
Abstract
Matrix metalloprotease 9 (MMP-9) is a 92 kDa type IV collagenase and a member of the family of endopeptidases. MMP-9 is involved in the degradation of extracellular matrix components, tissue remodeling, cellular receptor stripping, and processing of various signaling molecules. In the CNS, the effects of MMP-9 are quite complex, since it exerts beneficial effects including neurogenesis, angiogenesis, myelogenesis, axonal growth, and inhibition of apoptosis, or destructive effects including apoptosis, blood-brain barrier disorder, and demyelination. Likewise, in the periphery, physiological events, as the involvement of MMP-9 in angiogenesis, for instance in wound healing, can be turned into pathological, such as in tumor metastasis, depending on the state of the organism. Alzheimer’s disease is a neurodegenerative disorder, characterized by amyloid accumulation and deposition in the brain. Amyloidogenesis, however, also occurs in diseases of the periphery, such as type II diabetes mellitus, where an analogous type of amyloid, is deposited in the pancreas. Interestingly, both diseases exhibit similar pathology and disease progression, with insulin resistance being a major common denominator. Hence, combinatorial strategies searching new or existing molecules to apply for therapeutic use for both diseases are gaining momentum. MMP-9 is extensively studied due to its association with a variety of physiological and pathological processes. Consequently, meticulous design could render MMP-9 into a potential therapeutic target for Alzheimer’s disease and type 2 diabetes mellitus; two seemingly unrelated diseases.
INTRODUCTION
Matrix metalloproteinase-9 (MMP-9) structure and activation
MMP-9, an extracellularly operating member of the MMP family is a collagenase, whose action is dependent on Ca2+ and Zn2+ ions [1]. Its primary role is the degradation of extracellular matrix (ECM) in a wide range of physiological and pathological processes. MMP-9 is encoded in the human genome by a chromosome-based gene 20 [2] and is also known as gelatinase B due to its ability to cleave the gelatin substrate [3].
Human MMP-9 consists of an N-terminal region, a catalytic region, a binding region and a C-terminal hemopexin domain, which generate two fragments; a 92 kDa fragment constituting its inactive form (proMMP-9) and a 88 kDa fragment constituting its active form (MMP-9) [3]. Its catalytic region contains two Zn2+ ions and five Ca2+ ions, as well as three sequences of 58 amino acids homologous to the type II fibronectin motif [4]. The domain of fibronectin is essential for binding to denatured collagen or gelatin. One of the two zinc ions joins the catalytic domain with the cysteine domain of the propeptide in order to retain MMP-9 in its inactive structure while zinc ion catalysis is absolutely necessary for its activation [5].
At a translational level, the majority of metalloproteinases appear to be involved in the activation of MMP-9 by cleaving its propeptide. One of the proteases involved in the activation of MMP-9 is plasmin, the activation of which is catalyzed by the tissue plasminogen activator tPA (tPA) or uPA (urokinase plasminogen activator). Subsequently, plasmin activates the proteolytic system consisting of MMP-9 and the TIMP-1 (tissue inhibitor of metalloproteinases-1) inhibitor. In addition to TIMP-1, MMP-9 is also regulated by processes such as glycosylation and intracellular uptake [6].
AD and T2DM
Alzheimer’s disease (AD) is one of the most common neurodegenerative diseases, while type 2 diabetes mellitus (T2DM) is one of the most common metabolic disorders worldwide. Both AD and T2DM are diseases that exhibit a multitude of similarities, the major one of which involves the formation of toxic amyloid aggregates in the amyloid plaques of the brain and in the pancreatic islets of Langerhans, respectively [7]. Islet amyloid polypeptide (IAPP) or amylin presents similar properties and conformation to the Aβ peptide found in neurons. In particular, human amylin forms oligomer structures [8], but also fibrils are structurally analogous to those of Aβ [9]. Consequently, amylin oligomers show similar in vitro toxicity characteristics to Aβ oligomers [10], as they induce apoptotic pathways, whereas in vivo amylin accumulations and degeneration of pancreatic cells occur in 90% of patients with T2DM [11, 12]. Both the amyloidogenesis and the formation of neurofibrillary tangles are common pathological features of the two diseases, while further similarities include cellular degeneration, abnormal activation of kinases, such as GSK-3β, inflammation, and oxidative stress [13]. The above correlation of the two diseases is also reinforced by epidemiological studies showing that patients with T2DM are 2 to 5 times more likely to develop AD, while the association is further increased when risk factors like the ɛ4 allele of apolipoprotein 4 (APOE4) [14], hypertension, heart disease, or smoking [15] are taken into account.
One other major and more recently discovered connection involves the generation of an insulin-resistant state for both diseases. Earlier notion that the brain is not an insulin sensitive organ, gradually changed due to a bulk of studies proving that insulin is a crucial player in key metabolic events in the brain, as well as it confers vital neurotrophic properties [16]. Further connection of the two diseases was proven by pathological findings in the brains of patients with AD revealing that both insulin receptors and their substrates were significantly deregulated, hence indicating an extensive dysfunction in the central nervous system (CNS) insulin signaling pathway. Thus, the concept of “type III diabetes” for Alzheimer’s disease reflects the biochemical and molecular changes observed between AD and T2DM [17].
