
Abstract
Epigenetic mechanisms might be involved in Alzheimer’s disease (AD). Genetic polymorphisms in several genes, including APOE (Apolipoprotein E), PSEN1 (Presenilin 1), CR1 (Complement receptor 1), and PICALM (Phosphatidylinositol binding clathrin assembly protein), have been associated to an increased AD risk. However, data regarding methylation of these specific genes are lacking. We evaluated DNA methylation measured by quantitative bisulfite-PCR pyrosequencing in 43 AD patients and 38 healthy subjects (HS). In a multivariate age- and gender-adjusted model, PICALM methylation was decreased in AD compared to HS (mean = 3.54 and 4.63, respectively, p = 0.007). In AD, PICALM methylation level was also positively associated to Mini-Mental Scale Examination (MMSE) score (percent change 3.48%, p = 0.008). Moreover, a negative association between PICALM methylation and age was observed only in HS (percent change – 2.29%, p = 0.002). In conclusion, our data suggest a possible role of PICALM methylation in AD, particularly related to cognitive function. Given the small study sample and the associative nature of our study, further prospective investigations are required to assess the dynamics of DNA methylation in the early stages of AD development.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder of the elderly, characterized by memory impairment. Several estimates confirm that there are nearly 44 million people with dementia in the world, that are expected to almost double by 2030 and triple by 2050 [1].
AD is characterized by two neuropathological hallmarks: extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) [2], made of aggregates of hyperphosphorylated tau protein. Decreased levels of Aβ1-42 (Aβ 42) with increased levels of total tau protein (t-tau) and tau phosphorylated at position 181 (p-tau) have been observed in the cerebrospinal fluid (CSF) of patients affected by AD [3, 4] and are nowadays used in clinical practice for differential diagnosis with an accuracy of 90% for AD [5].
Familial forms of AD, which are generally early onset AD (<65 years), are often associated with known mutations in genes encoding for Amyloid Precursor Protein (APP), Presenilin 1 and Presenilin 2 (PSEN1 and PSEN2, respectively), involved in both synthesis and the processing of Aβ, AβPP, and principal constituent of the plaques [6]. However, familial ADs are only less than 3.5% of all AD cases [7], in fact the majority of AD cases (around 95%) are late-onset and sporadic (LOAD) and do not have well-defined genetic causes [8], as they are probably caused by the interaction between molecular and environmental factors [9].
In order to clarify the complex etiology of sporadic AD, several research has focused on detecting DNA sequence variation, mostly through genome-wide association studies; nonetheless, genetic variation have only estimated to account for ∼30% of phenotypic variance [10] supporting the hypothesis of non-genetic factors (e.g., epigenetic factors) operating in the development of AD. Among the genes which consistently resulted mutated in AD patients in several large genome-wide studies [11 –14], we focused on four genes (APOE, PSEN1, CR1, and PICALM) whose function appears particularly relevant in biological processes involved in AD development [15], with the purpose of investigating DNA methylation of their specific promoters. DNA methylation is a molecular mechanism which regulates genes expression. In particular, the methylation of promoter regions mediates gene silencing whereas the methylation of intragenic positions are generally linked to gene activation, possibly regulating the specificity of the correct transcript [16].
Apolipoprotein E (APOE) has been extensively studied for its role in lipoprotein transport related to AD. APOE exists in the population as a polymorphic gene with three alleles of variable frequency: APOE ɛ2 (27%), APOE ɛ3 (78%), APOE ɛ4 (15%). The APOE ɛ4 allele has been found to represent a factor of susceptibility, with a fourfold increase in the risk of AD development [17].
However, only approximately 40% of LOAD cases carry the APOE ɛ4 allele, indicating that other genetic risk factors might be involved. In this context, the search for new markers and molecular risk factors appears particularly relevant.
Presenilin-1 (PSEN1) is one of the four core proteins in the gamma secretase complex, which plays an important role in the generation of Aβ from AβPP [18]. More than 200 different PSEN mutations have been identified in AD patients [19].
