
Abstract
Background:
Dementia with Lewy bodies (DLB) is a heterogeneous disease in which clinical presentation, symptoms, and evolution widely varies between patients.
Objective:
To investigate the existence of clinical subtypes in DLB based on the initial clinical presentation.
Methods:
81 patients with a clinical diagnosis of probable DLB were consecutively included. All patients underwent a neurological evaluation including a structured questionnaire about neuropsychiatric symptoms and sleep, an assessment of motor impairment (Unified Parkinson Disease Rating Scale subscale III), and a formal neuropsychological evaluation. Onset of core symptoms (hallucinations, parkinsonism, and fluctuations) and dementia were systematically reviewed from medical records. We applied a K-means clustering method based on the initial clinical presentation.
Results:
Cluster analysis yielded three different groups. Patients in cluster I (cognitive-predominant, n = 46) presented more frequently with cognitive symptoms (95.7%, n = 44, p < 0.001), and showed a longer duration from onset to DLB diagnosis (p < 0.001) than the other clusters. Patients in cluster II (neuropsychiatric-predominant, n = 22) were older at disease onset (78.1±5 versus 73.6±6.1 and 73.6±4.2 in clusters I and III, respectively, both p < 0.01), presented more frequently with psychotic symptoms (77.3%, n = 17), and had a shorter duration until the onset of hallucinations (p < 0.001). Patients in cluster III (parkinsonism-predominant, n = 13) showed a shorter time from onset to presence of parkinsonism (p < 0.001) and dementia (0.008).
Conclusions:
Three clinical subtypes of DLB can be defined when considering the differential initial presentations. The proposed subtypes have distinct clinical profiles and progression patterns.
INTRODUCTION
Dementia with Lewy bodies (DLB) is a heterogeneous condition from a clinical and pathological point of view [1, 2]. Pathologically, DLB is defined by the presence of inclusions constituted by aggregates of α-synuclein, known as Lewy bodies and Lewy neurites, which are also hallmarks in Parkinson’s disease (PD). Alzheimer’s disease (AD)-related pathology is a common comorbid pathology in most patients with a pathological DLB diagnosis leading to a pathological heterogeneity [3]. In addition, it has been shown that different distribution patterns of Lewy body and AD pathologies in synucleinopathies influence the clinical phenotype [2, 4].
Clinically, DLB is characterized by a dementia associated with a variable combination of visual hallucinations, fluctuations, parkinsonism and REM sleep behavior disorder (RBD) [1, 6]. However, not all patients develop all symptoms along the course of the disease and the order of appearance and severity may vary across patients leading to a variety of different phenotypes that may be related to the underlying physiopathology. For example, DLB patients with early RBD had lower neurofibrillary and neuritic plaque burden compared with those with late or absent RBD [7] while patients with hallucinations were more likely to have a cerebrospinal fluid (CSF) biomarker profile suggestive of AD copathology [8]. Nevertheless, this variation in the clinical presentation of DLB and its underlying possible pathophysiology has been seldom investigated.
One of the current limitations in the diagnosis of DLB is the lack of established criteria for the prodromal phases of the disease [9]. The previous and current consensus diagnostic criteria of DLB [5, 6] require the presence of dementia. However, the main differences in the clinical picture in DLB are more evident during the first years of the disease and the features tend to be more homogeneous as the disease progresses, likely reflecting a more global synaptic and neuronal degeneration [10]. Therefore, in order to disentangle the high clinical heterogeneity observed in DLB is crucial to focus on the initial presentation.
Cluster analysis is a useful data-driven tool for deep phenotyping previously used in neurodegenerative diseases including PD [4, 11–14]. In DLB, there is just one study using a two-step clustering analysis to investigate the core DLB symptoms in a group of patients with mild dementia [15]. Nevertheless, the definition of clinical subgroups in DLB through data-driven tools has been barely investigated.
In this work, we used a cluster analysis to investigate clinical subtypes in DLB based on the initial clinical presentation and the predominant clinical features during the first years of the disease. Our hypothesis was that these measures could better reflect the different subtypes than measures obtained during later stages of the disease.
