part of our nascent biomarker discovery protocol [287, 288]. This will be complemented in the future with multiple layers of blood omics that can be related to antemortem imaging and brain omics. Further, we are still refining our ex vivo validation approaches [289, 290]. A forward-looking framework has been developed and we will focus on neural reserve or resilience as a high value target [291, 292]. We identified one high value module that nominates genes/proteins that drive resilience and others that drive amyloid-β [138]. The latter have been validated in an ex vivo model system. Finally, as we look forward to better clinical trial designs, we have been refining our cognitive phenotype that would best serve as a clinical trial outcome [293–295].
Resource sharing
The ROSMAP investigators are committed to timely data sharing of raw or processed data which can be found on our Resource Sharing Hub with relevant links to the AMP-AD Knowledge Portal [296, 297]. A sample of work done with ROSMAP data including as part of translational studies and consortia is provided to give the community a better sense of the range of work that could be done leveraging the resource [298–350].
Discussion
Ongoing for nearly a quarter of a century, ROSMAP has generated a wealth of data across a range of age-related phenotypes from the same individuals. It has served as a research resource for investigators around the globe who have generated about 400 publications over the past 6 years. The data support some general conclusions. Perhaps the over-arching theme running through the work is complexity. Complexity at multiple levels: 1) continuum of AD, 2) mixed and newly recognized pathologies, 3) neural reserve or resilience, and 4) non-cognitive phenotypes. Complexity has important implications for drug discovery.
Continuum of AD
Findings illustrate that cognitive decline as part of the AD dementia syndrome begins years or decades prior to AD dementia onset and MCI onset. We found that AD and other pathologies are common in persons without dementia and without MCI. Further, AD pathology is associated with change in cognition a decade or more prior to death, and that these associations occur long before dementia onset. These data complement data from other clinical-pathologic studies illustrating that AD pathology is present in persons without dementia [351–353]. However, there is much less information from other studies linking pathology to trajectories of cognitive change [354–357]. These data are complemented by clinical-imaging studies with amyloid and subsequently tau PET data [358–362]. More recently, amyloid and tau PET done on younger cohorts are finding that these pathologies appear to accumulate years prior to onset of overt cognitive impairment [363–366]. Together these data provide strong support for a new framework being proposed to capture the continuum of AD that allows a diagnosis of AD to be made in the absence of cognitive symptoms [367]. The framework is based largely on a recently proposed A/T/N classification scheme [368].
Mixed and newly recognized pathologies
We find that the clinical syndrome AD dementia cognitive impairment and dementia is a complex process that results from the additive and interactive effects of numerous pathologies. This is consistent with the results of several prior clinical-pathologic studies that have examined this issue [4, 369–377]. Similar data are emerging from neuroimaging studies as well [378–380]. At this point, we have documented 9 pathologies on more than 1000 brains and find nearly 250 combinations. Besides AD pathology, this includes TDP-43/hippocampal sclerosis, several measures of macro- and micro-vascular disease. We also have other pathologies on a subset of participants, including some measured with ex vivo imaging, which will further increase the number of combinations.
Neural reserve or resilience
There are several approaches to the concept of neural reserve or resilience [381–384]. Many researchers limit the concept of reserve or resilience to having a unidirectional beneficial effect. We take a complementary approach that assumes all cognitive systems have some reserve; however, some have more reserve and others less [385]. Persons with more reserve have a slower rate of cognitive decline and lower AD dementia risk, and those with less reserve have a faster rate of cognitive decline and higher AD dementia risk. We have found many risk factors associated with more (e.g., cognitive activity, purpose in life) or less (e.g., neuroticism, loneliness) reserve, and some biologic factors associated with more reserve (e.g., BDNF expression, neuron density, presynaptic proteins), and network modules with genes associated with more and others with less reserve [292]. Several other groups have reported on the ability of factors to buffer or augment the impact of pathology on cognition [386–392]. Similar findings have been reported with neuroimaging and CSF biomarkers of AD pathology [361, 392–398]. Functional imaging approaches are also being employed to explore the neural basis of reserve [399, 400]. Finally, similar to our findings of neurons and presynaptic proteins, several groups have reported other structural indices that underlie reserve [401–404].
