
Abstract
Sleep represents an active phenomenon regulated by a highly integrated network of cortical and subcortical structures. This complex model results in disruptions at various levels during physiological aging and more deeply during neurodegenerative disorders, thus leading to different sleep alterations. In Alzheimer’s disease (AD), sleep-wake abnormalities were described to occur even in the preclinical phase, thus suggesting they could be a possible AD biomarker. On the other hand, they also favor the progression of the disease. In this paper, we review current theories regarding sleep regulations and functions to highlight the pathophysiological mechanisms at the basis of the bidirectional relationship between sleep and AD. A better understanding of these complex interactions might also be useful to target both sleep disorder management and AD-related symptoms.
INTRODUCTION
Different evidence showed a bidirectional interaction between sleep disturbances and Alzheimer’s disease (AD) pathology [1–3]. Sleep alterations appear frequently during the early stage of dementia and have a significant negative impact on the quality of life of patients with AD dementia, representing one of the leading causes of institutionalization [4]. On the other hand, recent studies demonstrated how sleep disruption could increase AD dementia risk and progression [5].
Sleep represents a dynamic and active process regulated by highly integrated different systems [6]. Thus, an alteration of these processes at different levels can lead to a large spectrum of negative effects on both health and cognitive performance.
The aim of this review is to summarize the up-to-date theories regarding sleep regulations and functions to better understand the pathophysiology mechanism underlying the well-known “bidirectionality” between sleep and AD dementia. An in-deep knowledge of the inter-relationship between sleep and AD pathology might be also essential to better target both the sleep disorder management and the AD-related symptoms.
HOMEOSTATIC AND CIRCADIAN REGULATION OF SLEEP
The sleep-wake rhythm is regulated by the balanced interaction of homeostatic and circadian drives. Such an interaction is well depicted within the “two-process model” of sleep regulation where a homeostatic sleep/wake dependent process (Process S) and a circadian process (Process C) generates the timing of sleep and waking as well as the quality and quantity of sleep [7, 8] (Fig. 1, upper part). Process S reflects the increase in sleep propensity during a waking period and its decline during subsequent sleep [9]. The time-course of the homeostatic process during sleep is reflected by the dynamics of the electroencephalographic slow wave activity (SWA; 0.5–4.0 Hz) (considered the hallmark of deep sleep) across the consecutive NREM-REM sleep cycles. “Sleep need” is high at the beginning of sleep and gradually decrease with the progression of sleep. Accordingly, in young adults SWA is highest in the first NREM cycle of the night and then exponentially declines in intensity across successive NREM sleep cycles, reflecting a homeostatic dissipation of sleep pressure [10]. The longer an individual remains awake, the greater the pressure to sleep and the greater the amount of subsequent SWA during sleep [10]. Process S was originally assumed to reflect a global brain process, however different EEG study demonstrated that SWA shows a frontal predominance and its percentagewise increase after sleep deprivation is most prominent in frontal areas [11, 12]. Analogously, functional data found a main decrease of cerebral blood flow in prefrontal cortex during slow wave sleep [13].
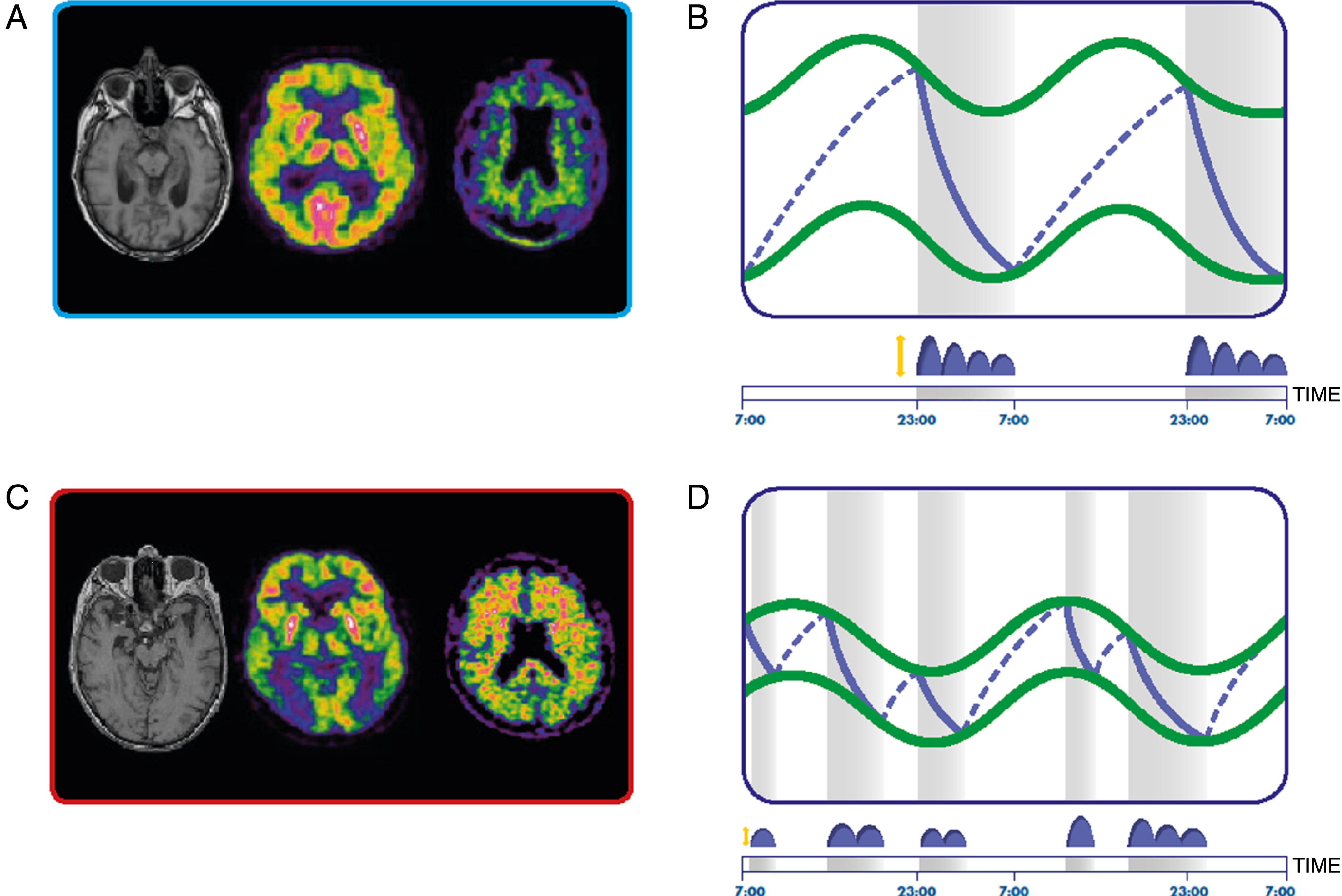
Homeostatic and circadian sleep regulation in healthy (A and B) and Alzheimer’s disease dementia (C and D) subjects. A, C) From the left, magnetic resonance image, Fluoro-2-Deoxy-D-glucose positron emission tomography (PET) and amyloid-β PET in a control (A) and AD dementia (C) subject. B, D) Circadian (green lines) and homeostatic (blue lines) processes in healthy (B) and AD dementia (D) subjects. The sleep-wake abnormalities in AD patients derive from a reduced amplitude of the circadian modulation and different dynamics of the accumulation and discharge of the homeostatic process. Grey bars represent sleep periods. Distribution of slow wave activity is represented in blue above the time scale.
