
Abstract
It is widely accepted that the endocannabinoid system (ECS) is a modulator of neuroinflammation associated with neurodegenerative disorders, including Alzheimer’s disease (AD). Thus, expression of the cannabinoid receptor 2 (CB2) is induced in plaque-associated microglia and astrocytes in brain tissues from AD patients and in genetic mouse models expressing pathogenic variants of the amyloid precursor protein (APP). However, the exact mechanism of CB2 signaling in this mouse model remains elusive, because the genetic deletion of CB2 and the pharmacological activation of CB2 both reduced neuroinflammation. Here, we demonstrate that CB2 deletion also improved cognitive and learning deficits in APP/PS1*CB2–/– mice. This was accompanied by reduced neuronal loss and decreased plaque levels and coincided with increased expression of Aβ degrading enzymes. Interestingly, plaque-associated microglia in APP/PS1*CB2–/– mice showed a less activated morphology, while plaques were smaller and more condensed than in APP/PS1 mice. Taken together, these results indicate a beneficial effect of CB2-deficiency in APP transgenic mice. CB2 appears to be part of a protective system that may be detrimental when engaged continuously.
INTRODUCTION
Alzheimer’s disease (AD) is one of the most prevailing neurodegenerative diseases in Western countries, affecting up to 70% of the more than 35 million people suffering from dementia [1, 2]. Beside neuropathological characteristics, such as amyloid-β (Aβ) plaques and neurofibrillary tangles, increased neuroinflammatory markers are main characteristics of the disease. Neuroinflammation in AD is described as enhanced microgliosis and astrocytosis induced by the presence of Aβ plaques. Microglia and astrocytes in turn respond by increased production of pro-inflammatory mediators such as chemokines, cytokines, nitric oxygen, or free radicals [3]. Release of the pro-inflammatory components is closely associated with neuronal dysfunction and cell death. However, the role of microglial cells in AD is controversial. For example, it has been shown that microglia can phagocyte Aβ [4] and constitute a protective barrier around toxic hotspots in Aβ plaques [5] This suggests both a beneficial and detrimental role for microglia in AD-dependent neuroinflammation.
Several studies have indicated that the endocannabinoid system (ECS) plays an important role in neuroinflammation [6, 7]. The ECS is a retrograde messenger system, consisting of lipid signaling molecules that bind to at least two G-protein-coupled receptors. One of those, CB2, is primarily expressed on immune cells such as B-cells, T-cells, macrophages, dendritic cells, and microglia [6, 7]. CB2 receptors are very weakly expressed in the healthy CNS, but they are readily induced under various pathological conditions, primarily in microglia and in infiltrating immune cells.
In the context of AD-dependent neuroinflammation, increased CB2 expression has been found in plaque-associated microglia in human brain tissue of AD patients [8] and AD mouse models [9, 10]. CB2 activation with JWH-133 in transgenic APP/PS1 mice improved cognitive performance, decreased microglial reactivity and the expression of pro-inflammatory cytokines [11]. In two other mouse models of AD, extended oral cannabinoid treatment led to a reduction of Aβ levels and plaque load [12, 13]. Moreover, rats that were intracerebrally injected with Aβ1 - 40 fibrils showed a decreased secretion of IL1β and diminished CB2 receptor expression after treatment with the CB2 receptor agonist MDA7 [14]. These data suggest an anti-inflammatory role for CB2 in AD-dependent neuroinflammation. We have used genetic mouse models to determine if a deletion of the CB2 encoding cnr2 gene also modulates disease vulnerability. This is of importance, because functional polymorphisms in the human CNR2 gene, which reduce the signaling efficacy of the receptor have been described and were associated with schizophrenia [15], postmenopausal osteoporosis [16], and low bone mass [17]. We found that the genetic deletion of CB2 in APP/PS1 mice also caused a significantly reduced neuroinflammation and a decrease in Aβ plaque load [18]. In the present study we further investigate the effects of CB2-deficieny in transgenic APP/PS1 mice using stringent spatial memory tests, a detailed analysis of neuronal loss and the interaction of microglial cells and Aβ plaques.
MATERIALS AND METHODS
Animals
The generation of CB2–/– mice has been described previously [19, 20]. APP/PS1 mice were purchased from Charles River Laboratories (B6.Cg-Tg (APPswe(K594 N/M595 L)/,PSEN1dE9)85Dbo/J; Charles River Laboratories, Germany GmbH) and crossed with mice on a CB2–/– genetic background to obtain APP/PS1*CB2–/– mice. Wildtype control mice (WT) are non-transgenic littermates on C57BL6/J background. All animals were bred and housed in a specific pathogen-free animal facility under standard animal housing conditions in a 12 h dark-light cycle with ad libitum access to food and water. Experiments were carried out with mice at the age of 9 and 14 months. Mice were genotyped by polymerase chain reaction (PCR) using DNA from tail tissue. Primers: CB2 common (5’-GTC GAC TCC AAC GCT ATC TTC-3’), CB2 wild-type (5’-GTG CTG GGC AGC AGA GCG AAT C-3’), CB2 knock-out (5’-AGC GCA TGC AGA CTG CCT-3’), PSEN1-F (5’-GGT CCA CTT CGT ATG CTG-3’), and PSEN1-R (5’-AAA CAA GCC CAA AGG TGA T-3’). Experimental procedures complied with all regulations for animal experimentation in Germany and were approved by the Landesamt für Natur, Umwelt und Verbraucherschutz in Nordrhein-Westfalen, Germany (AZ 87-51.04.2011.A041, 8.84- 02.05.20.11.101). The number of animals that were used for individual experiments are given in the corresponding figure legends.
Morris water maze
To assess spatial learning and memory, the Morris water maze (MWM) was used. Thereby, mice are trained to find a hidden platform in a large pool of turbid water using visual cues. Learning ability of the mouse is given by the escape latency, i.e., the time to find the hidden platform. In this study, mice were trained to find the hidden platform over 5 consecutive days with 4 trails per day and an intertrial time of 1 h. After MWM experiments, animals were sacrificed for molecular and immunohistochemical studies.