Given the clear connection between the two diseases and their similar underlying pathology, emerging studies are exploring combinatory treatments on utilizing molecules as drug targets that can affect similar pathways in order to target both diseases; an example being the use of anti-diabetic drugs to treat AD [18]. MMPs are considered to be attractive target molecules and modulators for therapeutic interventions for a variety of disease, such as inflammatory disease [19] and cancer [20]. MMP-9, in particular, is extensively studied as to its crucial role in the CNS and in the development of neurodegenerative diseases [21, 22]. As it is discussed below, current research is unravelling a positive implication for MMP-9 in both AD and T2DM (Fig. 1), rendering it plausible to be considered for therapeutic usage for both diseases.
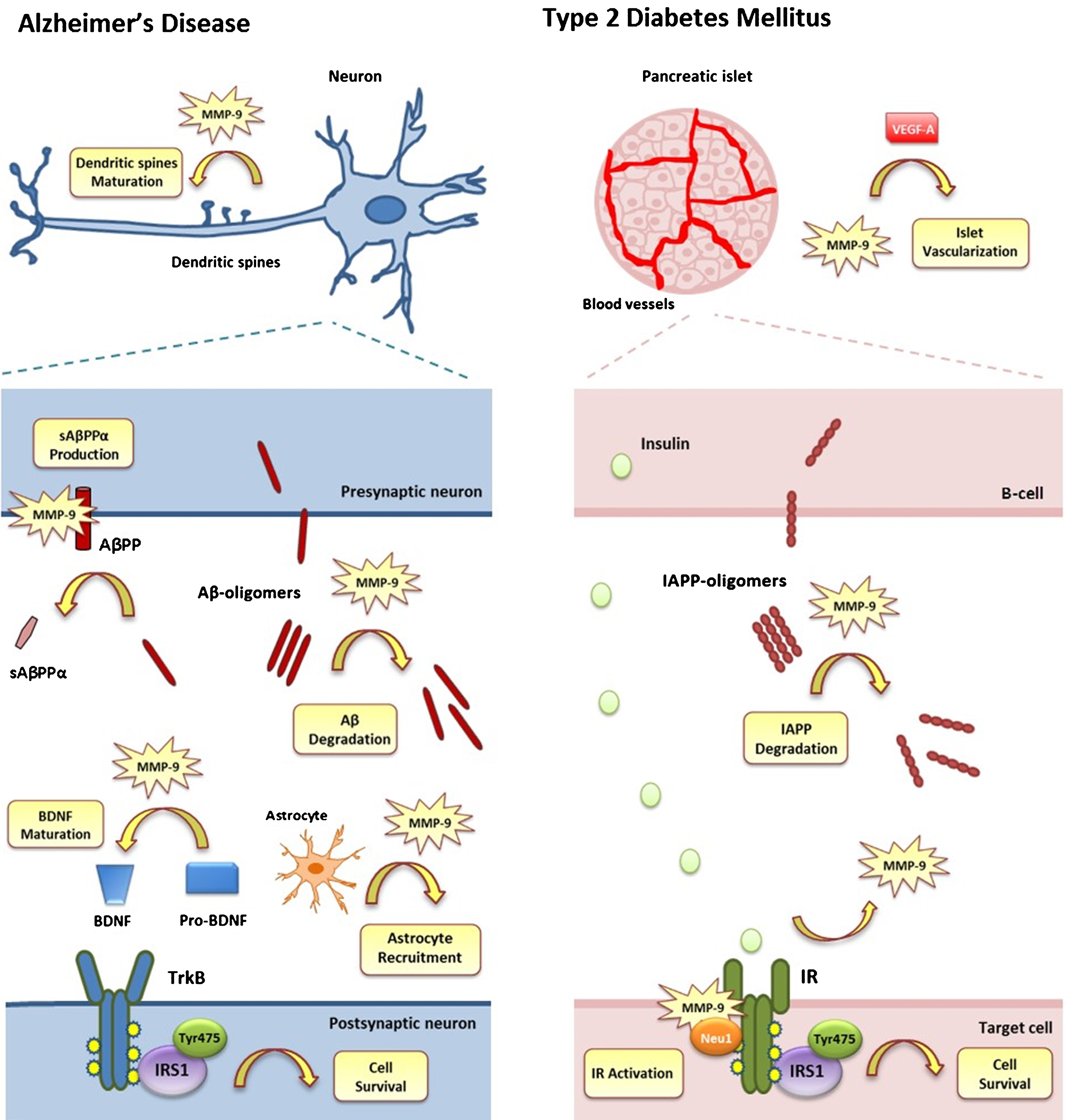
Protective action of MMP-9 against AD and T2DM. A) In AD, synaptic plasticity and cell survival are crucially affected. MMP-9 is involved in maintaining synaptic plasticity by affecting the morphology of dendritic spines (Dendritic Spine Maturation). MMP-9 also reduces the formation of toxic Aβ-oligomers (Aβ-Degradation), favors the increase of neurotrophic sAβPPα (sAβPPα Production) and stimulates astrocyte activation (Astrocyte Recruitment). Moreover, it is involved in maintaining the cell’s normal function by increasing BDNF levels (BDNF Maturation) and activating IRS-1 substrate via TrkB activation (Cell Survival). B) In T2DM, proper islet function and cell survival is compromised. MMP-9 is involved in maintaining the normal vascularization of the pancreatic islets via increasing VEGF-A levels leading to angiogenesis (Islet Vascularization). It is also involved in reducing the formation of toxic IAPP-oligomers (IAPP-Degradation). Insulin produced by β-cells binds to IR receptor of target cell (IR Activation), which is activated due to MMP-9/Neu1 binding, leading to the activation of IRS-1 substrate (Cell Survival).