Different structures and pathways responsible for sleep/wake alternation have been identified. Thalamocortical arousal (EEG desynchronization) is effected by a set of ascending projections from different wakefulness-promoting cell groups, including cholinergic neurons of the basal forebrain, histaminergic and hypocretinergic neurons of the posterior and lateral hypothalamus, and aminergic and cholinergic neurons of the rostral brainstem. The hypocretin system represent the main “wake-executive” system, responsible for coordinating the activities of the other arousal systems. Transition from wake to sleep is triggered by specific sleep-active neurons in the ventrolateral preoptic nucleus (VLPO). These neurons are tonically inhibited by wake-active basal forebrain neurons, and disinhibited after prolonged wakefulness; on turn, they maintain sleep by inhibiting activity of wakefulness-promoting systems. Finally, alternation between NREM and REM sleep states is controlled by reciprocal interactions between aminergic and GABAergic REM-off neurons and cholinergic and glutamatergic REM-on brainstem neurons [6].
Process C is driven by an endogenous biological pacemaker, the suprachiasmatic nucleus (SCN), located in the anterior hypothalamus [14, 15]. This master clock has a genetically determined endogenous period length of approximately 24 hours. The SCN is normally entrained by different stimuli that signal the time of day. These stimuli are known as “zeitgebers” (German for “time-giver”), of which light is the most important and potent stimulus. Light entrainment depends on classical photoreceptors but especially on a specialized set of photoreceptive retinal ganglion cells (melanopsin-containing ganglion cells). This latter light-detecting system has been recently identified in the mammalian eye and seems to mediate different sleep-related functions, such as photo entrainment of circadian rhythms, and pineal melatonin synthesis. The receptor cells, a small subpopulation of retinal ganglion cells (RGCs), are intrinsically photosensitive due to the expression of the photopigment melanopsin (mRGCs). The photic signals from the mRGCs are conveyed to the SCN via direct (retino-hypothalamic) projection [16]. In addition to light, feeding schedules, activity, and melatonin can also affect the circadian timing. In particular, the onset of melatonin secretion (approximately 2 hours before natural sleep time) by the pineal gland is regulated by the SCN [14, 15]. The SCN contains melatonin receptors (G protein–coupled receptors, termed MT1 and MT2), and the circadian pacemaker can be reset by melatonin through a feedback mechanism.
Human preferences in the timing of sleep and wake (called “chronotypes”) are based at least partly on genetics and represent one of the most common interindividual differences in circadian rhythmicity [17]. Circadian rhythmicity is determined genetically by polymorphisms in different clock genes, including Per1, Per2, Per3, Clock, Bmal1, Cry1 and, Cry2. These genes serve to control the near–24-hour regulation of gene transcription and ultimately to manifest circadian rhythmicity at the physiological level. It is estimated that approximately 10% of all expressed genes are under regulation of these clock genes.
At the beginning, one of the basic assumption of the “two process model” was the independence of C and S processes. However, recent findings indicate a considerable cross-talk between these two system at various levels of organization; moreover, a misalignment of these processes can have negative effects on health and cognitive performance [18].
SLEEP AND AGING
Physiologic aging is characterized by substantial sleep modifications that seem to reflect an alteration or modification of both mechanism of sleep regulation. Macro-sleep changes encompass longer sleep-onset latency, shorter overall sleep duration, increased sleep fragmentation and increased time spent awake throughout the night [19]. Moreover, excessive daytime sleepiness and daytime napping seem to increase during later life. Considering sleep stages, a reduction in deep sleep and a compensatory increase in the lighter stages of NREM sleep (N1 and N2) are observed. In particular, significant reductions in SWA are described in older adults when compared to young and middle-aged adults with maximal SWA decrease over the prefrontal regions and during the first NREM sleep cycle [19]. Thus, the exponential slope of SWA dissipation across the night is less pronounced in old subjects with respect to young adults suggesting an impairment of “process S” in aging. Analogously, homeostatic SWA increase in response to sleep deprivation is blunted in older in comparison with younger adults. On the other hand, a modification of “process C” has been also described. In particular, tendency to phase advancing are frequently observed during later life [20]. This change in circadian rhythmicity could result not only from behavioral factors but also from neuro-hormonal modifications, especially melatonin whose plasma concentration declines with age [21]. Nevertheless, it is worth to remind that a great inter-individual variability in the degree of these sleep modifications is observed in aging, probably also influenced by somatic and psychiatric pathologies and pharmacological treatments. Finally, the incidence of specific sleep disorders such as sleep-related breathing disorders and restless legs syndrome also increase significantly with age [20].
A still open question concerns whether older adults simply need less sleep, or rather, they are unable to generate the sleep that they still need. In particular, some evidence supports the hypothesis that less sleep need simply depends on a lower homeostatic sleep drive in aging while other findings seem to show that older adults have an impaired ability to generate that unmet sleep need, related to an impaired sensitivity to a still present homeostatic drive [19].
SLEEP FUNCTIONS
The question of the unique or multiple functions of sleep has been a topic for decades. Current evidence shows that the different neurophysiological phenomena that occurs during sleep fulfill multiple functions, encompassing neural maturation and growth; cerebral restitution; metabolic, hormonal and thermal regulations; learning and memory consolidation and regulation of personality traits and emotion.