Recent genome-wide studies reported Complement receptor 1 (CR1) and Phosphatidylinositol binding clathrin assembly protein (PICALM) as additional susceptibility genes [11, 12].
CR1 is the main receptor of the complement C3b protein, a key inflammatory protein activated in AD [20] predominantly involved in adaptive immunity that can act as a negative regulator of the complement cascade, mediate immune adherence and phagocytosis and inhibit both the classical and alternative complement pathways. Aβ aggregates are known to activate innate immune responses via complement pathways (including CR1), which have been associated with LOAD [15]. In addition, changes in the complement system can lead to programmed synaptic death [21].
PICALM encodes for the phosphatidylinositol-binding clathrin assembly protein, which is ubiquitously expressed in all tissue types, and most prominently in neurons, where it is non-selectively distributed in the pre- and postsynaptic structures. PICALM plays an essential role in the intracellular trafficking of large molecules including proteins and lipids [22, 23]. Loss of expression of CALM protein encoded by PICALM seems to reduce Aβ 42 production and deposition in vivo [24].
A growing number of studies showed an epigenetic contribution in AD [25]. Both global and gene-specific methylation alterations have been shown in affected postmortem brain regions [26, 27] or in peripheral blood mononuclear cells [28]. Moreover, epimutations due to increased DNA methylation in specific genes are very common somatic events, and can also arise in the germline and cause soma-wide transcriptional silencing of a gene [29]. Epigenetic mutations in DNA methylation, possibly linked with AD phenotype but not related to differences in the DNA sequence [30], might contribute in determining the fraction of AD risk not explained by genetic mutations.
In a previous study conducted on 43 AD patients and 38 healthy volunteers, we found a higher methylation level of LINE-1 elements in AD patients compared to healthy subjects [31], suggestive of a profound remodeling of chromatin.
In the present study, we further evaluated DNA methylation of four specific gene promoters (APOE, PSEN1, CR1, and PICALM) in the same study group. In AD patients we evaluated the association between specific gene methylation and known determinants of AD, such as APOE polymorphisms, serum homocysteine, serum B12 vitamin, serum folate, CSF Aβ, t-tau, and p-tau, along with dementia severity using the Mini-Mental State Examination (MMSE).
MATERIALS AND METHODS
Study subjects and sample collection
The study included 43 patients with AD, and without a familiar history of AD, consecutively recruited between October 2009 and January 2010 (response rate: 75%), at the Alzheimer Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, University of Milan, Milan, Italy. Subjects with heavy comorbidities (i.e., cancer, pneumonia, stroke, and major cardiovascular diseases) were excluded as these conditions might affect DNA methylation making the interpretation of results more difficult. Diagnosis of AD was done in accordance to McKhann et al. criteria [32] and subsequent revisions [5].
All patients were evaluated for medical history, physical and neurological examination, and neurocognitive evaluation and fulfilled the DSM IV and NINCDS-ARDA criteria as probable Alzheimer’s disease [33]. AD diagnosis was then supported by brain imaging: MRI and PET. Dementia severity was assessed by the MMSE at the time of blood drawing. The MMSE is the most commonly administered psychometric screening assessment of cognitive functioning: it is used to screen patients for cognitive impairment, track changes in cognitive functioning over time, and often to assess the effects of therapeutic agents on cognitive function [34]. Sensitivity and specificity of this instrument were investigated in a systematic review published in the Cochrane database [35]. As reference group we enrolled 38 healthy volunteers (>55 years old) among the personnel of the University of Milan and their relatives. For each control a complete medical history was collected, they were assessed to exclude the presence of cognitive disorders and MMSE was administered.
In accordance with our institutional guidelines, all study subjects signed an informed consent.
For all study subjects, we collected an EDTA blood sample stored at – 80°C until analysis.