Organ extraction
For immunohistochemistry, mice were anesthetized and perfused transcardially with PBS and subsequently with 4% w/v paraformaldehyde. Afterwards, brains were incubated overnight in 20% sucrose solution, embedded in TissueTek and stored at –80°C. For RNA extraction, mice were sacrificed by cervical dislocation. Both hippocampi were isolated, snap-frozen and stored at –80°C.
RNA isolation and cDNA synthesis
Hippocampal RNA was extracted by the TRIzol extraction protocol. Briefly, frozen tissue was homogenized in 1 ml or 800μl TRIzol (Invitrogen, Camarillo, CA). After precipitation with isopropanol, RNA was stored at –80°C. For cDNA synthesis, 400 ng RNA was incubated for 5 min at 65°C and then reverse transcribed at 42°C for 50 min. A total volume of 20μl included 4μl first-strand buffer (Invitrogen), 2μl 0.1 mol/L DTT, 1μl 10 mmol/L dNTPs, 1μl oligo(dT)20 primer (Invitrogen), and 200 U SuperScript II reverse transcriptase (Invitrogen).
Quantitative Real-Time PCR (qPCR)
qPCR of cDNA samples was performed using an ABI 7900 sequence detector (Perkin-Elmer, Waltham, MA) and Universal PCR Master Mix (Perkin-Elmer). A 25 ng quantity of cDNA was used per reaction. A standard program was applied as follows: step 1 (1x) 95°C, 10 min; step 2 (40x) 95°C, 15 s and 60°C, 1 min. TaqMan primer (all Applied Biosystems, Foster City, CA): adam10 (Mm00545742_m1), adam17 (Mm00456428_m1), ager (Mm01134790 g1), app (Mm01344172_m1*), bace1 (Mm00478664_m1), cnr1 (Mm01212171_s1*), cnr2 (Mm00438286_m1), daglα (Mm00813830_m1*), gapdh (Mm9999991_g1), marco (Mm00440265_m1*), magl (Mm00449274_m1*), mmp9 (Mm00442991_m1*), nape-pld (Mm00724596_m1*), nep (Mm00485028_m1*), tlr4 (Mm00445273_m1).
Immunohistochemistry
Sagittal brain slices of 14μm thickness were stained with primary anti-mouse antibodies diluted 1:500 overnight at 4°C. Incubation with secondary antibody was done for 1 h at room temperature, followed by staining with 4’,6-diamidino-2-phenyl-indole DAPI. Antibodies: Iba1 (AB_10641962), Lamp1 (AB_2134499), Parvalbumin (AB_369891), NeuN (AB_177621), Goat-anti-rabbit Cy3 (AB_10563288), Donkey-anti-rat AF647 (AB_2535864). For plaque analysis, slices were incubated with 0.025% (w/v) thioflavin S solution (1:2 EtOH/PBS) for 1 min. Pictures were taken with an inverse fluorescence microscope (10x/ 20x) (Zeiss Axiovert 40 CFL, AxiocamMRm) or a confocal laser scanning microscope (63x) (Leica TCS SP8). For microglial analysis, z-stacks were taken with a step size of 1μm. Neuronal loss was quantified by ImageJ software by either counting NeuN+/Parv+ cells or measuring immunopositive area and its average thickness (neuronal bound).
Microglial cell reconstruction and analysis of microglial ramification index
Microglial ramification index was analyzed in ImageJ, using custom-written plugins. Per animal, 6–9 plaque-associated microglial cells, i.e., microglial cells with direct contact to thioflavin-positive plaques – were randomly selected. For each selected cell, a single-cell image was semi-automatically generated by an investigator, who was blinded to the experimental groups and selected the microglial cell in all z-slices of a confocal stack using a custom-written ImageJ plugin. The resulting single-cell images were segmented using a custom-written image segmentation plugin, which, for each single-cell image: 1) calculated an intensity threshold (“Huang”-algorithm, implemented in ImageJ) in an 8-bit converted, 0.5-fold scaled and maximum-intensity projected copy of the original image, of which the specific single-cell image had been previously derived, and 2) applied the threshold to the unmodified single-cell image. Segmented single-cell images were subjected to a custom-written 3D particle filter that removes all 3D particles below 100 voxel. Next, volume and surface of the remaining particles in the image were determined. The 3D microglial ramification index was defined as:cellsurfacearea/(4π · (3 · cellvolume)/(4π)) 2/3, which describes the ratio of cell surface to cell volume and serves as a sensitive measure for cell shape complexity [21].
Plaque reconstruction and plaque-microglia interaction analysis
Above described custom-written image segmentation plugin was used to segment microglial cells (see settings above) and plaques from image background (threshold calculation in an 8-bit-converted, 0.5-fold scaled maximum-intensity projection; “Otsu”-algorithm, implemented in ImageJ). After removing 3D objects from segmented images below a certain size threshold (microglia channel: 100 voxel; plaque channel: 300 voxel), the plaque volume, plaque surface, plaque morphology index, microglia coverage, plaque-microglia co-localization, and maximum plaque radius were determined in the segmented images by a custom-written ImageJ plugin. The plaque shape index was determined as plaque surface area/(4π · (3· plaque volume)/(4π))2/3, as described previously [21]. The microglia coverage was calculated as the fraction of the plaque surface covered by microglia. The plaque-microglia co-localization was calculated as the fraction of plaque volume co-localized with microglial signal. The maximum plaque radius was calculated as the radius from the center of the plaque to the outer-most plaque point. Plaques were excluded from analysis when the largest diameter was at either end of the z-stack. The number of microglial cells associated to a plaque was manually quantified in maximum-intensity projection images by a blinded, trained investigator and results were re-evaluated by a second observer. A microglial cell was defined as plaque-associated when the distance of the cell soma contour to the plaque center was smaller or equal to the double maximum plaque radius. In total, 132 plaques of APP/PS1 and 151 plaques of APP/PS1*CB2–/– mice of 5 animals per genotype were analyzed.
Statistical analysis
Statistical analysis was performed using two-way analysis of variances (ANOVA), followed by Bonferroni post hoc or Fisher LSD tests if appropriate (version 6.0d, Prism software, GraphPad, USA). A value of p < 0.05 was considered significant.