One of the most intriguing aspects of sleep functions regards the hypothesis that sleep promotes learning and memory consolidation processes. Different models have been elaborated to explain the sleep-related memory consolidation at a neurophysiological level. Indeed, while initial theories hypothesized a passive role for sleep in consolidation processes mainly by providing a time of reduced interference from external stimuli, more recent findings highlight an active role for sleep in the process of long-term memory storage. In particular, “The Active System Consolidation Hypothesis” proposed that memory consolidation originates from the repeated reactivation of newly encoded memory representations during slow wave sleep. According to this theory, recently learned information is quickly stored and held temporarily in hippocampal areas and then repeatedly reactivated and transferred during deep sleep to the long-term store (i.e., the neocortex). This reactivation and integration led to a reorganization of the memory representation that needs to be stabilized in a synaptic consolidation process occurring during succeeding periods of REM sleep [22].
At variance, the “Synaptic Homeostasis Hypothesis-SHY” [23], starts from the observation that during learning, in wakefulness, synapses are repeatedly activated (synaptic potentiation) and this determines an increase in synaptic volume. This growth must be balanced in order to avoid synaptic saturation and abolition of memory storage. In the framework of SHY hypothesis, sleep would represent a peculiar moment for such a synaptic reorganization; SWA at the beginning of the night would represent a direct marker of synaptic potentiation during prior wakefulness. Conversely, the progressive SWA decline during sleep would reflect the process of synaptic strength downscaling, restoring synaptic plasticity and efficiency. Linear downscaling across synapses would improve the signal-to-noise ratio in learning related neuronal networks thus enhancing memory functions. A recent 3-D electron microscopy study seems to confirm this hypothesis showing that a few hours of sleep may lead to an 18% decrease in the size of the synapses thus confirming that the balance of synaptic size and strength is upset by wake and restored by sleep [24].
The contribution of REM sleep “per se” in memory consolidation remains unclear. Indeed, many animal studies strongly support the role of REM sleep in memory processing while such an evidence seems to be very inconsistent in humans [22]. In particular, in a recent study in mice, Boyce et al. found that REM sleep is critical for normal spatial and contextual memory consolidation [25]. Analogously neuroimaging findings support the role of REM sleep in processing memory traces [13, 26]. Finally, it has been hypothesized that REM sleep would subtend the consolidation of procedural (“non-hippocampus dependent”) memories [22] although controversial results have been obtained [27, 28].
Besides learning and memory functions, experimental studies suggest that sleep can also have a “restorative function” facilitating the clearance of degradation products of neural activity that accumulate mainly during wakefulness. Indeed, recent neuroimaging findings led to the conceptualization of a system (the so-called “glymphatic system”) responsible for the removal of interstitial fluid (ISF) into the cerebrospinal fluid (CSF), mostly but not exclusively around the veins [29–31]. This clearance pathway arises from the bulk flow of ISF and flushes out macromolecules and hydrophilic compounds from the brain parenchyma into the CSF. The paravascular circulation of ISF is induced by arterial pulsation along the basement membrane of the blood-brain barrier towards the leptomeninges and is driven in part by smooth muscle cells but especially by astrocytes. In turn, the CSF recirculates from the brain ventricles to the subarachnoid meningeal space: the major part of its aqueous content drains through arachnoid granulations while a second route for removal of CSF macromolecules is along the olfactory bulb across the ethmoid plate to reach the lymphatic network associated with the nasal mucosa [30]. Intriguingly, the glymphatic system acts mainly during sleep and is largely disengaged during wakefulness. Specifically, in vivo 2-photon imaging of glymphatic function found, that this CSF-interstitial fluid exchange increases during sleep [32] and favors the removal of metabolites that usually accumulate during wakefulness, including amyloid-β (Aβ).
ALZHEIMER’S DISEASE
AD [33] is the main cause of dementia worldwide A long pre-dementia period is acknowledged representing an important phase for pathophysiological and preventive therapeutic studies (for terminology adopted in the text see Box 1 [33, 34]).
Terms used in the text, accordingly with the new lexicon for AD [33]
During the last years, many studies have highlighted the possible molecular mechanisms at the basis of AD and several hypotheses have been postulated to explain the AD physiopathology. However, none of these, as taken alone, has been able to clarify the entire pathological aspects of AD. The original ‘amyloid hypothesis’ postulated a linear causality in the cascade started by Aβ deposition, leading progressively to Tau pathology, synaptic dysfunction, inflammation, neuronal loss and, eventually, dementia [35]. However, the process that leads to the accumulation and deposition of Aβ is still poorly understood. The inflammatory process, both at the level of the brain parenchyma and at the barriers of the brain, seems to be relevant not only for the initiation and progression of amyloid pathology, but most of all for the appearance of cognitive deficits [36]. The AD lesions proliferate in a predictably systematic manner with peculiar distribution pattern within the CNS. Indeed, the developmental pattern and progression of the lesions repeat in reverse the phylo- and ontogenetic maturational phases of the CNS [37]. Therefore, late-maturing structures are frequently the ones that display the disease-associated tau lesions particularly early. In converse, phylogenetically older and ontogenetically early-maturing components prove to be resistant. However, the simple linearity of the amyloid cascade hypothesis is debated and a complex, multicausal pathogenesis of AD is increasingly recognized. Indeed, AD pathology affects both neuronal and non-neuronal cells, including microglia, astrocytes, oligodentrocytes and endothelial cells [38], leading to a network dysfunction that contributes to cognitive deficit.
Whichever the causal mechanisms/pathophysiology, this results in multiple neurotransmitter system failure. Cholinergic system dysfunction was the first evidence for network dysfunction in AD, leading to the postulation of the so-called ‘cholinergic hypothesis’ [39]. This finding, together with the demonstration of the role played by acetylcholine in cognitive functions, has provided a rationale for the majority of treatment strategies and symptomatic drug development approaches, although with disappointing results [40]. Current and future trials in presymptomatic individuals could offer the hope that earlier intervention might yet succeed where trials in patients with established dementia have failed. Moreover, considering the multifactorial nature of AD, long-term, multidomain interventions, including anti-Tau agents, lifestyle management, diet and cognitive training aimed at risk reduction might be needed.