MMP-9 AND ALZHEIMER’S DISEASE
AD is the most common form of dementia worldwide. It is characterized by extensive nerve loss, attributable to abnormal protein deposition, inside and outside the neurons. These deposits create two distinct pathological features, amyloid plaques and neurofibrillary tangles. Amyloid or neuritic plaques, in particular, are extracellular deposits of amyloid protein, which consists of insoluble amyloid-β peptides (Aβ) [23]. Aβ, first isolated in 1984 [24], is a 38–42 amino acid peptide and the product of the sequential proteolytic breakdown of the amyloid-β protein precursor (AβPP), a type I transmembrane protein, whose physiological role remains largely unclear [25]. Some of the properties attributed to AβPP concern its involvement in neuritic growth and synaptogenesis, protein transport of vesicles along the axis, cell adhesion, calcium metabolism, and others [26].
AβPP is synthesized in the endoplasmic reticulum (ER) and then transferred to the Golgi system, where post-translational modifications take place [27]. It is then transported via vesicles to the cytoplasmic membrane, where it undergoes proteolytic degradation by α-, β-, and γ-secretases. While proteolytic processing of AβPP occurs physiologically throughout life, during AD, an increase in sequential cleavage by β- and γ-secretases creates a pathological imbalance that results in the generation of more Aβ peptides compared to the soluble neurotrophic fragment N-terminal fragment AβPP (sAβPPα), which results from cleavage by α-secretases [28]. The production of Aβ peptides followed by their subsequent oligomerization results in a cascade of catastrophic events extracellularly, as well as intracellularly. Some of these events include destabilization of internal cell membrane organelles, induction of oxidative stress, disruption of synaptic plasticity, formation of pores in the cell membrane, which thereby disrupts cell homeostasis and inhibition of normal function for a plethora of signaling paths, which ultimately lead to apoptosis [29].
Because of their role in the production of Aβ, the AβPP secretases have been under intense investigation, especially members of the MMP family, such as MMP-9. MMP-9 has beneficial or detrimental effects, depending on the levels of expression and activation of the enzyme, as well as the given state of the organism. In terms of expression, some studies report elevated MMP-9 levels in the plasma of AD patients [30–32], indicating that MMP-9 may contribute to AD pathology, while other studies are conflicting on this matter reporting decreased [33] or unaltered MMP-9 levels in the plasma levels of AD patients [34]. More recent studies show that MMP-9 levels do not differ in the cerebrospinal fluid of patients with AD in comparison to the levels observed in normal samples [35, 36]. Hence, regarding the pathogenesis of AD, several studies have correlated MMP-9 with either pathological or neuroprotective properties.
Amyloid formation
Amyloid formation is a key pathological feature in AD and therefore enzymes that have shown Aβ-degrading properties are considered important therapeutic targets. Some of these enzymes include proteases such as neprilysin, endothelin converting enzyme, insulin degrading enzyme, as well as members of the MMPs family [37, 38]. Probably the most well-studied MMPs are the two members of the gelatinase subfamily, gelatinase A (MMP-2) and gelatinase B (MMP-9). Studies in AD animal models using genetic or pharmacological inhibition of MMP-2 or MMP-9 activity have indicated that amyloid deposition is increased when gelatinase activity is decreased [39, 40]. Active MMP-9 in particular has been reported to degrade in vitro synthetic Aβ1–40 at the α-secretase cleavage site between Lys 16-Leu 17 [41] and in contrast to various proteases involved in AβPP processing, MMP-9 has been shown to be the only Aβ-degrading enzyme capable of also degrading Aβ fibrils in vitro and Aβ plaques in situ [42].