MATERIALS AND METHODS
Participants
We included 81 patients with a clinical diagnosis of DLB who were recruited consecutively in the Memory Unit of the Hospital de la Santa Creu i Sant Pau, between July 2013 and December 2015. Patients were referred by primary care physicians and evaluated in the Memory Unit by neurologists with expertise in dementia. Patients with suspected DLB were then referred to one of the investigators (EMR) for a specific evaluation. All patients were at a stage of mild dementia at inclusion, according to a consensus agreement between two experienced neurologists and one expert neuropsychologist based on core clinical criteria for dementia [16] and the score of the Clinical Dementia Rating scale Sum of Boxes (CDR-SB) [17] and were diagnosed using criteria for probable DLB [5, 6]. The time from first symptom onset until the specific evaluation was 5±3.2 years. All patients underwent a general neuropsychological assessment [18] and a specific neurological evaluation directed at detecting DLB signs and symptoms. The neurological evaluation included a structured questionnaire to interrogate about the features and onset of psychotic symptoms (hallucinations in different sensory modalities, sense of presence and passage hallucinations, illusions, misidentifications and delusions) and sleep, including clinical symptoms suggestive of RBD and its onset. The visit also included the assessment of motor impairment by means of the Unified Parkinson Disease Rating Scale subscale III (UPDRS-III). Fluctuations were quantitatively evaluated using the Clinician Assessment of Fluctuations (CAF) and the One Day Fluctuation Assessment Scale (ODFAS) questionnaires [19].
The following variables were collected in all patients in the structured questionnaire and the retrospective review of the medical records and during the first evaluation in the Memory Unit: first neurological or psychiatric symptom attributable to the underlying neurodegenerative process, date of onset of each core diagnostic feature (hallucinations, parkinsonism and fluctuations) and date of fulfilment of criteria for dementia [5]. The date of disease onset was considered as the date of the first symptom reported. Data were coded as missing value when information was absent or inconsistent. We also reviewed the date in which each patient met probable DLB criteria considering all collected and reviewed variables and data.
To investigate the clinical heterogeneity during the first years of the disease we generated a paradigm to obtain a quantitative measure that could capture the length of each predominant core symptom before the diagnosis of DLB. Due to the difficulty for determining accurately the beginning of RBD (lack of bedpartner and/or polysomnography in most cases) and fluctuations, we excluded these core symptoms in this quantification. The earliness of each selected core feature (hallucinations and parkinsonism) was estimated as the relative duration of each core symptom respective to the total disease duration from onset to fulfilment of probable DLB criteria as follows:
Time-to-dementia = the time from the date of first symptom to fulfilment of criteria for dementia.
TimeToDLBCriteria = time from the date of first symptom to the fulfilment of probable DLB criteria. Hallucinations duration = TimeToDLBCriteria –(time from the date of first symptom to the beginning of hallucinations). Parkinsonism duration = TimeToDLBCriteria — (time from the date of first symptom to the beginning of motor symptoms/signs).
Then, to avoid the possible differences in the length of the clinical follow up, the following variables were calculated.
Earliness of hallucinations = (Hallucinations duration/TimeToDLBCriteria)*100 Earliness of parkinsonism = (Parkinsonism duration/TimeToDLBCriteria)*100
These calculated variables represent the percentage of the prodromal phase of the disease in which each single clinical feature was present.
Statistical analyses
We applied the K-means clustering method [15] to classify the subjects assuming three underlying subgroups based on their progression until the patient met probable DLB criteria and the core symptoms of the disease: early presence of hallucinations, early presence of parkinsonism and none of those. The absence of correlation between the selected variables in the cluster analysis (time to dementia, earliness of hallucinations and earliness of parkinsonism) was tested in order to avoid redundant information.
Then, we explored the descriptive features of each group and analyzed their differences. Normality of the quantitative variables was explored with Shapiro-Wilk test. Variables with a non-normal distribution were analyzed by means of a Kruskal-Wallis test, while those with a normal distribution were analyzed by an ANOVA test. A chi-square test was used for categorical variables. Post-hoc analyses were performed also by means of a chi-square test for categorical variables, a Mann-Whitney U test in the case of non-normally distributed quantitative variables and a t-test in the case of quantitative variables with a normal distribution. Bonferroni correction for multiples comparisons was applied when more than two comparisons were made simultaneously.