RESULTS
Differential ECS expression in control and APP/PS1 mice
To determine if the expression of components of the ECS was altered in APP/PS1 transgenic mice, we performed qPCR from hippocampal tissue of middle-aged (9 months old) and aged (14 months old) wild type (WT), CB2–/–, APP/PS1, APP/PS1*CB2–/– mice.
Figure 1 shows that expression of cnr1 was altered in aged APP/PS1 mice compared to WT mice as well as middle-aged APP/PS1 mice. Expression of cnr1 was significantly downregulated in aged APP/PS1*CB2–/– mice compared to both middle-aged APP/PS1*CB2–/– mice as well as to aged WT and CB2–/– mice (Fig. 1A). For cnr1 expression, we observed a genotype, age, and interaction effect (Supplementary Table 1A). For cnr2 expression, a genotype and age effect as well as an interaction effect was detected (Fig. 1B, Supplementary Table 1A). However, the genotype and interaction effect was due to the data obtained from APP/PS1*CB2–/– mice. The genetic mutation in our CB2–/– mouse model disrupts the coding region so that no functional protein can be generated, but transcripts are still produced from the mutant gene locus that are detected by the expression assay [18]. The data for cnr2 in CB2–/– and APP/PS1*CB2–/– mice can therefore not be compared to WT animals. Expression of the main ligand 2-AG synthesizing enzyme daglα (Fig. 1C) was enhanced in aged APP/PS1*CB2–/– compared to aged WT and CB2–/–, as well as to middle-aged APP/PS1*CB2–/– mice. Genotype and age effects, as well as an interaction effect were detected (Fig. 1C, Supplementary Table 1A). Expression of degrading enzyme magl (Fig. 1D) was not significantly altered in any of the four middle-aged and aged genotypes when compared to control (WT) mice of the equivalent age group. However, an age-dependent increase in magl expression in aged mice was detected (Supplementary Table 1A). Finally, expression of the AEA synthesizing and degrading enzymes faah (Fig. 1E) and nape-pld (Fig. 1F) were evaluated. Both enzymes were expressed at significantly lower levels in CB2–/–, APP/PS1, APP/PS1*CB2–/– mice than in WT mice at the age of 14 months (aged mice) Expression of both genes was not altered in middle-aged mice (Fig. 1E, F).

Expression levels of endocannabinoid system components in middle-aged and aged mice. Shown are gene expression levels of endocannabinoid receptors cnr1 (A) and cnr2 (B), 2-AG synthesizing and degrading enzymes daglα (C) and magl (D) and AEA synthesizing and degrading enzymes faah (E) and nape-pld (F). RNA was analyzed from middle-aged (9 months old) and aged (14 months old) WT, CB2–/–, APP/PS1 and APP/PS1*CB2–/– mice. N = 5–8 mice per genotype; samples were run in triplicates. Data were analyzed by two-way ANOVA followed by Bonferroni Post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001, # = significance to corresponding middle-aged (9 months old) genotype.
APP/PS1*CB2–/– mice show morphological differences of microglia and Aβ plaques
It has been suggested that interaction of microglial cells with Aβ plaques may alter Aβ plaque morphology and growth by providing a protective barrier around them [5], which in turn influences Aβ plaque toxicity. As our previous studies revealed a more pronounced effect of CB2 deletion on AD pathology in aged (14 months old) mice, we focused our following studies on this age group [18].
First, we analyzed the ramification index of plaque-associated microglial cells, which is a sensitive marker for the complexity of the cellular shape of microglia. Microglial shape complexity decreases during microglial activation and is reduced in Alzheimer’s disease mouse models [22]. Ramification of plaque-associated microglial cells in APP/PS1*CB2–/– mice was significantly increased as compared to APP/PS1 mice (Fig. 2A, B), suggesting that plaque-associated microglial cells in APP/PS1*CB2–/– mice showed reduced signs of morphological activation.

Analysis of microglia and plaque morphology by MultiQ. Representative confocal image (A) and 3D ramification index (B) of Iba-1 + microglial cells associated with thioflavin + Aβ plaque in aged (14 months old) APP/PS1 and APP/PS1*CB2–/– mice. Microglial cells were analyzed using a custom-written ImageJ analysis tool [21]. Representative confocal image of Iba-1 + microglial cells associated with thioflavin + Aβ plaque and specification of microglia-plaque interaction parameters (C). Plaque shape index (D), microglia coverage (E), plaque-microglia co-localization (F), and number of microglia per plaque (G) in aged (14 months old) APP/PS1 and APP/PS1*CB2–/– mice. A low plaque morphology index corresponds to round and condensed plaques. n = 131 plaques in APP/PS1 and 151 plaques in APP/PS1*CB2–/– mice from 5 animals per genotype. Significance of data with normal distribution was tested using the unpaired t-test (B). Significance of data with non-normal distribution was tested using the Mann-Whitney test (D), *p < 0.05, **p < 0.01, ***p < 0.001. Grouped data (E, F, G) were analyzed by two-way ANOVA (Fisher’s LSD test). Scale bars = 20μm.
Next, we investigated Aβ plaque morphology in more detail (Fig. 2C). Plaques in APP/PS1*CB2–/– mice had a significantly lower plaque shape index, indicating that they are more rounded and condensed than plaques in APP/PS1 mice. Since a more condensed plaque morphology may be induced by an increased microglial plaque shielding [5, 23], we next analyzed the plaque surface fraction that is covered by microglial cells in APP/PS1*CB2–/– mice (Fig. 2C). Additionally, we determined the co-localized volume of plaque and microglial signals, which indicates the degree of plaque penetration by microglial processes. The covered plaque surface (Fig. 2E) and the co-localized volume (Fig. 2F) were unchanged between APP/PS1 and APP/PS1*CB2–/– mice. Thus, plaque shielding by microglia is unlikely be responsible for the more condensed morphology of plaques in APP/PS1*CB2–/– mice. This is supported by the fact that the total number of plaque-associated microglia did not differ between APP/PS1 and APP/PS1*CB2–/– mice (Fig. 2G).