Along the same lines, our group further elucidated MMP-9 role in AβPP processing by showing that its activity relates to that of an α-secretase. In particular, we demonstrated that MMP-9 overexpression or treatment with recombinant MMP-9 in vitro results in enhanced secretion of soluble AβPP (sAβPPα), a product of α-secretase cleavage, and reduction of Aβ release [43]. Furthermore, we demonstrated in vitro that MMP-9 actively participates in NGF-induced α-secretase cleavage of AβPP, precluding the formation of neurotoxic Aβ peptides [44]. This shifting of AβPP processing towards the non-amyloidogenic pathway was further proven in vivo, where elevated levels of sAβPPα were reported in the hippocampus and cortex of transgenic mice, generated to overexpress full-length MMP-9 in the CNS [45].
In order to clarify whether the increase in MMP-9 activity can interfere with the pathology of AD our group reported that overexpression of MMP-9 in AD mice models induced an increase in sAβPPα levels, decreased the generation of Aβ oligomers and prevented the cognitive deficits displayed by AD mice, without however, affecting the amyloid burden or activation of astrocytes in the brains of 5xFAD mice [46].
Synaptic plasticity
Synaptic plasticity is an activity-dependent mechanism by which preexisting neurons strengthen or modify synaptic transmission. Synaptic plasticity plays, therefore, a central role in brain’s development, as well as in learning and memory formation. Concerning AD, emerging studies support the notion that cognitive dysfunctions occurring early in the disease are due to defective synaptic function caused by the action of Aβ oligomers, which occurs well before the extensive synaptic loss and neurodegeneration which characterizes the final stages of the disease [47].
MMP-9 is the most characterized metalloproteinase of the CNS, shown to be involved in neuronal plasticity. It is expressed in the adult brain mainly by neurons and is released in response to increased nerve activity in both normal and pathological states [48]. In addition to neurons, its proteolytic activity is also found in astrocytes [49], microglia [50], and oligodendrocytes [51]. Higher levels of MMP-9 are detected in the early stages of development and are diminished in adulthood [52], leading to an increase in the maturation of dendritic spines [53]. In the adult brain, a large number of studies showed that MMP-9 is involved in neurogenesis mechanisms, cell migration and differentiation [54–56]. Both the expression pattern and the dendritic localization of MMP-9 in the stimulatory brain neurons suggest a vital role in synaptic plasticity, learning and memory [57, 58]. An increasing number of studies suggest that MMP-9 plays a critical role in maintaining long term potentiation (LTP), the main mechanism for memory formation and storage, by affecting the morphology of dendritic spines [59, 60].
Consistent with the above observations, the transgenic mice generated by our group to overexpress MMP-9 in the CNS, displayed enhanced performance in cognitive tasks, which was accompanied by elevated levels of sAβPPα, followed by increased dendritic spine density in the hippocampus and cortex, as well as prolonged maintenance of LTP in hippocampal slices [45]. Additionally, a multitude of in vivo studies from other groups advocates the functional significance of endogenous sAβPPα in nerve plasticity, but also in the creation of memory [61–63]. In a similar study, it has been shown that in rats of advanced age, sAβPPα levels in the cerebrospinal fluid are clearly reduced, which is associated with spatial memory malfunctions [64]. Conversely, decreased levels of sAβPPα have been reported in familial or cerebrospinal fluid [65] or in the sporadic form of the disease [66], a decrease which is partly related to altered cognitive functions observed in AD [67].
Moreover, a follow-up study from our group demonstrated that when mice models of Alzheimer’s disease were crossed with mice overexpressing MMP-9 in the CNS, their offspring performed better in diagnostic behavioral procedures, compared to AD mice. This improvement was consistent with elevated levels of sAβPPα and synaptic proteins, such as synaptophysin or with increased maturation of the brain derived neurotrophic factor (BDNF) [46].
Insulin signaling
Insulin is the main anabolic hormone of the body and has been found to play a critical role not only in the regulation of central and peripheral metabolism, as originally believed, but also in the proper function of the CNS [68]. Insulin acts beneficially at synapses, by controlling synaptic plasticity and cell survival. In vitro studies have shown that insulin inhibits the amyloidogenic pathway, by enhancing the production of soluble sAβPPα and reducing the formation of Aβ peptides [69, 70]. Moreover, Aβ peptides were found in vivo to block sAβPPα-mediated prosurvival properties acting via insulin receptors (IRs) in a transgenic mouse model for AD [71].
Under physiological conditions, binding of insulin to its receptor causes activation of the insulin receptor-1 substrate (IRS-1), a substrate involved in learning and memory mechanisms [72]. Activation of IRS1 at tyrosine residues leads downstream to signal transduction through the PI3K/Akt kinase, a signaling pathway that induces the growth and survival of nerve cells [73, 74]. One of the protective mechanisms of action of Akt involves the inactivation of GSK-3β kinase, which is responsible for hyperphosphorylation of the tau protein [75].