In addition, for AD patients only, CSF samples were obtained from lumbar puncture at the L4/L5 or L3/L4 interspace, centrifuged at 4°C and stored at ≤– 80°C until analysis. CSF cell counts, glucose, and proteins were determined. Albumin was measured by nephelometry rate. To evaluate the integrity of the brain-blood barrier (BBB) and the intrathecal IgG production, the albumin quotient (CSF albumin/serum albumin)×103 and the IgG index (CSF albumin/serum albumin)/(CSF IgG/serum IgG) were calculated [36]. Aβ 42, total tau (t-tau), and tau phosphorylated at position 181 (p-tau) were also measured. CSF levels were determined with human specific ELISA kits (Fujirebio, Gent, Belgium), as previously reported [37].
Data on serum homocysteine, vitamin B12, and folate were also collected for AD patients during the clinical work-up.
Apolipoprotein E genotyping was performed according to the methods previously described [31].
DNA extraction, bisulfite treatment, PCR, and Pyrosequencing
Genomic DNA was extracted using a Flexigene Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Extracted DNA was resuspended in sterile deionized water and DNA concentration was calculated using Nanodrop at a wavelength of 260 nm. DNA integrity was verified by gel electrophoresis. Extracted DNA was aliquoted and stored at – 80°C.
1μg DNA (concentration 50 ng/μl) was treated using EZ DNA Methylation-Gold™ Kit (Zymo Research, Orange, CA, USA) according to the manufacturer’s protocol. Final elution was performed with 30μl of M-Elution Buffer. Bisulfite-treated DNA was stored at – 20°C and used shortly after treatment.
PCR assays were specifically designed to amplify as many CpG positions as possible, located in the specific gene promoters. Predicted promoter sequences were located using the Genomatix software (Genomatix Software Inc., Ann Arbor, MI, USA). PCR primers were chosen to target CpG-free regions, to avoid the specific amplification of methylation-dependent molecules, therefore the PCR product would proportionally represent the methylation characteristics of the source DNA. Sequencing primers were chosen to allow the sequencing of ‘target’ CpG dinucleotides. The analysis of a non-CpG cytosine was included in each assay in order to give an internal control of the completeness of bisulphite treatment.
Analysis of DNA methylation was performed using previously published methods [38, 39] with minor modifications. Briefly, a 50μl PCR was carried out in 25μl of GoTaq Green Master mix (Promega, Madison, WI, USA), 1 pmol of the forward primer, 1 pmol of the biotinylated reverse primer, 50 ng of bisulfite-treated genomic DNA and water. The biotin-labelled primers were used to purify the final PCR product using Sepharose beads. Table 1 reports, for each investigated gene, the chromosomic location, promoter location, number of investigated CpGs and PCR primers and conditions.
Genomic location of the assayed CpGs, PCR primer sequences and conditions
The PCR product was bound to Streptavidin Sepharose HP (Amersham Biosciences, Uppsala, Sweden) and the Sepharose beads containing the immobilized PCR product were purified, washed, denatured using a 0.2 M NaOH solution, and washed again using the Pyrosequencing Vacuum Prep Tool (Pyrosequencing, Inc., Westborough, MA), as recommended by the manufacturer. Then, 0.3μM pyrosequencing primer was annealed to the purified single-stranded PCR product and pyrosequencing was performed using the PyroMark MD System (Pyrosequencing, Inc.). The degree of methylation was expressed as percentage of methylated cytosines divided by the sum of methylated and unmethylated cytosines (% 5mC). Every sample was measured three times and the average of the three replicates was used in statistical analyses.
Statistical analysis
We used standard descriptive statistics to summarize data. Graphical inspections of the main variables of interest were performed to examine their distribution. Methylation markers were log-transformed to achieve normal distribution.
In order to evaluate mean differences in promoter gene methylation between AD patients and healthy subjects we performed univariate linear regression models and age and gender adjusted models. A linear regression model adjusted for gender was applied to evaluate the association between age and gene methylation stratifying by disease status. In AD patients we used linear regression models adjusted for age and gender to evaluate the relationship between DNA methylation and laboratory parameters or MMSE. Data were presented as percent change (Δ%) in DNA methylation for 1-unit increase in the independent variable. All analyses were performed in SAS 9.4 (SAS Institute Inc., Cary, NC).