CIRCADIAN AND HOMEOSTATIC DYSREGULATIONS IN MCI AND AD DEMENTIA PATIENTS
As discussed in the previous paragraph, there is an urgent need to detect biomarkers that are accurate to identify individuals at greater risk for developing AD dementia. An early detection could offer the chance for preventive measures and for treatment during the initial phase of disease process. Recent evidence seems to suggest that sleep alterations could represent a candidate as a biomarker of preclinical/subclinical stage of AD [41]. To date there are evident limits to consider sleep abnormalities as potential early AD marker due to their lack of specificity. Indeed, differently from alfa-synucleinopathies, where REM-behavior disorder is a quite specific marker, sleep disorders associated with AD seem to be unspecific as they occur with high prevalence also in the elderly population. However, undoubtfully, during the full-blown stages of the disease, sleep disturbances have a significant negative impact on the quality of life of AD patients. Moreover, for caregivers, sleep disturbances is a source of physical and psychological burden and is often cited as a reason for a family’s decision to institutionalize a patient [4].
Prevalence of sleep disturbances has been documented to range from 8.8% to 45.5% in AD-MCI subjects and from 25% to 60% in AD dementia patients according to the categorization of sleep disturbances [42]. Different recent studies demonstrated that sleep abnormalities are present in the earliest stages of AD, even prior to the manifestation of any cognitive impairment. For instance, neuropsychological studies have identified associations between increased sleep latency, short sleep duration or poor sleep quality and concurrent [43] or prospective [44] cognitive test performance. More recently, Ju et al. [45] found that amyloid deposition, as assessed by CSF Aβ levels, was associated with worse sleep quality, as measured by sleep efficiency, in a group of cognitively normal subject at high risk for AD.
The etiology of sleep disturbances in AD is thought to be multi-factorial. First, pathophysiological changes resulting from the disease itself could interfere with the processes that regulate normal sleep (Fig. 1). Moreover, AD patients often suffer from medical and psychiatric comorbidities, which can disrupt the sleep-wake cycle. Furthermore, medications used to treat these comorbidities or to lessen the negative behavioral symptoms of dementia and to slow disease progression are often associated with side effects that negatively affect sleep/wakefulness quality. Finally, environmental factors, such as the chronic bed rest, can in itself contribute to sleep diseases.
The main sleep alterations of AD-MCI and AD dementia patients consist in: 1) sleep-wake cycle alterations; 2) alterations of micro- and macrostructure of sleep; and 3) sleep disorders. In this chapter, we will discuss the main features of every category.
Sleep-wake cycle alterations
Several studies have shown an alteration in circadian rhythmicity in AD patients as expressed by increased nocturnal activity, decreased diurnal activity, fragmented nighttime sleep and a higher frequency and duration of nighttime awakenings and daytime sleep episodes (naps). Actigraphic studies showed disturbed activity profiles, with an increased intradaily variability, and reduced interdaily stability and relative amplitude of the rest–activity rhythm in AD dementia [46]. Moreover, it has been estimated that 2.4–25% of AD dementia patients suffer from a sundown syndrome, consisting in the appearance or increment of neuropsychiatric symptoms in the late afternoon, evening, or night [47]; this manifestation correlates with a phase delay of core body temperature in AD dementia patients [48].
The high frequency of sleep-wake cycle alterations in AD patients seems to be caused by different variables, related in part to environmental factors and in part to a dysregulation of process C. Poor quality of social networks in nursing home residents, low-level assistance by staff in institutions or absence of available caregivers at night at home, caregiver burden and overstimulation by the environment are the main external factors favoring circadian disturbances [49]. Moreover, animal and human studies demonstrated that AD neurodegenerative process involves different brain structures responsible of the circadian sleep regulation, such as the SCN and the pineal gland. Indeed, postmortem studies found neuronal loss and neurofibrillary tangles in the SCN from patients with severe AD. Moreover, overall SCN volume and the expression of the neuropeptide vasoactive intestinal polypeptide and arginine vasopressin, both playing multiple roles in circadian time keeping, have been reported to decrease in AD dementia patients [50]. The neurodegenerative process affects also the circadian-pineal system, resulting an alteration in melatonin production. Indeed, different studies demonstrate that in AD dementia patients melatonin levels are selectively decreased during the night while are increased during the day [51]. Moreover, melatonin levels has been observed to be significantly reduced in postmortem CSF of AD dementia patients, correlating with the progression of neuropathological process [52].
Besides the well-known occurrence of neurodegeneration in the brain, different studies in AD dementia demonstrated that also retinal and optic nerve tissue are affected by the pathological process of AD. For instance, a reduction of the retinal nerve fiber layer thickness [53] and accumulation of Aβ in the retina have been described in AD dementia patients [54]. Moreover, a recent study found an age-related decrease of optic nerve axons and a significant loss of mRGCs in postmortem AD retinal specimens, associated with Aβ deposition [55]. These findings support the hypothesis that the degeneration of mRGCs—the photoreceptors driving circadian photoentrainment—contributes to circadian rhythm dysfunction in AD.
Finally, also the cycles of clock gene expression seem to be affected in AD patients. Indeed, in healthy subjects, clock gene expression is rhythmic in different brain structures, such as the bed nucleus of stria terminalis, the cingulate cortex and the pineal gland [56]. In the brains of AD dementia patients, most of these clocks also exhibited well pronounced 24-hour rhythmicity; however, a state of desynchrony in oscillation between these brain structures in AD dementia patients was found, possibly caused by the degeneration of the SCN cells in AD brains [56]. Further studies are necessary to confirm and better understand these results.
Alterations of micro- and macrostructure of sleep
Sleep changes observed in AD patients are quite similar, but more pervasive, to those found in healthy elderly. Specifically, AD dementia patients have shown to exhibit an increased number and duration of awakenings, an increased arousal index, an increased sleep latency, a decreased sleep efficiency and total sleep time, an increased light sleep and a decreased SWS as well as a reduction of REM sleep [3, 58]. In addition, a recent study showed that also sleep microstructure manifests evident modification in AD dementia patients. In particular, by analyzing Cyclic Alternating Pattern (CAP) parameters (a measure of sleep “resilience”) in amnestic MCI and AD dementia patients, Maestri et al. [59] found a decrease of CAP rate that was correlated with the cognitive decline. Despite the modifications of sleep structure seem to worsen with the progression of the disease, few cross-sectional studies described an inverted u-shaped of the sleep disturbances profile, with moderately demented persons showing more impaired sleep than persons who are in the early and advanced stages of disease [60]. Moreover, sleep structure alterations are supposed to occur years before the clinical AD onset; MCI patients show more evident sleep modification (decrease of REM sleep and increase of SWS fragmentation) in comparison with age-matched healthy subjects [59].