RESULTS
Demographics
Demographics of the study participants
aCluster I versus II, p = 0.003; Cluster II versus III, p = 0.009; Cluster I versus III, p = 0.7. bCluster II versus Cluster III, p = 0.008; Cluster II versus Cluster I, p = 0.058. cTime from the disease onset until the dementia stage. Cluster I versus III: p = 0.003; Cluster I versus II: p = 0.08. dTime from the disease onset until meet probable DLB criteria. Cluster I versus Cluster III, p = 0.001; Cluster I versus Cluster II, p = 0.009. eTime from the disease onset until the specific evaluation. Cluster I versus Cluster II, p = 0.02; Cluster I versus Cluster III, p = 0.015. fNumber of patients with each initial symptom across diagnoses.
Cluster solution and their clinical features
Clinical features of resultant clusters
aCluster I versus Cluster II and III and cluster II versus Cluster III, p < 0.001. bCluster II versus Cluster I and III, both, p < 0.001. cCluster I versus Cluster II, p < 0.001; cluster II versus cluster III, p = 0.001; Cluster I versus Cluster III, p = 0.09. dCluster I versus Cluster II and Cluster I versus Cluster III, p < 0.001. eCluster I versus Cluster II, p = 0.008. fCluster I versus Cluster II, p = 0.03. gCluster I versus Cluster II, p = 0.03. iCluster I versus Cluster II, p = 0.03. UPDRS, Unified Parkinson Disease Rating Scale; Pk, parkinsonism; VH, visual hallucinations; ODFAS, One Day Fluctuation Assessment Scale; CAF, Clinician Assessment of Fluctuations; Del, delusions; Mis, misidentifications; RBD, REM Sleep Behavior Disorder.
Cluster I or cognitive-predominant (n = 46)
The first symptom of this cluster was mostly cognitive impairment with only two patients presenting with psychotic symptoms. Visual hallucinations (54.3%), parkinsonism (97.8%), and fluctuations (84.8%) appeared latter than in other clusters (all comparisons, p < 0.001, Table 2). Other psychotic and perceptive symptoms were less frequent, with a later onset when compared with cluster II. Two patients in this cluster referred isolated auditory hallucinations.
This cluster showed a longer time to dementia than cluster III (p = 0.003) and a tendency for a longer evolution until the dementia stage than cluster II (p = 0.08, Table 2).
Cluster II or neuropsychiatric-predominant (n = 22)
This group had an older age at disease onset compared with the other two clusters (both p < 0.01, Table 1). The most common first symptom in this cluster was psychosis (n = 17, 77.3%). All patients had visual hallucinations and their mean onset was 0.3±0.6 years after their first symptom, significantly earlier than patients in the other clusters (p < 0.001).
Misidentifications were more frequent in this cluster when compared with cluster I (p = 0.03). The frequency of sense of presence was lower than 35% in all clusters. However, the onset of this feature tended to be earlier in cluster II compared to the other clusters (Table 2). The onset of passage hallucinations (26.2%) was earlier in cluster II compared to Cluster I (p = 0.03). We did not find significant differences in the frequency of delusions between clusters (Table 2).
Cluster III or parkinsonism-predominant (n = 13)
The mean age at onset of the disease in this cluster was 73.6±4.2 years, younger than those patients in Cluster II (p = 0.009) and comparable with those in cluster I. The most common first symptom was cognitive impairment (six patients), followed by motor symptoms (four patients) and psychosis (three patients). All patients in cluster III had parkinsonism, with an onset of 0.3±0.6 years from the first symptom. Mean III-UPDRS score in cluster III was 26.1±10.6 without reaching statistical significance when comparing with the other two clusters. At inclusion, 11 patients out of 13 had a clinical suspicion of RBD. The time-to-dementia in this group was shorter compared to Cluster I (p = 0.003).
Differential evolution from the disease onset of these three clusters is represented in Fig. 1.