Reduced amyloidosis in APP/PS1*CB2–/– mice
To determine if Aβ processing was altered, we analyzed plaque load as well as gene expression levels of APP secretases, amyloid receptors and degrading enzymes in aged APP/PS1 and APP/PS1*CB2–/– mice.
Thioflavin S staining in cortex and hippocampus from middle-aged and aged animals revealed a significant reduction of Aβ plaque load in the hippocampus of APP/PS1*CB2–/– mice compared to WT mice in both age groups (Fig. 3A, lower graph). Similar results were observed in the cortex, but this effect was only significant in 14-month-old mice (Fig. 3A, upper graph). Therefore, further experiments on CB2-dependent effects on amyloidosis were conducted in aged (14 months old) mice.

Amyloidosis in middle-aged and aged mice. Quantification of amyloid β plaques in cortex (A) and hippocampus (B) of middle-aged (9 months old) and aged (14 months old) APP/PS1 and APP/PS1*CB2–/– mice as evaluated by thioflavin S staining. Aged APP/PS1*CB2–/– mice show reduced plaque load compared to APP/PS1 mice in both brain areas. N = 15–22 mice per genotype. Data were analyzed by one-way ANOVA followed by Bonferroni Post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001. Gene expression of APP cleavage enzymes (B), Aβ specific degrading enzymes (C) and Aβ receptors (D) were normalized to expression levels of the corresponding reference gene glycerin-aldehyde-phosphate-dehydrogenase (GAPDH). Expression was analyzed in hippocampal tissue of aged (14 months old) WT, CB2–/–, APP/PS1 and APP/PS1*CB2–/– mice. N = 5–8 mice per genotype; samples were run in triplicates. Data were analyzed by two-way ANOVA followed by Bonferroni Post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001, # = significance to corresponding wildtype control.
Gene expression levels of APP secretase adam17 were significantly enhanced in aged APP/PS1*CB2–/– mice compared to WT mice, whereas expression levels of adam10 and bace were not altered (Fig. 3B). We also observed slightly enhanced expression levels of both adam genes when comparing APP/PS1*CB2–/– mice to APP/PS1 mice, but this increase was not significant. Expression levels of amyloid-degrading enzymes ide and mmp9 were significantly increased in APP/PS1*CB2–/– mice compared to APP/PS1 and CB2–/– mice. In contrast, we could not observe any differences in nep expression when comparing both genotypes (Fig. 3C). Nevertheless, nep expression was significantly reduced compared to CB2–/– control mice. Finally, gene expression levels of Aβ receptors tlr4 and ager were significantly enhanced in APP/PS1 mice when compared to WT mice. APP/PS1*CB2–/– mice displayed similar expression levels as WT mice. This reduced expression was significantly different in APP/PS1 mice for ager, but not for tlr4. Reduced levels of Aβ plaque load are in line with lowered expression levels of APP secretases and amyloid receptors and suggest an impact of CB2 receptors on Aβ processing.
Absence of CB2 improves cognitive behavior in APP/PS1 mice
Based on the observed beneficial effects of CB2 deletion on AD pathology, we evaluated cognitive behavior in APP/PS1*CB2–/– mice. Neurodegenerative processes as well as chronic expression of neuroinflammatory mediators and increasing Aβ accumulation have been linked to deficits in spatial learning and memory in APP/PS1 mice [24–26]. We have previously shown an improvement of cognitive function in young (6 months old) APP/PS1*CB2–/– mice, but not in middle-aged and aged mice [18] by using MWM as an established paradigm to investigate spatial learning and memory. Therefore, we now chose a more stringent paradigm by increasing the intertrial time and investigated cognitive performance of middle-aged (9 months old) and aged (14 months old) WT, CB2–/–, APP/PS1 and APP/PS1*CB2–/– mice in the MWM test.
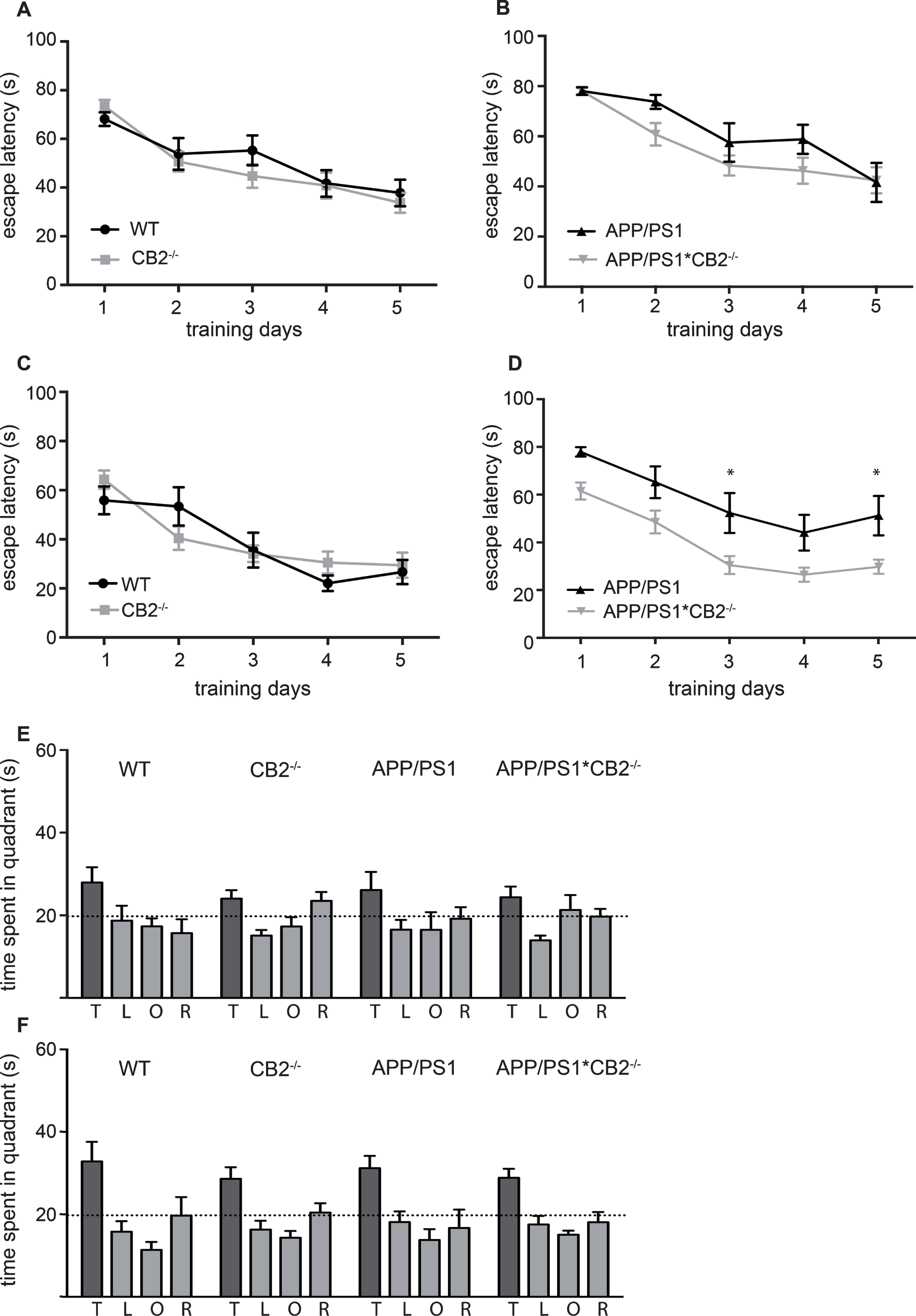
Cognitive performance of middle-aged and aged mice. Learning curves show mean escape latency of middle-aged (9 months old) (A,B) and aged (14 months old) mice (C,D). Statistical comparison was conducted between WT (black circles) and CB2–/– (grey squares) (A,C) as well as APP/PS1 (black triangles) and APP/PS1*CB2–/– (grey triangles) (B,D). Probe trial of middle-aged mice is shown in (E) and aged mice in (F). N = 8–16 mice per genotype; data were analyzed by two-way ANOVA followed by Bonferroni post hoc test, *p < 0.05, **p < 0.01, ***p < 0.001