Changes in sleep architecture are also accompanied by modifications in sleep EEG activity and power. For instance, a quantitative EEG study found that in AD dementia patients the delta frequency power did not show the dynamic reduction across NREM/REM sleep cycles; moreover, AD dementia patients exhibited a reduction of slow delta waves (0.4–1.6 Hz) and an increase of fast delta waves (2.0–3.6 Hz) with respect to control subjects [61]. Different studies have focused on the frequency and distribution of spindles in AD, finding a selective decrease of the number of fast spindles. More specifically, Gorgoni et al. [62] in both AD dementia and amnestic MCI patients found a reduction of count and density of fast spindles, that correlated with cognitive status. Moreover, AD dementia patients exhibit a progressive EEG slowing in both wake and REM sleep that increases with disease severity [63]. Finally, a recent study found that density of K-complex is significantly reduced in a full-blown phase of AD-where is positively associated with Mini-Mental State Examination (MMSE) scores, but not during the early stage of the pathology (AD-MCI) [64].
As previously illustrated for sleep/wake disturbances, also macro/microstructural changes seem to result from multiple factors. In particular, these sleep alterations could derive from an early vulnerability to neurodegeneration of specific neuronal populations important for regulation of process S. For instance, decreased galanin-immunoreactive neurons in the VLPO have been related to fragmented sleep in AD dementia patients [65]. Considering the sleep promoting function of VLPO, loss of these neurons is a possible mechanism for decreased NREM sleep and increased awakenings in AD dementia [53]. Moreover, early neurodegeneration of the basal forebrain cholinergic system and brainstem cholinergic neurons, as shown by a significant volume reduction of the nucleus basalis of Meynert even in amnestic MCI subjects [66], could not only affect cognition but also sleep (particularly REM sleep) regulation. Interestingly, recent evidence indicates that β-amyloid causes the degeneration only of the cholinergic neurons which depend on nerve growth factor for their survival, namely the forebrain cholinergic neurons, thus causing attention and learning and memory impairment. Contrarily, it is not fully understood the mechanism that causes the damage of the midpontine cholinergic neurons, implicated in sleep regulations, which express no specific nerve growth factor receptors [67].
SLEEP DISORDERS IN MCI AND AD DEMENTIA PATIENTS
In 2012, the Sleep Study Group of the Italian Dementia Research association (SINDem) published an Italian multicenter study on 431 patients with different sub-type of dementia and MCI showing a high proportion of patients with sleep disorders [68]. Interestingly, patients with AD dementia and amnestic MCI had almost the same frequency of any sleep disturbances without any predicting value on the progression from MCI to AD dementia. The most frequent sleep disorder in AD dementia was sleep-disordered breathing (53.9%), followed by insomnia (48.5%) and excessive daytime sleepiness (44.5%) and, more rarely, REM-behavior disorder (21.6%) and restless legs syndrome (6.4%) [68]. A detailed description and analysis of specific sleep disorders in MCI and AD patients go beyond the aim of this paper (for a review on this topic we suggest [3, 68].
SLEEP AND ALZHEIMER’S DISEASE: A BIDIRECTIONAL RELATIONSHIP?
As discussed above, sleep alterations show a high prevalence in AD patients and frequently precede the onset of cognitive symptoms. Sleep quality and circadian function seem to decline in parallel with the progression of AD pathology and cognitive dysfunction, influenced by both neurodegenerative process and lifestyle changes. Therefore, given the critical role of sleep in multiple functions, such as memory consolidation and “cerebral restitution”, sleep disturbances may represent in itself a promoting or aggravating factor for AD-related manifestation.
Sleep-wake cycle and brain Aβ deposition
The key early event in the pathophysiological cascade of AD consists in Aβ peptide accumulation [69]. CSF Aβ abnormalities are present up to 20 years before AD symptom onset [70]. Thus, Aβ accumulation represents an “upstream” event in the cascade that is associated with “downstream” alteration, such as synaptic dysfunction, neurodegeneration, and eventual neuronal loss. Along with the amyloid cascade, other specific host factors, such as brain and cognitive reserve, or other brain diseases have been demonstrated to influence the response to Aβ and/or the pace of progression toward the clinical manifestations of AD. Indeed, multiple health factors play an important role in increasing the risk of developing cognitive decline. For instance, vascular risk factors (such as hypertension, hypercholesterolemia, and diabetes) and psychiatric disturbances (depressive symptomatology, apathy, and chronic psychological distress) have been linked to increased risk of manifesting MCI and dementia, contributing directly to the effect of AD pathology on the aging brain [69]. Accordingly, sleep disturbances might be one of the factors able to accelerate the progression of AD cascade. Indeed, an increasing number of studies show that different sleep alterations and disturbances in older adults (short sleep duration, impaired sleep consolidation, insomnia, circadian rhythm disorders and sleep-disordered breathing) are associated with an increased risk of pathological changes that precede AD symptoms (e.g., increased amyloid burden or neurofibrillary tangles), as well as an increased risk of AD [5, 71]. Interestingly, although with conflicting results, different studies in elderly found an association between excessive daytime sleepiness and increased risk of cognitive decline [72–74]. Moreover, neuroimaging investigations showed that subjects with somnolence have greater amyloid burden in AD-sensitive brain regions [75].