Differential evolution from disease onset of the different resultant clusters, focused on core symptoms in DLB.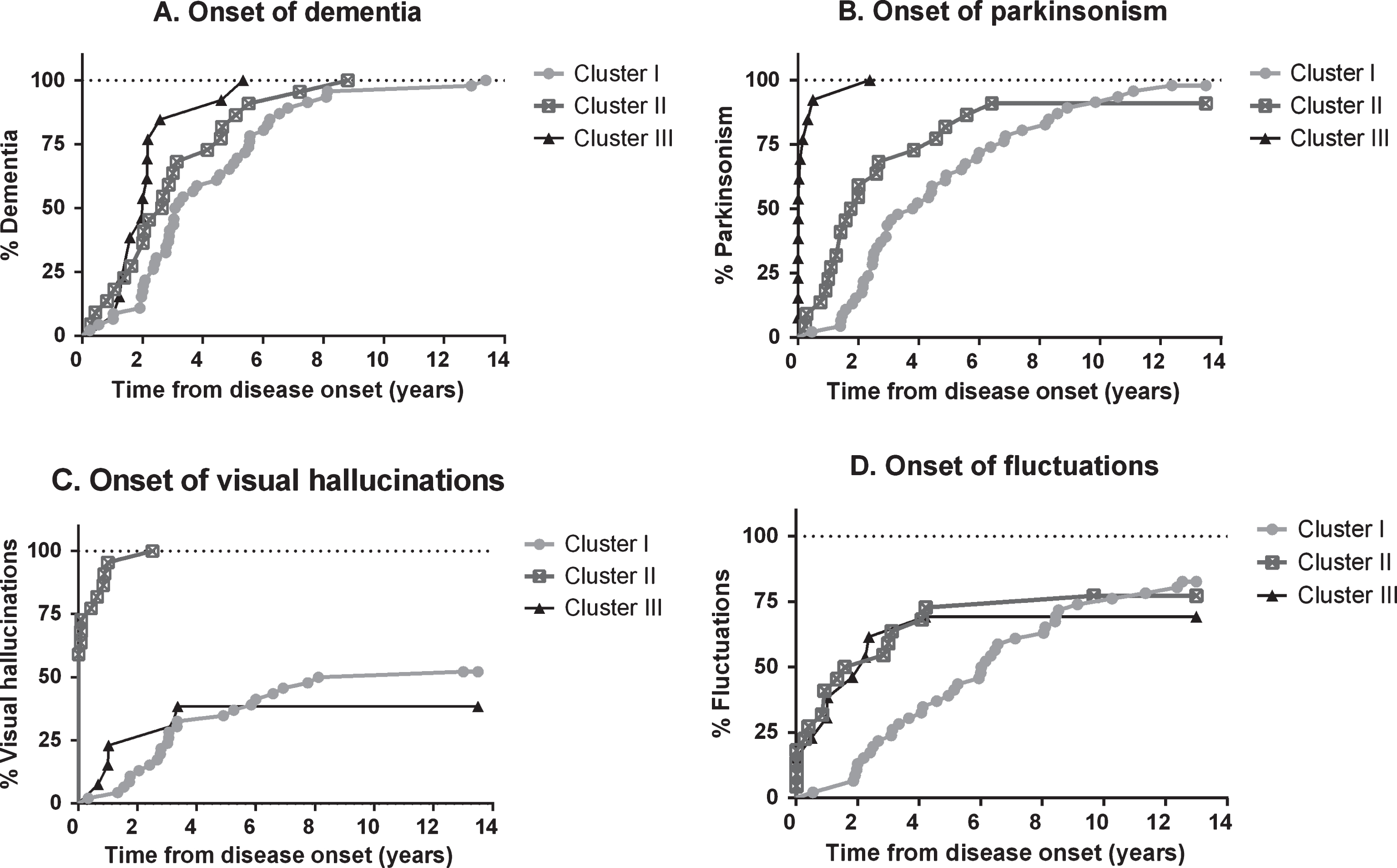
Neuropsychological assessment
Neuropsychological data of the resultant clusters
aCluster I versus Cluster II, p = 0.02; Cluster I versus Cluster III, p = 0.06. *Scalar scores according to age and educational level. MMSE, Mini-Mental State Examination; WAIS, Wechsler Adult Intelligence Scale; TMT-A, Trail Making Test A; FCSRT, Free and Cued Selective Reminding Test; VOSP, Visual Object and Space Perception battery; TMT-B, Trail Making Test B.
DISCUSSION
In this study, we used for the first time a cluster analysis to characterize the different clinical subtypes of DLB based on the main clinical features during the prodromal phase of the disease. We obtained three clusters: a cognitive-predominant (cluster I), a neuropsychiatric-predominant (cluster II), and a parkinsonism-predominant (cluster III). These clusters differed in their clinical presentation, but also in the disease course. The cognitive-predominant cluster was characterized by a long prodromal phase in which cognitive symptoms, mainly memory complaints, predominated. Patients in this cluster had hallucinations and other psychotic symptoms less frequently than patients in other clusters and a later onset. Parkinsonian features also appeared later as compared to the other clusters. The neuropsychiatric-predominant cluster was characterized by an early onset of hallucinations and a higher frequency of psychotic symptoms as the presenting feature. Patients in this cluster had also a late disease onset compared to the other two. The parkinsonism-predominant cluster was featured by predominant motor symptoms during the first years of the disease, a faster progression from symptom onset to dementia compared to the cognitive-predominant cluster, and a low frequency of hallucinations compared to the neuropsychiatric-predominant cluster.