In AD however, the action of Aβ oligomers has been shown to trigger pro-apoptotic pathways activation, compromising the IRS1/AKT signaling. JNK (C-Jun-Terminal kinase) activation by Aβ oligomers results in phosphorylation of the IRS1 substrate at serine residues resulting in decreased IRS-1 activation, as it was shown in vitro in cultured hippocampal neurons after treatment with Aβ oligomers [76]. Similar results were obtained in the hippocampi of both an AD mouse model, monkeys after intracerebroventricular (i.c.v.) stereotactic injection of Aβ oligomers [77], as well as in the cortex of AD patients [78]. Further inhibition of the pathway occurs due to the abundant accumulation of Aβ oligomers postsynaptically as the disease progresses which results in the removal and redistribution of insulin receptors from the cell surface to the cell body, thus weakening insulin signaling, both in vitro in primary hippocampal cultures after Aβ oligomer treatment [79] and in vivo in postmortem AD human brains [80].
Our group described a protective role of MMP-9 in preventing the impairment of the insulin survival pathway. More specifically, overexpression of MMP-9 has been shown to prevent insulin impairment in the 5XFAD model of AD, in vitro in primary hippocampal cultures and in vivo in hippocampal extracts, in a dual manner; by increasing BDNF levels, thus resulting in an increase of the inductive IRS1 tyrosine phosphorylation and by reducing Aβ oligomers, thus resulting in a reduction of the inhibitory IRS1 serine phosphorylation [81].
Inflammation
AD is an inflammatory disease, while matrix metalloproteinases are considered key regulators of proinflammatory mechanisms [82]. Studies showed that MMP-9 is increased in postmortem brain tissues of patients with the disease, particularly in the cytoplasm of neurons, neurofibrillary tangles, amyloid plaques, and vascular walls of hippocampal and cerebral cortex of AD patients [83–86], thus suggesting a possible link between MMP-9 and neuroinflammation. In fact, some studies categorize the elevated levels of MMP-9 and its inhibitor TIMP-1 as biomarkers for the pathogenesis of the disease together with hyperphosphorylated tau levels and the increase of the Aβ1–42 peptide [87–89]. Moreover, it was shown that intracerebroventricular injections of Aβ peptides in mice led to an increase in MMP-9 levels in their hippocampi, whereas in vitro inhibition of MMP-9 or MMP-9 gene deletion in vivo alleviated Aβ-neurotoxicity and cognitive impairments, respectively [90].
However, a significant number of studies attribute neuroprotective properties to the increased levels of MMP-9. In addition to neurons, MMP-9 is expressed in the normal brain by astrocytes [49], which are part of the body’s defensive line in inflammatory conditions, as in the case of AD [91–93]. Astrocytes are able to degrade amyloid both in vitro and in situ [94], while studies have shown that levels of MMP-9 are significantly induced in response to amyloid peptide in cultured rat astrocytes [95, 96]. Furthermore, in in vivo mice models for AD, astrocytes surrounding amyloid plaques showed enhanced expression of MMP-2 and MMP-9, whereas astrocyte-conditioned medium (ACM) degraded synthetic human Aβ peptide [39].
MMP-9 AND TYPE 2 DIABETES MELLITUS
T2DM is a disorder characterized by desensitization to insulin, hyperglycemia and progressive β-cell loss, resulting in pancreatic failure and as in AD, its prevalence increases with age [97]. β-cells are found in pancreatic islets and their main role is to synthesize and secrete the anabolic hormone insulin in order to maintain circulating glucose levels within physiological range [98]. Along with insulin, β-cells co-excrete islet amyloid polypeptide, IAPP, a 37 kDa polypeptide. Its physiological function remains to be elucidated, although findings correlate IAPP’s role in suppressing glucose uptake and insulin secretion [99]. An additional role of IAPP in the regulation of food intake has also been reported [100].
Under physiological conditions, IAPP is maintained in a monomeric and soluble conformation, which is aided by its interaction with insulin and its processing intermediates. However, under pathological conditions, such as in T2DM, amylin can self-aggregate and eventually form insoluble IAPP amyloid plaques. Insulin desensitization combined with prolonged hyperglycemia results in the upregulation and secretion of IAPP. IAPP amyloids in turn, result in β-cell toxicity both intacellularly and extracellularly. Some of their toxic effects include ER stress, disruption of intracellular organelles and inhibition of the proteasome, increased oxidative stress and inflammation [101].
Although the presence of matrix metalloproteinases in pancreatic islets was described more than a decade ago, their exact role has not fully been elucidated [102]. As with AD, MMP-9 is viewed in a positive and negative light due to its involvement in both normal physiology and disease, with a number of studies linking it’s dysregulation with the pathophysiology of diabetes. Hence, earlier studies correlated elevated plasma levels of MMP-9 as a feature of Type 2 diabetes [103, 104] whereas, increased urinary levels of MMP-9 in T2DM patients were strongly associated with age and the progression of diabetes [105]. However, other studies detected either low or unchanged MMP-9 serum levels between T2DM patients and non-diabetic controls [106, 107].