RESULTS
The main characteristics of the study subjects are reported in Table 2. The mean age at the time of enrollment was 75 years (Min-Max: 55–87; SD 8.1) for AD cases and 67 years (Min-Max: 56–87; SD 8.6) for healthy subjects. Twenty AD patients were males (46.5%) and 23 females (53.5%) while 24 males (64.9%) and 13 females (35.1%) were recruited among healthy subjects.
Characteristics of the study population
*Corrected for age and education.
We evaluated gene specific methylation levels expressed as % 5mC (percentage of cytosines that are methylated over unmethylated cytosines at a given CpG position) and mean methylation levels in AD and healthy subjects are shown in Table 3. In both the unadjusted and gender-age adjusted models, PICALM methylation was significantly decreased in AD patients compared to healthy volunteers while PSEN1 methylation was slightly significantly increased.
Gene-specific methylation (% 5mC) in AD patients (n = 43) and healthy subjects (n = 38)
*Adjusted model for age and gender. **Geometric mean.
As age is the main risk factor for AD, we investigated the association between age and candidate gene methylation in AD patients and healthy subjects. As shown in Table 4 and Fig. 1, we found that PICALM methylation was inversely associated with age (Δ% = – 2.29%, 95% CI – 3.64; – 0.93, p = 0.002) in healthy subjects but not in AD patients (Δ% = 0.55%, p = 0.4383) (Fig. 1). No association was found between age and CR1, APOE, and PSEN1 methylation.

Association between PICALM Methylation and age (years).
Associations between methylation markers and age, adjusted for gender
* Δ% = (exp(β) – 1)*100 Percentage change in methylation for an increment of 1 years in age.
In AD patients we evaluated the association between gene specific methylation and blood levels of homocysteine, vitamin B12, and folate levels (Table 5) and CSF levels of Aβ, t-tau, and p-tau (Table 6). None of the blood and CSF markers was related to gene methylation.
Association between gene-specific methylation and blood markers (n = 43). The models are corrected for age and gender
* Δ% = (exp(β) – 1)*100 Percentage change in methylation for an increment of 1 unit in blood marker.