We have identified only one study using a data-driven approach to investigate the features of DLB in a cohort of patients with mild dementia [15]. The authors found that the classical features of DLB aggregated together in their cluster solution supporting the diagnostic clinical criteria of DLB. However, clinical heterogeneity in DLB has not been specifically investigated by a clustering method. In contrast, previous studies have applied different clustering methods in PD avoiding subjective biases [4, 20–24]. Similar to our work, the studies performed in PD suggested subgroups differing in their age-at-onset and rate of progression [21, 24]. In agreement with our data, previous studies have described that patients with earlier onset of parkinsonism are younger than those with earlier dementia or psychosis [25, 26]. It is remarkable that in some studies [21, 24] the late-onset subgroup in PD was characterized by a faster clinical decline, differing from our late-onset cluster (cluster II, neuropsychiatric-predominant), defined by the presence of early hallucinations, but not a faster progression. Another described predictor for a faster progression in PD and DLB is the presence of concomitant AD pathology measured by CSF or amyloid-PET [8, 27]. It will be important to determine in future studies if those rapid evolution DLB clusters may have more frequency of concomitant AD.
Selection of variables in cluster analysis is critical for the obtained solutions. In our study we focused on the clinical features present during the prodromal phase of the disease. These features may capture better the initial topography of neuronal dysfunction and loss [23, 28]. As the disease progresses, symptoms and signs tend to converge, reflecting more severe and widespread Lewy body pathology and neurodegeneration resulting in a more homogeneous syndrome. Although criteria for prodromal DLB have been proposed [9, 30], they are not completely accepted or validated. In fact, one of the challenges in defining criteria in the prodromal phase of DLB is the high heterogeneity observed during this disease stage [30, 31]. Our findings clearly support the notion that clinical heterogeneity is larger during the prodromal than the dementia stage. Our results showed that the neuropsychiatric and the parkinsonian-predominant clusters presented with clinical features that are characteristic of an underlying synucleinopathy. However, the cognitive-predominant cluster consisted of a syndrome of amnesic mild cognitive impairment (MCI) with a very slow progression. At the clinical level, this cluster partially overlaps with the typical amnestic MCI of AD [32, 33]. In fact, previous clinicopathological studies have suggested that Lewy body disorders should be considered in the differential diagnosis of MCI [31], as a subset of subjects convert to DLB during follow-up [34]. The clinical heterogeneity in the prodromal phase of DLB and the potential overlap with a subset of patients with prodromal AD reinforces the need of developing specific CSF and/or imaging biomarkers for the initial stages of the disease.
It will be important to investigate how clinical heterogeneity in DLB relates to specific neuropathological profiles. Differences in the initial clinical syndrome could be the result of the different regional burden of α-synuclein pathology [3, 34]. A higher disruption of brainstem integrity could translate into more severe motor features, while a predominant cortical distribution may translate into predominant cognitive or perceptive disturbances [3, 4]. In addition, the common presence of coincident pathologies such as AD [4, 35], could also influence the clinical manifestation of Lewy body disorders. The main strength of this study is the detailed characterization of the predominant clinical features during the prodromal phase by a replicable equation. This has been possible due to a structured chart review and questionnaire specifically oriented to detail the main clinical features during the prodromal phase of DLB. This data-driven approach allowed us to avoid multiple possible biases by defining particular time points.
The main limitations of the study are that part of the data was collected retrospectively, only a small subset of patients had imaging and/or CSF biomarkers, there was no validation in an independent clinical cohort and the absence of neuropathological findings to validate these clusters. In addition, in this study patients were recruited entirely in a memory unit, and this bias could underestimate the proportion of DLB patients with an initial motor presentation of the disease.
In summary, we propose three subtypes of DLB that differ in their clinical manifestations and progression patterns. It will be important to validate these subtypes in independent clinical cohorts with autopsy confirmation to investigate differences in biomarkers and neuropathological traits among the proposed clusters.