Candidate mechanisms underlying the pathophysiological association between sleep-wake disruptions and AD pathology are different and not completely clarified [76]. One of the main hypothesis concerns the role of sleep on the clearance of AD-promoting metabolites from the brain. The concentrations of Aβ show diurnal variation with the levels rising during wakefulness and falling during sleep. As illustrated above, it has been suggested that sleep facilitates the removal of different degradation products of neural activity, encompassing Aβ [32]. In particular, natural sleep induces a 60% increase in the interstitial space, leading to a significant improvement of the convective exchange of cerebrospinal fluid with interstitial fluid and, in turn, of the rate of Aβ clearance during sleep [32]. Conversely, awakenings significantly reduced the para-arterial and parenchymal influx of interstitial fluid, suggesting that sleep disruptions could reduce the ability of the glymphatic system to remove Aβ from the brain [32]. Another experimental work, using in vivo microdialysis, showed a correlation between interstitial fluid Aβ amount and wakefulness in mice [77]. In particular, the acute effect of sleep deprivation was an increase in Aβ concentrations while chronic sleep deprivation accelerates Aβ deposition into insoluble amyloid plaques in both wild-type mice and in a transgenic mouse model of amyloidosis [77]. This finding suggested that diurnal Aβ fluctuations are intrinsic to normal cellular physiology and not strictly related to pathology. Moreover, it has been proven that interstitial fluid Aβ levels fluctuate according to the sleep-wake cycle, and not light or dark exposure [77], thus suggesting a specific link between sleep and Aβ deposition.
The presence of Aβ diurnal fluctuation was confirmed also in humans by assessing CSF Aβ levels in healthy subjects via lumbar catheters over a 33–36 hours period [77, 78]. Following these results, it has been postulated that Aβ deposition decrease during SWS, by showing a temporal association with homeostatic process S [76].
Another proposed mechanism by which sleep disturbances could increase the risk of AD is that different sleep alterations increase synaptic activity and facilitate the accumulation of Aβ. Indeed, deep sleep (N3) is the sleep stage in which synaptic activity is most greatly decreased. Thus, poor-quality sleep with a decrease in SWS induces a consequent increased synaptic activity with a higher Aβ release into brain interstitial fluid [1, 42]. Moreover, it is worth to remind that Aβ accumulation is promoted by oxidative stress and deep sleep actively reduces inflammation and oxidative stress and promotes cellular repair [41]. In this perspective, inflammatory immune cascades related to sleep problems, hypoxia related to sleep-disordered breathing or the sympathetic nervous system response to sleep loss could further contribute to promote or quicken the pathological process of AD.
Taken together, these findings suggest that CNS Aβ levels fluctuation is specifically associated with the sleep-wake cycle, both in normal condition and in the presence of AD pathology. Therefore, the disruption of the sleep-wake cycle might play a role in the pathological deposition of Aβ by several mechanisms of action.
Sleep fragmentation and Tau-protein hyperphosphorylation
It has to be noted that brain Aβ deposition alone is not enough to cause AD. Indeed, brain Aβ accumulation represents the upstream phenomenon that triggers AD pathogenesis. However, according to Bloom hypothesis [79] the trigger (Aβ deposition) fires a ‘bullet’ that is represented by the conversion of tau protein from a normal to a toxic state. Moreover, toxic tau enhances Aβ toxicity via a feedback loop [79]. In rodent models of AD, sleep deprivation altered tau phosphorylation [80, 81]. Moreover, tau-deficient mice showed major abnormalities in sleep-wake cycle, including high sleep fragmentation and reduced NREM sleep time [82]. Thus, sleep-wake cycle modifications seem to have reciprocal effect on tau pathology, suggesting that sleep dysfunction might be related with both the ‘trigger’ and the ‘bullet’ of AD pathogenesis (Fig. 2). Indeed, self-report of poor sleep has been associated with both Aβ and tau CSF markers of AD-pathology in humans [83].
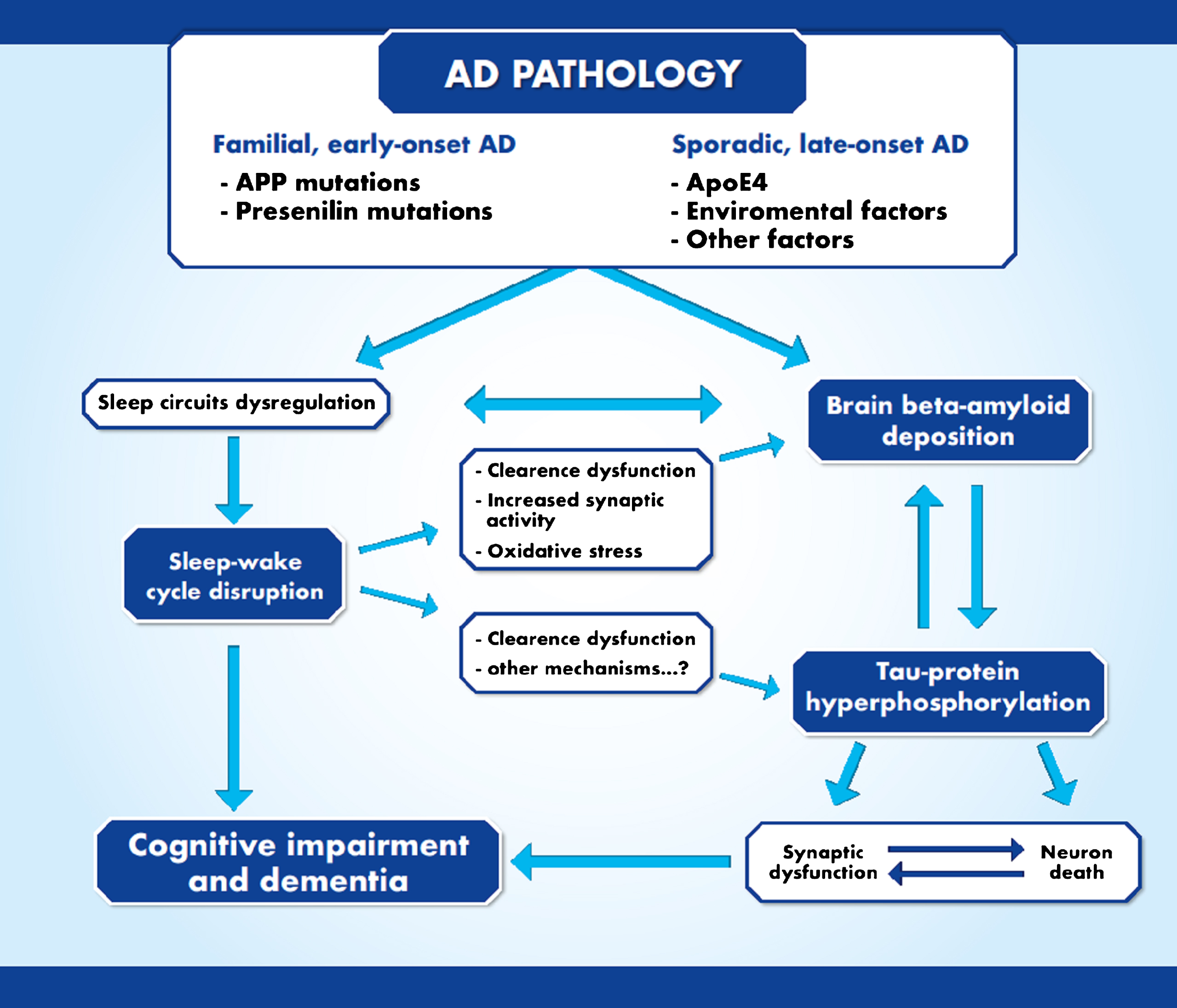
Bidirectional interaction between sleep and Alzheimer’s disease pathophysiology. Sleep disturbances may represent the triggering factor able to increase both brain Aβ deposition and tau protein hyperphosphorylation.