Association between gene-specific methylation and CSF markers (n = 43). The models are corrected for age and gender
* Δ% = (exp(β) – 1)*100 Percentage change in methylation for an increment of 10 unit in CSF markers.
MMSE average score was 20.9 point and ranged from 7.99 to 28. Our data (Table 7 and Fig. 2) showed a positive association between PICALM methylation and MMSE score (Δ% = 3.48, 95% CI 0.94–6.10, p = 0.0084).
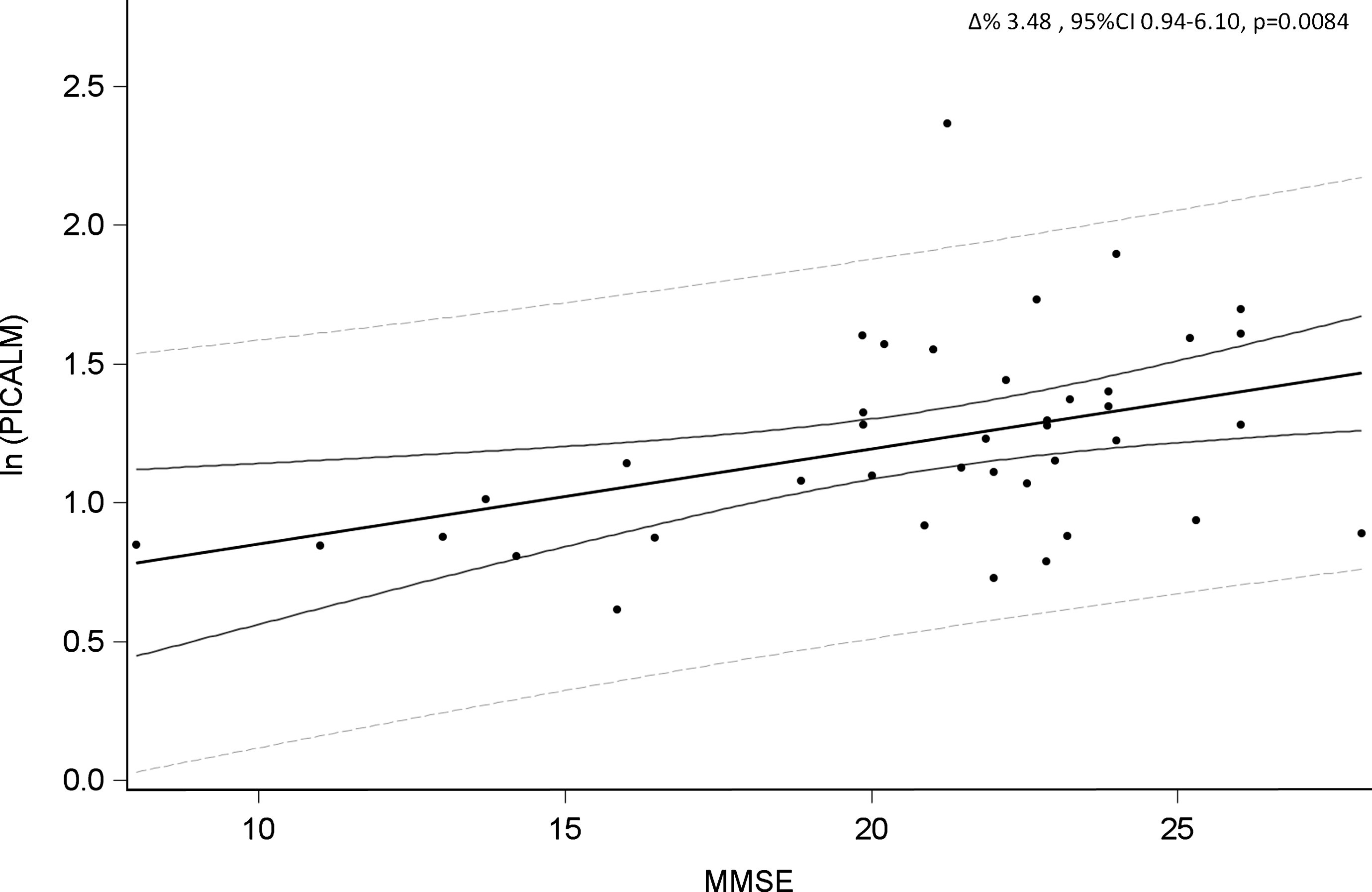
Association between PICALM methylation and MMSE score in AD patients.
DISCUSSION
In the present study, we analyzed the methylation status of APOE, CR1, PICALM, and PSEN1 in blood samples of 43 AD patients and 38 healthy subjects. When identifying candidate genes to be assessed for DNA methylation, we focused on genes with established roles in AD, as their altered expression caused by genetic alterations has been consistently reported. On the contrary, studies investigating their methylation in blood are currently missing.
Association between gene-specific methylation and MMSE score in AD patients. The models are corrected by gender
* Δ% = (exp(β) – 1)*100, Percentage change in methylation for an increment of 1 unit in MMSE score.
Previous studies investigating DNA methylation in AD have been mainly conducted in human postmortem brain tissues, showing hyper or hypomethylation of several candidate genes [40]. Nevertheless, this type of study does not allow to investigate the disease while it is still developing and thus thoroughly understand the underlying mechanisms.
As DNA methylation is a tissue-specific mechanism, the absolute methylation percent in blood and brain is expected to differ. However, genome-wide studies repetitively showing a high correlation of methylation among different tissues (see for example, Whal et al. [41]) support the hypothesis that our observations in blood may reflect changes occurring in the brain tissue and, more specifically, blood and brain methylation levels can be related. In addition, blood biomarkers also take into account the key role of the adverse inflammatory processes occurring in AD.