First, we wanted to know if the deletion of CB2 itself has any effect on spatial learning and memory. Swim speed was not affected by the genotype (data not shown). CB2–/– mice showed similar escape latencies as WT mice during the acquisition phase in both age groups, middle-aged (Fig. 4A) and aged mice (Fig. 4C). APP/PS1 mice showed an increased escape latency already at the age of 9 months when compared to APP/PS1*CB2–/– mice (p = 0.0033, genotype effect; Fig. 4B, Supplementary Table 1D). At the age of 14 months, this effect was more pronounced, a genotype-dependent effect was detected between APP/PS1 and APP/PS1*CB2–/– mice (p < 0.0001; Fig. 4D, Supplementary Table 1D). Thus, deletion of CB2 rescues the cognitive impairment of APP/PS1 mice back to baseline levels in aged mice (Fig. 4B, D). The probe trial showed a preference for the target quadrant in both age groups independent of the genotype (Fig. 4E and F, Supplementary Table 1D), but no difference in the overall performance in middle-aged (Fig. 4E) or aged (Fig. 4F) mice.
Reduced neuronal loss in APP/S1*CB2–/– mice
Next, we wanted to know if the improved cognitive behavior after CB2 deletion in APP/PS1 mice was by accompanied by changes in AD-associated neuronal loss. We analyzed NeuN immunoreactivity in cortex and hippocampus of aged APP/PS1 and APP/PS1*CB2–/– mice. As the beneficial effect of CB2 deletion on spatial learning was most prominent in 14-month-old mice, we investigated the neuronal loss only in this age group.
Aged APP/PS1 mice showed a significant reduction of NeuN-positive (NeuN +) cells in both cortex (Fig. 5B) and hippocampus (Fig. 5D) when compared to WT control mice. Deletion of CB2 itself had no influence on the number of NeuN-positive cells (Fig. 5B, D). However, APP/PS1*CB2–/– mice showed significantly increased levels of NeuN immunoreactivity in the cortex when compared to APP/PS1 mice (Fig. 5B). Here, the number of NeuN-positive cells was comparable to baseline levels (Fig. 5B).
Finally, we stained sections for parvalbumin to reveal if this specific subtype of hippocampal interneurons (Parv +) was affected by the CB2 deletion in APP/PS1 mice. Several studies reported a specific loss of interneurons in different AD mouse models [27, 28]. We observed a significant reduction of Parv + cells in APP/PS1 mice compared to WT control mice (Fig. 6B), which matches our observations for the NeuN staining. This effect was partially rescued by the deletion of CB2 (Fig. 6B). However, the numbers of Parv + interneurons in APP/PS1*CB2–/– were still significantly reduced when compared to WT control mice.
DISCUSSION
In the current study, we have shown that CB2 deletion has a significant effect on AD-associated disease characteristics in an AD mouse model. The expression of ECS components is changed in an age-dependent manner in both, control and APP/PS1 mice. We further observed reduced plaque load in APP/PS1*CB2–/– mice, which was accompanied by enhanced expression of APP secretases in aged APP/PS1*CB2–/– mice. Interestingly, microglia in APP/PS1*CB2–/– mice were more ramified and plaques were smaller and more condensed than in APP/PS1 mice. These changes in AD pathology after CB2 deletion were associated with beneficial effects on learning and memory behavior and reduced neuronal loss in aged APP/PS1*CB2–/– mice.
APP/PS1 mice are a common model to study AD-related pathologies. They start to develop Aβ plaques at the age of 3 months [29] and show enhanced neuroinflammation, such as increased microgliosis and the expression of inflammatory components, which subsequently leads to cognitive impairment [30]. However, it is not clear if neuroinflammation is a cause or consequence of Aβ plaque development. A recent study by Lopez-Gonzales et al. showed that expression of inflammatory genes is not altered in APP/PS1 mice at the age of 3 months when the development of Aβ plaques begins, but only later at the age of 6 months when [31]. These data suggest that neuroinflammation goes align with Aβ plaque formation or might be slightly delayed but is not observed before plaque formation. Previous data demonstrated that inflammatory molecules such as TNFα or IFNγ increased the production of Aβ and impaired the ability of microglia to phagocyte Aβ, thus indicating that neuroinflammation contributes to Aβ pathology [32].