Interestingly, recent research showed that genetic deletion of MMP-9 in diabetic mice worsens diabetic pathology [108] and results in poor wound healing, a side effect of diabetes [109], indicating that the expression of MMP-9 appears to have protective, rather than pathological effect in T2DM.
Islet amyloid formation
Along with insulin, β-cells co-excrete islet the amyloid polypeptide, IAPP. IAPP is a normal product of the pancreatic islet β-cell and it is released in response to nutrient stimuli, as a propeptide, which is then converted to active IAPP by the same enzymes that convert proinsulin to insulin. Its sequence is highly conserved among species and its ability to self-aggregate is restricted to humans, primates, and cats, but not in rodents [110].
In T2DM, there is in vitro evidence that intermediates formed during the aggregation process itself may actually initiate cell death via apoptosis [111, 112] while in vivo, mice transfected with human IAPP together with diabetic traits develop IAPP oligomers and fibrils in the islet and show more severe diabetic symptoms [113]. Similar to AD, amyloid deposits are found in the islets of most patients with type 2 diabetes [114] and in a lesser degree in non-diabetic controls. Abnormal IAPP deposition results in reduced β-cell mass, as well as increased β-cell apoptosis [115].
In a recent in vitro study it was demonstrated that in a cell-free system, synthetic human IAPP (hIAPP) was cleaved by MMP-9 into non-amyloidogenic fragments, whereas, adenoviral overexpression of MMP-9 in amyloid-prone islets reduced amyloid deposition and β-cell apoptosis [116]. This finding is further supported by a previous study where in an islet culture model where hIAPP transgenic mouse islets develop amyloid, inhibition of MMP-9 activity increased amyloid formation and the resultant β-cell apoptosis [117]. Furthermore, it was demonstrated in the same study that MMP-9 mRNA levels were found to be reduced in islets of subjects with T2DM, compared to non-diabetic controls.
Vascularization of pancreatic islets
Unperturbed islet blood flow is essential for proper islet function in order for hormones and nutrients to reach the islet vasculature from distant tissues via the bloodstream. MMP-9 has been reported in aiding proper islet function, by remodeling ECM and thus by regulating the availability of chemokines and growth factors [118].
In adulthood, the β-cells are reported to endogenously produce high levels of the growth factor vascular endothelial growth factor A (VEGF-A) in order to maintain the fenestration of their microvascular endothelium [119], whereas during development VEGF-A is crucial for regulating islet vascularization formation and capillary density [120]. MMP-9 has been shown to exhibit a predominant proangiogenic effect due to the release of VEGF-A, in the initial stages of islet tumor angiogenesis, during matrix degradation ex vivo [121]. This was further supported by in vivo studies, where revascularization of islets transplanted to MMP-9– deficient mice was impaired [122], whereas MMP-9 pretreatment of islets improved revascularization of the transplanted islets [123], showing that MMP-9 is critical for revascularization of transplanted pancreatic islets. More recently it was shown that genetic deletion of MMP-9 in adult mice resulted in disrupted β-cell function and islet vascularization in vivo, as well as in vitro in isolated islets, underlying the importance of proper matrix remodeling, exerted by MMP-9 [124].
Insulin signaling
Insulin signaling regulates glucose, lipid, and energy homeostasis by acting on central and peripheral tissues. Physiologically, insulin binds to its receptor IR and activates IRS substrates in a similar fashion, as it was described for insulin signaling in the CNS. While inductive phosphorylation at tyrosine residues results in metabolic actions of insulin, inhibitory phosphorylation at serine residues by activation of JNK kinase mediated by tumor necrosis factor-α (TNF-α), blocks insulin action and contributes to peripheral insulin resistance [125]. As a result, β-cells produce more insulin in order to compensate for the lack in insulin responsiveness, which in turn causes hyperglycemia [126].
An earlier in vitro study demonstrated a connection between insulin and MMP-9, where insulin stimulated MMP-9 activation in monocytes in a time and concentration dependent manner via IR activation [127], whereas a more a recent study discovered a novel mechanism for normal insulin receptor activation, which involves a crosstalk between MMP-9 and Neu1 protein [128]. In this mechanism of action MMP-9 binds along with Neu1 to IRβ units, creating a G-protein coupled receptor (GPCR) signaling platform that is essential for IR activation upon binding to insulin. Therefore, as in the CNS, MMP-9 could be used as an enhancer of insulin signaling in order to improve peripheral insulin resistance observed in T2DM.
COMBINATORIAL APPROACHES FOR THE TREATMENT OF AD AND T2DM
Since disturbed insulin signaling is one of the most important risks to the development of AD and a common denominator for both diseases, enhancing the insulin-dependent survival pathway is a reasonable therapeutic approach. Current anti-diabetic drugs aim to ameliorate insulin resistance either by stimulating insulin survival pathway itself by direct administration of insulin or insulin analogs [129] or by bypassing the insulin pathway and activating alternative pathways that exert similar functions [130].