Non-cognitive phenotypes: Motor function and decision making
Our work is congruent with work by other groups that a wide range of motor phenotypes are related to cognitive decline and to AD [405–410]. In addition, like others we find that cognitive function and many risk factors for AD are also related to change in motor structure and function [411, 412]. Our work extends these findings by showing that simultaneous change in cognitive and motor decline is highly correlated [239]. Few studies address whether cognitive or motor decline begins earlier [413]. The idea that both late-life cognitive and motor impairment may share a common neurobiology is supported by our postmortem results and work by others [404, 414–418]. Our studies extend these findings by showing that AD and other pathologies are related to level as well as progressive decline of several motor phenotypes [234, 239]. Together the clinical and postmortem findings are consistent with accumulating evidence that both cognition and motor function may rely on similar underlying neural systems essential for planning and monitoring goal—directed behavior and both may be affected by AD and other common brain pathologies [367, 419–422].
We also showed that cognition, AD, and other pathologies negatively impact health and financial decision making [254–258, 270]. This work is consistent with prior studies that have reported impaired decision making among persons with overt cognitive syndromes [423–427] and some small studies of non-demented persons [428–432] but extends prior work by showing that impaired decision making among cognitively intact persons is in fact a consequence of preclinical cognitive decline. Few studies have examined the association of decision making and related behaviors with subsequent cognitive or other health outcomes [423, 431]. Our work suggests that decision making and related behaviors predict several adverse health outcomes including incident AD, incident mild cognitive impairment and mortality [252, 268]. Moreover, whereas some prior studies have examined the neural underpinnings of select aspects decision making in older persons using neuroimaging approaches [433–439], we expanded this work by examining multiple aspects of decision making using a variety of imaging approaches [265–274] Finally, we found that age-related changes in decision making are associated with common neuropathologies such as AD pathology [264, 265]. Together, findings suggest that impaired decision making is an early manifestation of AD and other neuropathologies and a harbinger of adverse cognitive and other health outcomes.
Implications of complexity for drug discovery
It has been a bleak 15 years in the AD drug discovery space. Other than re-formulations, no new drug has been approved by the Food and Drug Administration (FDA) since 2003. The string of failed studies is long despite the investment of billions of dollars from the public and private sectors, the participation of many tens of thousands of people in clinical trials, and the efforts of thousands of researchers and study staff [440]. There are many reasons for these failures. A recent analysis pointed to complexity, low signal-to-noise, and recruitment/retention [441]. As many people have and others will develop AD dementia, more robust symptomatic treatment is urgently needed. However, symptomatic therapies will not reduce the overall human and economic toll of AD [442]. This can only be accomplished by prevention.
There are currently about 100 drugs in the AD pipeline in the USA with an additional 100 in development in the European Union with participants ranging from those with moderate to severe AD dementia to asymptomatic persons [443, 444]. The therapies are relatively evenly divided into three buckets. The first is small molecules for therapeutic treatment. The other two are disease modifying agents, one of which is small molecules and the other immuno-therapies.
The majority of the disease modifying agents target amyloid and tau. It has been argued that reducing complexity and creating more homogenous populations for clinical trials will improve the signal to noise ratio improving the likelihood of success. Thus, many trials now enrich studies by enrolling those at genetic risk or with a positive amyloid PET [445–447]. Perhaps this will be a successful approach. However, failure will only inform on subpopulations. A drug that fails to slow cognitive decline among persons at genetic risk or those who have amyloid might still work on those not at genetic risk of those who have not yet developed amyloid. Estimates suggest that studies to slow cognitive decline in asymptomatic persons may need to be much longer than is currently being done [448]. Further, requiring multiple spinal taps and/or PET scans likely increases the healthy volunteer effect by excluding people with non-cognitive factors that predict cognitive decline and are associated with AD pathology such as gait disturbance and frailty [235, 449]. We also found that amyloid-β does not predict cognitive decline after controlling for tangles and that amyloid-β and tangles together only account for about 25% of the variance of cognitive decline [118, 450]. Are anti-amyloid studies adequately powered to impact such a small component of the trajectory? In addition, we found that the impact of nine common pathologies, e.g., pathologic AD, on cognitive decline varies widely depending on the presence of other pathologies, many of which are beyond the resolving power of extant biomarkers. Finally, is developing a biomarker for each pathology and a cocktail to treat each pathology really scalable? This could result in multiple cocktails over a long period of time in older persons with aged livers and kidneys, at a cost that is likely beyond what can be paid.