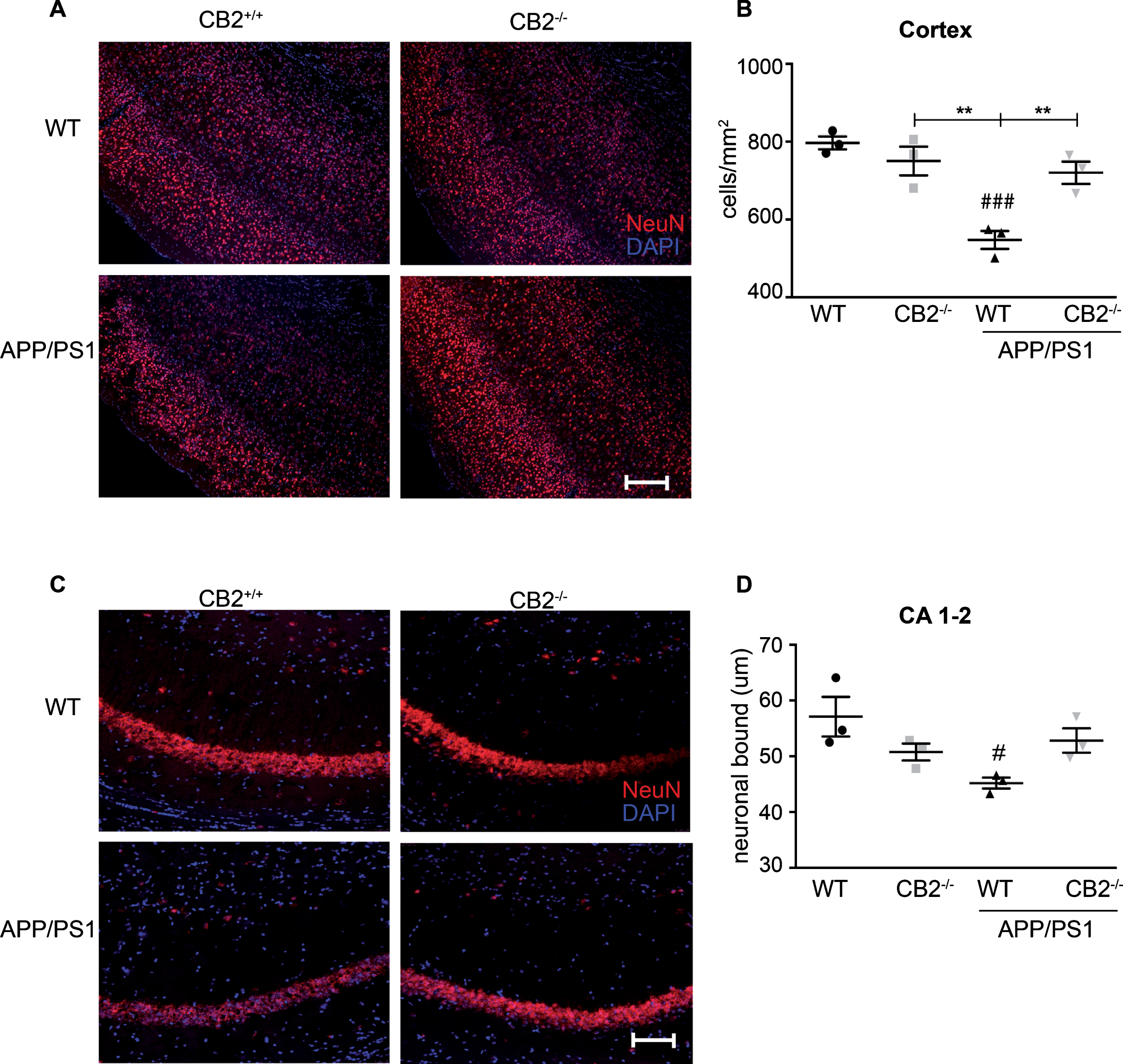
Evaluation of neurodegeneration in aged AD and control mice. Representative images of NeuN immunoreactivity in aged (14 months old) WT (upper left), CB2–/– (upper right), APP/PS1 (lower left) and APP/PS1*CB2–/– (lower right) mice in the cortex (A) and CA1-2 (C) show the cellular distribution (scale bar = 100μm). The quantification of neuronal bound (D) and number of NeuN positive cells per mm2 (B) revealed decreased immunoreactivity in APP/PS1 mice. N = 3 mice per genotype, mean of 6 images per animal; Data were analyzed by one-way ANOVA, followed by Bonferroni post hoc test, *p < 0.05, **p < 0.01, ***p < 0.001, # = significance to corresponding WT control.