One particular strategy towards this direction is intranasal administration of insulin to AD and T2DM patients at the early stages of the disease. Because patients with AD, as well as with T2DM, exhibit cognitive decline, due to insulin desensitization, and reduced ability of insulin to enter the blood-brain barrier (BBB), intranasal administration has the advantage of bypassing any barriers, thus leading to immediate effect. Indeed, clinical studies have shown that this mode of administration of insulin has resulted in an improvement in cognitive dysfunctions in both healthy subjects and patients with AD and T2DM without affecting the levels of insulin or glucose in the circulation [131, 132]. It is worth mentioning that positive effects of intranasal insulin have been observed not only in its direct administration to patients but also after a long period of time without serious side effects [133]. However, these beneficial effects were not observed in patients bearing the e4 allele of APOE4, which suggests that the design of treatment should take into account the different risk factors for AD [134].
Another antidiabetic drug, metformin, has been long used for treating T2DM due to its effect in decreasing glucose production and increasing insulin sensitivity [135]. Concerning AD, it has been shown in vitro that metformin is able to enhance the reduced neuronal susceptibility to insulin that is observed in hyperinsulinemia, while improving AD-resulting pathology [136].
Beneficial effects of MMP-9 in Alzheimer’s disease and Type 2 Diabetes Mellitus
AD, Alzheimer’s disease; Aβ, amyloid-β; NO, nitric oxide; AβPP, amyloid-β protein precursor; IAPP, islet amyloid polypeptide; T2DM, type 2 diabetes mellitus; MMP-9, matrix metalloproteinase-9; IR, insulin receptor.
Other strategies include use of insulin sensitizers, thiazolidinediones (TZDs) and glucagon likepeptide-1 (GLP-1) agonists. TZDs are as peroxisome proliferator-activated receptors (PPARs) agonists that exert their antidiabetic effects by activating the PPARγ receptor, which alters the transcription of metabolism regulation genes. TZDs have been associated with improvement on insulin desensitization observed in AD and T2DM patients, as well as with the enhancement of impaired cognitive abilities, as demonstrated by successful clinical studies [137, 138]. GLP-1Rs receptor agonists, on the other hand, trigger common pathways to insulin by signaling through G protein coupled receptors. Exendin-4 and liraglutide are two medicines that have already been approved for the treatment of T2DM [139]. Studies in mice have shown that GLP-1R analogs are stable in the circulation and can greatly penetrate the blood-brain barrier, and have been associated with enhancement of synaptic plasticity in hippocampus, cell survival and cognitive functions [140, 141]. Recent data have also shown that Exendin-4 has the property of inhibiting desensitization observed in AD by Aβ oligomers [77].
Some of the strategies applied for boosting insulin signaling in T2DM and AD include upregulation of the IR receptor, insulin receptor substrates and downstream kinases [142, 143]. To this respect, MMP-9 could be a common therapeutic agent for both diseases. As described earlier, MMP-9 has the ability to interfere with AD pathology by reducing Aβ-peptide burden both in vitro and in vivo, by acting as α-secretase [43, 46]. As a result, it favors the production of the neuroprotective fragment sAβPPα, which improves synaptic plasticity and thus cognitive abilities that are crucially affected in AD [45, 46]. It also stimulates astrocyte activation in order to reduce amyloid plaque burden [94]. Thus, MMP-9 could be applied to counterbalance the disrupted synaptic plasticity mechanisms that occur in AD, as well as to act as a mediator of innate amyloid degradation by CNS defensive mechanisms. New evidence suggests an additional neuroprotective role of MMP-9 in ameliorating insulin survival pathway indirectly by increasing BDNF levels and stimulating an alternative to insulin’s survival pathway [81]. It was also found to be pivotal for the activation of IR receptor [128], while MMP-9 expression is insulin-mediated [127]. These properties could, henceforth, constitute MMP-9 as a novel modulator of insulin pathway that can have immensely effect in ameliorating AD, as well as T2DM deficiencies.
MMP-9 AS A THERAPEUTIC AGENT
Given the promising reported protective effect of mild overexpression of MMP-9 in ameliorating AD and T2DM pathology, summarized in Table 1, MMP-9 administration could constitute a viable option for therapeutic purposes. Such an approach would require the design of an effective way of administering MMP-9 or possible modulators of its activity, following extensive in vitro and in vivo research to determine the enzyme’s safety levels. Additionally, in vivo imaging of MMP-9 activity is essential in order to fully elucidate MMP-9 role in various physiological and pathological processes [144].
Delivering MMP-9 to the target
In order to ameliorate the pathogenesis of T2DM and AD, MMP-9 needs to be delivered to the affected target-organs; the pancreas and the brain. Although administration in the periphery is without restrictions for targeting the pancreas, this is not the case for targeting the brain. Since MMP-9 is a high molecular weight protein, one of the toughest challenges concerns the effective crossing of the enzyme into the CNS. Most drug delivery techniques focus on either penetrating the blood-brain barrier via drug binding with BBB diffusible molecules, such as liposomes or nanoparticles, while others focus on bypassing the BBB via directly administrating drugs in the CNS, such as with intranasal, intrathecal and intracerebroventricular delivery [145].