An alternate therapeutic strategy would be to target resilience itself. All physiologic systems have reserve or resilience. In some cases, it is simply an extra organ, e.g., lung, kidney. However, with the brain it is plasticity that allows it to tolerate and/or recover from injury and disease. There is no evolutionary pressure to develop these systems from age related disease. Thus, there are likely few such systems and they are, as we have found, relatively agnostic to specific age-related disease. We are currently using our drug discovery pipeline to find novel targets for reserve. To determine if they are druggable, the field needs to develop and validate an ex-vivo model for high throughput drug screens. In other words, we will need to model cognitive decline in a dish.
ADDENDUM
To ensure the review is up to date, we add a list of manuscripts published or accepted for publication since the manuscript was last submitted. Dawe RJ, Leurgans SE, Yang J, Bennett JM, Hausdorff JM, Lim AS, Gaiteri C, Bennett DA, Buchman AS. Association between quantitative gait and balance measures and total daily physical activity in community-dwelling older adults. Journal of Gerontology: Medical Sciences 2018;73:636-642. Buchman AS, Nag S, Leurgans SE, Miller J, VanderHorst VV, Bennett DA, Schneider JA. Spinal lewy body pathology in older adults without an antemortem diagnosis of Parkinson’s disease. Brain Pathology 2017;doi: 10.1111/bpa.12560. Barral S, Habeck C, Gazes E, De Jager PL, Bennett DA, Stern Y. A dopamine receptor genetic variant enhances perceptual speed in cognitive healthy individuals. Alzheimers Dementia 2017;3(2)254-261. Klaver AC, Coffey MP, Bennett DA, Loeffler DA. Specific serum antibody binding to phosphorylated and non-phosphorylated tau in non-cognitively impaired, mildly cognitively impaired, and Alzheimer’s disease subjects: an exploratory study. Translational Neurodegeneration 2017;6:32. Arvanitakis Z, Leurgans S, Fleischman D, Schneider JA, Kumar KB, Pruzin J, Shah RC, Evans DA, Barnes LL, Bennett DA. Memory complaints, dementia, and neuropathology in older blacks and whites. Annals of Neurology 2018;83:718-729. Benedet AL, Yu L, Labbe A, Mathotaarachchi S, Pascoal TA, Shin M, Kang MS, Gauthier S, Rouleau GA, Poirier J, Bennett DA, Rosa-Neto P. CYP2C19 variant mitigates Alzheimer s disease pathophysiology in vivo & post mortem. Neurology: Genetics. 2018:4:e216. Ramos-Miguel A, Jones AA, Sawada K, Barr AM, Bayer TA, Falkai P, Leurgans SE, Schneider JA, Bennett DA, Honer WG. Frontotemporal dysregulation of the SNARE protein interactome is associated with faster cognitive decline in old age. Neurobiology of Disease 2018;114:31-44. Wang X, Philip V, Ananda G, White C, Michalski P, Malhotra A, Karuturi D, Chintalapudi S, Acklin C, Sasner M, Bennett DA, De Jager PL, Howell GR, Carter GW. A Bayesian framework for generalized linear mixed modeling identifies new candidate loci for late-onset Alzheimer’s disease. Genetics. 2018;209:51-64. Smith RG, Hannon E, De Jager PL, Chibnik L, Lott SJ, Condliffe D, Smith AR, Haroutunian V, Troakes C, Al-Sarraj S, Bennett DA, Powell J, Lovestone S, Schalkwyk L, Mill J, Lunnon K. Elevated DNA methylation across a 48-kb region spanning the HOXA gene cluster is associated with Alzheimer’s disease neuropathology. Alzheimer’s & Dementia. 2018; doi: 10.1016/j.jalz.2018.01.017. Wilson RS, Capuano AW, Yu L, Yang J, Kim N, Leurgans SE, Lamar M, Schneider JA, Bennett DA; Boyle PA. Neurodegenerative disease and cognitive retest learning. Neurobiology of Aging. 