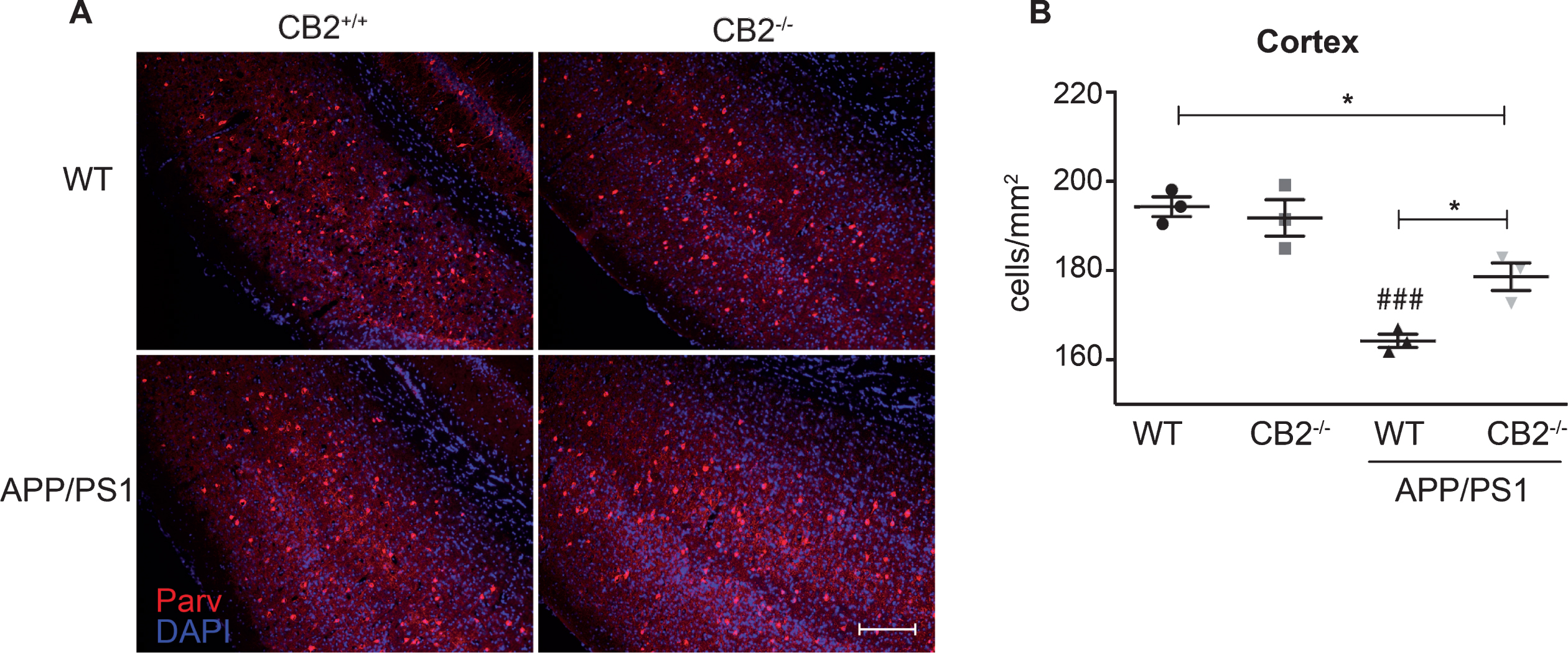
Distribution of parvalbuminergic interneurons in aged AD and control mice. Representative images of parvalbumin immunoreactivity in aged (14 months old) WT (upper left), CB2–/– (upper right), APP/PS1 (lower left) and APP/PS1*CB2–/– (lower right) mice in the cortex (A) (scale bar = 100μm). The quantification of Parv + cells per mm2 (B) revealed decreased immunoreactivity in APP/PS1 mice. N = 3 mice per genotype, mean of 6 images per animal; Data were analyzed by one-way ANOVA, followed by Bonferroni post hoc test, *p < 0.05, **p < 0.01, ***p < 0.001, # = significance to corresponding WT control.
Several previous studies have shown that the ECS, especially CB2, is linked to neuroinflammatory processes in AD and therefore might be a promising target for new therapeutic approaches. For example, CB2 expression is enhanced in plaque-associated microglia in brain tissue of human AD patients [10]. Pharmacological activation of the ECS, particularly by CB2 agonists, diminished microglial activation [11, 33] and improved cognitive function in animal models of AD-related pathologies [10, 14]. Administration of the CB2 agonist JWH-133 to a APP/PS1 transgenic mouse model also ameliorated tau hyperphosphorylation in the vicinity of Aβ plaques [11]. It was therefore surprising that we observed a similar effect when CB2 is genetically deleted. Mice lacking CB2 often showed reduced inflammatory and immune responses when compared to control mice [19, 34]. However, a genotype effect on learning and memory was previously detected only at the age of 6 months, but not in older animals [18]. Interestingly, a study by Koppel et al. (2014) observed enhanced microgliosis and increased levels of soluble Aβ in their AD J20-mouse model lacking the CB2 receptor [35]. As they used a different mouse model of AD, effects of CB2 deletion might vary between different AD mouse models. Nevertheless, in general CB2–/– mice show an immune response phenotype to brain inflammatory stimuli [18, 36].
In the present study we first focused on analyzing the expression of different ECS components in middle-aged (9 months old) and aged (14 months old) APP/PS1 and age-matched control mice. We observed age-dependent differences in cnr1, cnr2, daglα, magl, and faah expression. Although several studies reported an upregulation of CB2 in human and murine AD tissue [8–10], we observed only a tendency toward an upregulation of cnr2 expression when comparing expression in WT and APP/PS1 mice, due to a high variance in the aged group. The obtained results might be also influenced by blood-derived signals which might alter or cover cnr2 signals in brains from APP/PS1 mice. However, the detected cnr2 expression was very low. In aged mice, we observed a significant downregulation of cnr1 and faah in APP/PS1 mice, whereas magl was slightly upregulated. Previous studies on CB1 expression in the context of AD showed decreased expression of CB1 protein in mouse models of AD and tissue of human AD patients [37, 38] which matches our observations of decreased cnr1 expression in APP/PS1 mice. Furthermore, CB1 deletion in AD APP23- mice enhanced cognitive impairment without influencing plaque load [39] suggesting that CB1 also plays an important role in the interaction of the ECS with AD pathology. Interestingly, genetic deletion of both, CB1 or CB2 in AD mouse models, led to reduced neuroinflammation as evaluated by microglial Iba1-immunohistochemistry [18, 39]. These data suggest differential effects of the ECS on AD pathology.
Thus, in our present study we focused on the role of CB2 during AD-mediated inflammation. We previously described CB2 deletion reduced the plaque load in aged mice [18]. We now detected a significant reduction of Aβ plaque load in both cortex and hippocampus of aged APP/PS1*CB2–/– mice when compared with APP/PS1 mice. This supports the idea that microglial activity is reduced in APP/PS1*CB2–/– mice. These findings are in contrast to another study, which also investigated the influence of CB2 deletion in a different AD mouse model (J20 APP) [34]. That mouse model showed enhanced amyloid pathology when CB2 was deleted. These data demonstrate that different mouse models for Aβ plaque deposition develop distinct pathologies, which probably depend on the nature of the Aβ mutation as well as the expression levels. The divergent data emphasize the importance of studying different animal models, because the etiology and pathomechanisms of AD in humans are also highly diverse. Another study with younger APP/PS1 transgenic mice lacking CB2 at the age of 3 or 6 months [40] showed an increased plaque load, indicating that CB2 affects not only the severity of the symptoms, but rather the entire dynamic disease pathology. This is consistent with the notion that age and stage of AD pathology in AD mouse models are critically important factors in the evaluation of gene effects. Age and disease progression might be also of importance when investigating other signaling pathways and their impact on AD pathology [41, 42].