Studies with amyloid degrading enzymes towards this purpose, such as neprilysin and insulin-degrading enzyme have showed that it is feasible to deliver enzymes of high molecular weight to the CNS [146]. One strategy for targeted gene delivery to the CNS includes coupling with red blood cells for blood to plasma transport [147], while another includes peripheral expression of amyloid degrading enzymes via lentivirus-transduced bone marrow cells [148] or gene binding with lentiviral vectors that express apolipoprotein genes which facilitate transport via the BBB [149]. Moreover, stereotactic injections of lentiviral vectors expressing neprilysin in the hippocampus and frontal cortex of AD mice [150] or ex vivo gene delivery utilizing primary fibroblasts to introduce neprilysin into the brains of AD mice has shown promising results in reducing amyloid deposition and improving impaired cognition [151]. Additionally, it is possible to directly administer microRNA lentiviral vectors regulating gene expression in specific brain areas via stereotactic injections [152]. Interestingly, a more global approach of successfully delivering a neuronal specific adenoviral gene to the CNS after peripheral administration has been reported [153]. Furthermore, immune cells, such as monocytes, can be used as gene carriers combined with viral vectors, due to their ability to migrate to affected brain areas [154, 155].
Another promising strategy includes the use of neural stem cells (NSCs). NSCs can be transplanted and integrate into affected brain areas resulting in amelioration of cognitive functions [156–158]. NSCs can be furthermore modified, prior to transplantation, to express amyloid degrading enzymes, such as neprilysin, in order to ameliorate AD pathology, as it was demonstrated in transgenic AD-mouse models [159]. Additionally, it has been demonstrated that MMP-9 expression is upregulated after NSC transplantation in mice brains and that is indispensable for NSCs migration [54]. Lastly, a reported study of a successful intranasal delivery of a lysosomal enzyme, of a similar molecular weight to MMP-9, offers a novel and promising approach [160].
Utilizing MMP-9 enhancers
A milder and possibly safer approach would be through increasing endogenous MMP-9 expression levels. Since MMP-9 is regulated by specific inhibitors, such as TIMP-1, blocking their activity could be a plausible way to result in increased MMP-9 activity. For instance, increasing serine elastase activity, which results in suppressed TIMP-1 activity, would ultimately lead to increased MMP-9 activity [161]. Another option would be to directly activate MMP-9 by increasing the proteolytic activity of tPA or uPA [162, 163]. Other reported enhancers of MMP-9 include E-26 (Ets) transcription factors, NF-κB, polyomavirus enhancer A-binding protein-3 (PEA3), activator protein-1 (AP-1), specificity protein 1 (Sp-1), and serum amyloid A-activating factor (SAF)-1 [164].
Nevertheless, utilizing such molecules still remains a difficult task, as it has been described in existing literature concerning inhibitors of MMP-9; a plethora of synthetic and endogenous MMP-9 inhibitors has been tested both in vivo and in vitro extensively, without however being able to reach clinical trial testing [165]. The main reason for failure refers to the lack of specificity of MMP-9 inhibitors, as well as the increased risk of side-effects, since an enzyme, such as MMP-9, is involved in a complex spectrum of physiological processes [20]. Thus, it is critical to characterize selective substrates for modulating MMP-9 expression. For instance, peripheral administration of MMP-9 substrates of low molecular weight, such as neurotrophic factor BDNF, would be a more viable approach [166], due to the advantage of overcoming undesirable side effects following administration of the enzyme itself. However, BDNF is not a selective substrate of MMP-9, hence its administration would not result in upregulation of MMP-9 solely. Therefore, extensive research is still needed in order to develop more sophisticated enhancers for strict and controlled MMP-9 upregulation, while determining a safe and effective way of targeting its endogenous levels still remains the greatest challenge.
CONCLUSION
AD and T2DM are two epidemiologically linked diseases with common pathological mechanisms. Amyloidogenesis, as well as insulin resistance are two of the most prominent characteristics, which are common to both diseases. MMP-9 could represent a pathophysiological link between T2DM and AD. Direct usage of MMP-9 or enhancers of its activity could be used for new therapeutic interventions for both diseases. However, because MMP-9 is subjected to strict regulation, it is necessary to meticulously design a safe route of administration, as well as selective targeting to avoid unnecessary side effects. Overall, promising research supports the fact that MMP-9 could, in small and controlled amounts, constitute a promising target in order to interfere with the development and progression of AD and T2DM.
Footnotes
ACKNOWLEDGMENTS
We acknowledge support of this work by the project “Target Identification and Development of Novel Approaches for Health and Environmental Applications” (MIS 5002514) which is implemented under the Action for the Strategic Development on the Research and Technological Sectors, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) and co-financed by Greece and the European Union (European Regional Development Fund).