2018;66:122-130. Sekiya M, Wang W, Fujisaki N, Sakakibara Y, Quan X, Ehrlich ME, De Jager PL, Bennett DA, Schadt EE, Gandy S, Ando K, Zhang B, Iijima KM. Integrated biology approach reveals molecular and pathological interactions among Alzheimer’s A?42, Tau, TREM2, and TYROBP in drosophila models. Genome Medicine. 2018;10:26. Buchman AS, Dawe RJ, Lei Y, Lim A, Wilson RS, Schneider JA, Bennett DA. Brain pathology is related to total daily activity in older adults. Neurology. 2018;90:e1911-e1919. doi: 10.1212/WNL.0000000000005552. Nag S, Yu L, Boyle PA, Leurgans SE, Bennett DA, Schneider JA. TDP-43 pathology in anterior temporal pole cortex in aging and Alzheimer’s disease. Acta Neuropathologica Communications. 2018;6(1):33. Li P, Yu L, Lim AS, Buchman AS, Scheer F, Shea S, Schneider JA, Bennett DA, Hu K. Fractal regulation in motor activity and incident Alzheimer’s disease in elderly individuals. Alzheimer’s & Dementia. 2018;doi: 10.1016/j.jalz.2018.03.010. Rodriguez JJ, Capuano AW, Barnes LL, Bennett DA, Shah RC. Effect of antidepressant medication use and the level of depressive symptoms in community-dwelling, older African Americans and whites with dementia. Journal of Aging and Health. 2018;doi: 10.1177/0898264318772983. Oveisgharan S, Buchman AS, Yu L, Farfel JM, Hachinski V, Gaiteri C, De Jager PL, Schneider JA, Bennett DA. APOE ?2?4 genotype, incident AD and MCI, cognitive decline, and AD pathology in older adults. Neurology. 2018;doi: 10.1212/WNL.0000000000005677. Knight JE, Bennett DA, Piccinin AM. Variability and coupling of olfactory identification and episodic memory in older adults. Journals of Gerontology B Psychological and Social Sciences. 2018;doi: 10.1093/geronb/gby058. Arvanitakis Z, Capuano A, Lamar M, Shah RC, Barnes LL, Bennett DA, Schneider JA. Late-life blood pressure association with cerebrovascular and Alzheimer’s disease pathology. Neurology. In press. Dawe RJ, Yu L, Schneider JA, Arfanakis K, Bennett DA, Boyle PA. Postmortem brain MRI is related to cognitive decline, independent of small vessel disease in older adults. Neurobiology of Aging. Pending revisions. De Jager PL, Ma Y, McCabe C, Xu J, Vardarajan BN, Felsky D, Klein HU, White CC, Peters M, Lodgson B, Nejad P, Tang A, Mangravite L, Yu L, Gaiteri C, Mostafavi S, Schneider JA, Bennett DA. A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Scientific Data. In press. James BD, Wilson RS, Shah RC, Yu L, Bennett DA, Boyle. Association of financial literacy with hospitalization in community-dwelling older adults. Medical Care. In press. Kapasi A, Leurgans S, James BD, Boyle PA, Arvanitakis Z, Nag S, Bennett DA, Buchman AS, Schneider JA. Watershed Microinfarct Pathology and Cognition in Older Persons. Neurobiology of Aging. In press. Yu L, Petyuk VA, Gaiteri C, Mostafani S, Young-Pearse T, Shah RC, Buchman AS, Schneider JA, Piehowski PD, Sontag RL, Fillmore TL, Shi T, Smith RD, De Jager PL, Bennett DA. Targeted proteomics of human neocortex uncover multiple paths to Alzheimer’s dementia. Annals of Neurology. In press.
Footnotes
Acknowledgments
This work is dedicated to the participants in the Religious Orders Study and Rush Memory and Aging Project. We thank the faculty and staff of the Rush Alzheimer’s Disease Center, and our collaborators around the world. Work was supported by grants from the National Institutes of Health: P30AG10161, RF1AG15819, R01AG17917, RF1AG22018, R01AG33678, R01AG34374, R01AG36042, R01AG40039, R01AG042210, U01AG46152, R01AG47976, R01AG43379, RF1AG54057, R01AG56352, R01NS78009, and UH2NS100599, and the Illinois Department of Public Health. Authors’ disclosures available online (![]() ).
).