To determine if the reduction in Aβ plaque load in APP/PS1*CB2–/– mice is based on differential processing of Aβ, we analyzed gene expression levels of different Aβ degrading and synthesizing enzymes as well as Aβ receptors in aged mice. We found a significant upregulation of ide and mmp in brain lysates of APP/PS1*CB2–/– mice when compared with APP/PS1 mice, whereas ager was significantly downregulated. Hickman et al. (2008) observed a significant decrease in expression of mmp, ide and nep in CD11b + microglial cells of APP/PS1 mice [43]. These differences might be explained by the fact that we analyzed total brain lysates and therefore observed a dilution of effects on gene expression when compared to gene expression of CD11b + microglial cells. The genes that were differentially regulated in APP/PS1*CB2–/– mice are closely linked to microglia activation and inflammation. Ide, mmp9 and nep are secreted by microglia and degrade Aβ [43]. Therefore, enhanced expression of these genes in APP/PS1*CB2–/– mice support our previous findings that deletion of CB2 promotes an anti-inflammatory phenotype in APP/PS1 mice [18]. This is further supported by a reduced expression of tlr4 in APP/PS1*CB2–/– mice compared to APP/PS1 mice, as deletion of tlr4 has been shown to inhibit microglia and macrophage activation and therefore promotes a reduction in neuroinflammation [44]. Our data indicate an influence of CB2 activity on Aβ degradation, which may be connected to microglial activation.
Indeed, we found that plaque-associated microglia in APP/PS1*CB2–/– mice have a more ramified shape. Ramified microglial cells are classified as surveying and non-activated, but they rapidly change to an activated form in the presence of inflammatory stimuli [45] or in AD mouse models [22]. This is in agreement with other studies that demonstrated that microglial phagocytic capacity was decreased in activated microglial cells in the vicinity of Aβ plaques [46, 47] and a decreased internalized Aβ load was found in activated TNF-α – or IL-β-positive microglial from AD mouse models as compared to TNF-α – or IL-β-negative microglia [48]. Thus, we propose that the higher ramification index of plaque-associated microglia in APP/PS1*CB2–/– mice is closely linked to a reduced activation status, which is influenced by CB2 deletion. This is in line with our previous study, in which we observed reduced microgliosis in APP/PS1*CB2–/– mice and a limited response from CB2–/– microglia to inflammatory stimuli.
In addition, we found that plaques in APP/PS1*CB2–/– mice are smaller and more condensed than in APP/PS1 mice, which has been correlated to a reduced plaque toxicity. Condello et al. (2014) reported for the AD mouse models 5xFAD and TgCRND8 that reduction in plaque size and more condensed plaque morphology may be induced by a protective barrier that is formed around plaques by microglial cells and their processes, i.e., by an enhanced shielding role of microglia [5]. However, we could not observe any difference in plaque coverage by microglia between APP/PS1 and APP/PS1*CB2–/– mice, suggesting that the change in plaque size and plaque shape in APP/PS1*CB2–/– mice is not caused by an altered microglia-plaque interaction.
Finally, we investigated whether the reduction in plaque load and the reduced inflammatory phenotype of microglia in APP/PS1*CB2–/– mice subsequently leads to an improvement of cognitive performance. In our previous study, a genotype effect in learning and memory tasks was detectable between young mice, but not between older APP/PS1 and APP/PS1*CB2–/– mice [18]. We therefore increased the level of difficulty of the paradigm by increasing the intertrial time. Under these conditions, we now observed a significant improvement of cognitive performance during the acquisition phase in the MWM in middle-aged and aged APP/PS1*CB2–/– mice compared to APP/PS1 mice. These data support our notion that an overall reduction of neuroinflammation as a consequence of CB2 deletion in an AD mouse model may have a beneficial effect on spatial learning and memory. The probe trial of the MWM revealed neither a difference in performance between the four genotypes and nor between age groups. Thus, in our paradigm APP/PS1 mice needed more time to learn the spatial learning task than controls. This was rescued by the CB2 deletion in APP*CB2–/– mice. However, all mice independent of the genotype eventually learned the task and showed a preference for the target quadrant during the probe trial.
Lastly, we demonstrated that the improvement in cognitive performance in APP/PS1*CB2–/– mice is linked to a reduction in neuronal loss, as evaluated by the quantification of NeuN + cells and by a reduction of the loss of Parv + interneurons. This might also be connected to the reduction in microglia activity by a diminished expression of neurotoxic factors such as TNFα as previously observed in APP/PS1*CB2–/– mice [18]. Earlier studies reported a severe loss of Parv + interneurons in APP/PS1 mice [49]. CB2 activation by WIN55,212-2 in an AD rat model reduced microglial activation and prevented neuronal loss [50].
In summary, our data show that CB2 modulates Aβ plaque-induced neuroinflammation and that its genetic deletion has beneficial effects on AD pathology. We further suggest that CB2 deletion reduces microglial activity, which subsequently leads to a reduced neuroinflammation, increased expression of Aβ-degrading enzymes as well as α-secretases and a diminished plaque load. This in turn causes reduced neuronal loss, possibly through a decreased release of microglial neurotoxic factors, which together with the diminished neuroinflammation and reduced Aβ plaque load cause a clear improvement of cognitive behavior.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank Hanna Schrage, Blanca Randel, and Kirsten Krengel for technical support. This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) to AZ (FOR926 SP6, CP2), a research grant by Boehringer Ingelheim Pharma GmbH & Co. KG to AH and start-up grant by the Medical Faculty of the University of Bonn “Bonfor” to ACS. ACS, AH and AZ are members of the Excellence Cluster Immunosensation.