Only a few studies have investigated DNA methylation in peripheral blood leukocytes in AD patients to date, obtaining heterogeneous findings [42]. In a previous work, conducted on the same study population, we observed that LINE-1 methylation was increased in AD patients compared with healthy volunteers. Moreover, the group with best performances in MMSE showed higher levels of LINE-1 methylation compared to the group with worst performances, suggesting a possible use of LINE-1 as risk stratification tool [31]. In agreement with these findings, a recent work by Di Francesco and colleagues showed an increased global DNA methylation measured by LUMA assay in AD patients; however, they observed an inverse relationship between global methylation and MMSE score [28]. On the other hand, a study conducted by Hernandez and colleagues in peripheral blood samples did not find differences in LINE-1 methylation levels between AD patients and controls [43]. An additional target considered in the context of AD pathogenesis is TNF-α promoter methylation, that was found to be hypomethylated in AD patients’ brain compared to controls, but not in blood [44].
The main finding of the present study is the observed decrease of PICALM methylation in AD patients compared with healthy subjects. Moreover, PICALM methylation was negatively associated with age in the healthy subject group, but not in AD patients. Finally, PICALM methylation level was associated with MMSE score, showing that patients with lower methylation had lower MMSE score.
PICALM protein is essential for neurotransmitter release at the presynaptic membrane, being important for memory formation and neuronal function [45]. An overexpression of PICALM, potentially linked to a lower methylation, is in fact associated with an increase of AβPP in the hippocampus, leading to an increase of Aβ in plaques [46] thus altering memory formation [45]. As PICALM polymorphisms have been consistently associated to an increased AD risk, it is possible that both genetic and epigenetic changes in PICALM expression cause synapse perturbations, contributing to neurodegeneration.
When we evaluated the correlation between PICALM methylation and age, we observed a negative and significant correlation in healthy subjects only. This finding could be explained by a physiological loss of PICALM methylation in healthy subjects due to aging, which is by itself the main risk factor of senile dementia and is characterized by a progressive decrease in DNA methyl transferase activity [47]. On the other hand, in AD patients, this modulation is lost, being the neurodegeneration pathologically fast evolving. We might speculate that AD subjects prematurely reach low levels of PICALM methylation, resulting in the observed lack of association with age.
Further supporting the pathological role of PICALM in AD development, we found an association between PICALM hypermethylation and increased MMSE score. This also means that subjects with worse performance at the MMSE score were characterized by a lower methylation levels of PICALM gene, supporting a role for PICALM both in the aging process and AD related neurodegeneration.
The present study has some limitations. The study population is relatively small, and the age and gender distribution slightly differed between cases and controls, thus potentially confounding the results of the comparison between the two groups. We overcame this criticism by performing adjusted analyses and presenting both unadjusted and adjusted results which were highly concordant.
In addition, although the observed methylation differences were small when expressed as a percentage of the total number of cytosines in the examined position, these differences involved many DNA molecules and correspond to a large number of blood cells in which candidate gene function might be altered. Moreover, we focused our investigation only on four candidate genes selected a priori on the basis of their functional role: being the present study associative in nature, we cannot exclude that the observed modification represents a consequence of a fully developed AD phenotype rather than causative alterations, and that other genes are involved in the pathogenesis of AD. Finally, another potential limitation is that we did not evaluated the possible association between DNA methylation and gene expression, due to the lack of a suitable RNA sample. We are planning to conduct a larger investigation, involving a larger number of subjects, and an extensive quantification of gene expression to support our findings on DNA methylation.
In conclusion, our data suggest a possible role of PICALM methylation in AD, particularly related to cognitive function. Due to the above-mentioned limitations, further prospective studies are required to assess the dynamics of DNA methylation in the early stages of AD development. In addition, further investigations of epigenetic mechanisms implicated in the onset of the disease, could potentially help in the identification of new markers to be used for AD risk stratification.