Orexin system and AD pathology
Orexin is an important regulator of the sleep-wake cycle. The hypothesis that hypothalamic orexin dysfunction may actively contribute to AD pathophysiology is supported by the finding that orexin infusion induced an increase of interstitial fluid Aβ amount while dual orexin receptor antagonist significantly decreased the Aβ plaque deposition in transgenic mice [77].
Moreover, individuals carrying a polymorphism of an orexin receptor gene show increased AD risk [84]. Orexin levels have been found to be correlated to specific CSF biomarkers for AD, such as Aβ and tau-protein. For instance, Slats et al. [85] found a correlation between orexin CSF levels and Aβ in AD dementia patients. Moreover, Liguori et al. reported that orexin CSF levels were positively correlated with total tau protein levels and strictly related to sleep impairment, hypothesizing that increased orexin levels may be related to a faster and more marked tau-mediated neurodegeneration occurring in AD dementia patients [86]. In the same study, authors found that decreased MMSE scores correlated with decreased sleep efficiency and REM sleep. Nevertheless, different studies investigated the relationship between the orexinergic system and the pathological changes involved in AD neurodegenerative process, showing contrasting results. Indeed, some studies found a decrease of CSF orexin concentration, while other reported normal or higher level of orexin in AD dementia patients in comparison with healthy subjects [85, 88]. These different findings could depend on the fact that orexin changes across AD stages are not linear, with higher orexin levels in AD-MCI [87] and lower levels in late AD dementia stages [88]. Indeed, in the early stage of the disease, orexin levels may be increased in response to orexinergic neurodegeneration while in later AD stages, degeneration of orexinergic neurons may have overtaken compensation [41]. Considering that the orexin cells are degenerated in AD patients [88–90], it could be hypothesized that AD pathology may induce orexin system degeneration which—through the disruption of the sleep-wake cycle—reinforce AD pathology.
Clinical implication of sleep-wake cycle dysfunction in AD
Other than its restorative function in the brain, sleep has also a critical role in memory and cognitive functions. Sleep deprivation as well as different fragmented sleep–wake patterns have been associated with impairment in memory functions. Moreover, cognitive impairment in AD-MCI and AD dementia correlates with quantitative measures of poor sleep quality, especially NREM sleep impairment. Further, CSF levels of Aβ, tau, and orexin levels have been demonstrated to be associated with both sleep and cognitive alterations, suggesting that sleep may be linked to both disease pathology and the memory decline of ADD [41].
In summary, although Aβ aggregation represent the first identifiable pathological event in patients with AD, clinical signs do not occur until 10–15 years after its initial deposition. Thus, sleep disorders may represent a relevant modifiable factor able to affect AD pathogenesis over the entire course of the disease, from the preclinical phase to advanced phases of AD dementia and, consequently, to promote a faster cognitive decline. Conversely, an aggravated AD pathogenesis deteriorates sleep, forming a vicious cycle during the progression of AD.
TREATMENT OF SLEEP DISTURBANCES IN AD PATIENTS
Treatment of sleep disturbances in elderly and specifically in AD patients represents an important issue for different reasons. Firstly, considering the relevant role of sleep alterations as a modifiable risk factor for dementia, the treatment of sleep problems in cognitive healthy subjects may be a valuable and unavoidable instrument in the prevention of neurodegenerative disorders. Moreover, the early identification and management of specific sleep disorders in AD patients may help counteracting the progression of AD pathology and the consequent cognitive decline. Finally, treatment of sleep alterations may delay or even prevent institutionalization, resulting in significant cost savings and improving the quality of life of AD patients and their caregivers.
For all these reasons, assessment of sleep should be part of clinical evaluation of patients presenting with cognitive impairment. To this end, the sleep SINDem study group has recently elaborated recommendations for the clinical assessment and management of sleep disorders in individuals with mild cognitive impairment and dementia [91]. According with these recommendations, sleep disorders should be carefully investigated using an in-depth sleep history, physical examination, questionnaires and clinical scales and should be validated with the support of a direct caregiver. Moreover, clinician should be aware that sleep disorders tend to occur almost invariably in association in patients with cognitive decline [68]. Before the choice of a specific sleep alteration treatment, sleep assessment in AD patients should include screening and management of secondary causes, including medical (e.g., pain) and psychiatric conditions (e.g., depression) and medication side effects. Non-pharmacological treatment should be considered the first line of therapy, due to the risk of sedation, cognitive symptoms, falls, injuries, and medication interactions with other pharmacological treatments in this specific group of patients. However, pharmacological treatment can be used in some cases, though with particular caution [92, 93]. Considering the aim of this review, here we summarize only the main therapeutic options for insomnia and circadian rhythm sleep disorders.
Non-pharmacological treatment
Different studies have previously focused on sleep hygiene education or light for improving sleep in patients with dementia, however to date there is a paucity of methodologically rigorous research on non-pharmacological sleep interventions in this group of patients [94].
During daytime, AD patients should be encouraged to exercise regularly and to minimize the intake of stimulants and the duration of naps. Time in bed should be reduced and the sleep/wake schedule should be regular. Moreover, nighttime noise and light exposure and sleep disruptions should be reduced also in nursing homes [93]. Although studies on the efficacy of sleep hygiene education in AD patients report contrasting results [95, 96], Ooms et al. [92] suggests that since there are no anticipated adverse effects, good sleep hygiene could represent a first line of treatment of sleep disturbances in this population.
Bright light therapy (BLT) is an intervention used to treat circadian disturbances: the exposure to light activates the retino-hypothalamic tract to the SNC, thus entraining circadian phase. A systematic Cochrane meta-analysis in 2014 [97] examines the BLT efficacy in improving cognition, sleep and quality of life in demented patients and concluded that there is insufficient evidence of its effectiveness. The authors hypothesized that the lack of evidence could have been due to the heterogeneity of the studies (light intensity, duration of the exposition per day, treatment duration, etc). Conversely, a more recent meta-analysis [71] reported a more significant benefit of BLT for sleep disturbances in dementia, for sleep onset latency, total sleep time, time in bed, and sleep efficiency.
Pharmacological treatment
The drugs usually used for sleep disturbances are melatonin, benzodiazepines/z-hypnotics, antidepressants and antipsychotics. Usually benzodiazepines are avoided considering their side effects such as increased daytime sleepiness, rebound insomnia and a worsening of cognitive function. In 2015, McCleery et al. [98] conducted a review of the evidence for drug treatments for sleep disorders in people with AD, founding eligible randomized controlled trials for only three drugs: melatonin, trazodone, and ramelteon.
Endogenous melatonin is a regulator of the biological clock and sleep tendency. Its secretion, halted during the light phase, starts in the evening and reach a peak in the middle of the night. Melatonin exerts both a chronobiotic effect that facilitates the synchronization of the circadian clock with the ambient day–night cycle, and a soporific effect attenuating the wake-promoting signal of the circadian system [99]. Melatonin has also adjunctive cytoprotective, antioxidant, and antiamyloidogenic effects. Studies analyzing the effect of melatonin on sleep and cognition reported discordant results. Cardinali et al. [100] found that fast-release melatonin preparation given in addition to the standard medication improves the cognitive performances and the quality of sleep of patients with MCI. A 2015 meta-analysis [101] concluded that melatonin therapy may be effective in improving sleep efficiency and prolonging total sleep time in patients with dementia; however, no evidence of cognitive function improvement was found. Melatonin does not have major side effects in AD patients and only one study in the above mentioned meta-analysis reported that melatonin therapy was associated with worse withdrawn behavior and depression and that the behavioral effects were ameliorated by BLT [102]. A more recent meta-analysis [103] found that treatment with exogenous melatonin in AD patients has positive effects on sleep quality as assessed by questionnaire, but not for objective sleep outcomes. The authors hypothesized that lack of therapeutic efficacy of melatonin among AD patients could be due to the fact that AD patients have such a severe disruption of normal sleep patterns and diurnal rhythms that exogenous melatonin is not effective or that sleep alteration in AD patients could involve the dysregulation of other different pathways, such as orexin transmission. Another possible explanation is that dosages, release forms and timing (which affects phase shifting action) of melatonin as well as the duration of the treatment are notably variable between studies, thus not allowing a definitive interpretation of the results.
In the last years a prolonged-release melatonin (PRM) formulation (2 mg), has been approved for primary insomnia characterized by poor quality of sleep in people aged≥55 [104, 105]. It represents the only licensed medication containing melatonin and its prolonged release formulation mimics the internal melatonin secretion profile by releasing melatonin gradually. Such a formulation has been shown to preserve sleep structure [106] without withdrawal effects [107] in the absence of negative impacts on psychomotor functions, memory recalls and postural stability in older adults [108, 109]. In a 6-month randomized controlled study, Wade et al. [110] investigated the effects of add-on PRM (2 mg) to standard therapy on cognitive functioning and sleep. They reported that patients treated with PRM had significantly better cognitive performance and sleep efficiency than those treated with placebo, as measured by the Instrumental Activities of Daily Living, MMSE, and Pittsburgh Sleep Quality Index.
Among antidepressants, trazodone is the only drug with some evidence of efficacy in AD patients. In particular, in a double-blind, randomized, controlled trial [111], trazodone 50 mg demonstrated an improvement in total sleep without significant daytime somnolence or negative effects on cognition or functionality.
Ramelteon is a melatonin receptor agonist with high affinity for the melatonin receptors MT1 and MT2 used to treat insomnia. However, the recent review cited above [98] concluded that there was no evidence of any effect of ramelteon on sleep in patients with mild to moderate dementia due to AD.
CONCLUSION
A great amount of studies underline how sleep represents an active phenomenon regulated by a highly integrated network of cortical and subcortical structures. This complex model results to be disrupted at various levels during physiological aging and more deeply during neurodegenerative disorders, thus leading to different sleep alterations. The etiology of sleep disturbances in AD is complex and multi-factorial, involving environmental, clinical and pharmacological factors. In particular, neuropathophysiological changes resulting from the disease itself could actively interfere with the processes that regulate normal sleep. Although the pathological hallmarks of AD have been extensively known and described, several theories have been postulated to explain the physiopathology of this form of dementia. Accordingly with a recent hypothesis [79], Aβ accumulation could represent the “upstream phenomenon” in AD pathogenesis that acts promoting the conversion of tau from a normal to a toxic state; conversely, toxic tau could enhance Aβ toxicity via a feedback loop. In this vicious circle, sleep disturbances may represent the triggering factor able to increase the deposition of both Aβ and tau proteins. Anyway, Aβ deposition is also part of the normal aging process and the main sleep abnormalities observed in AD patients are also present in elderly sleep. In particular, since Aβ deposition and sleep abnormalities seem to have a similar course, there is a significant overlap between prodromal AD and aging. Therefore, to date there is an evident limit to consider sleep abnormalities as potential early AD marker due to lack of specificity of sleep abnormalities in AD. We may speculate that sleep abnormalities, when noted to increase in severity beyond that expected for age, could be a surrogate marker reflecting pathophysiological processes related to neurodegenerative disorders. Therefore, further longitudinal studies examining specific sleep disturbances in the conversion of AD-MCI to AD dementia are needed to confirm this hypothesis and to identify potential markers.
The main current theories reviewed in this paper may help in better understanding the complex interaction between sleep physiology and AD pathophysiology and could provide also some hints regarding sleep disorders treatment in AD patients. Indeed, the management of sleep disorders in AD patients must be based on both deep knowledge and clinical experience, thus to tailor every pharmacological or non-pharmacological intervention taking into account every clinical, environmental and psychological aspect of the single patient.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0041r1).