
Abstract
This study was designed to determine whether the levels of renin-angiotensin system (RAS) components are associated with Alzheimer’s disease (AD) pathology. Cerebrospinal fluid levels of Angiotensin (Ang) II, Ang-(1-7), angiotensin-converting enzyme (ACE), ACE2, Amyloid-β (Aβ)40, Aβ42, total tau (hTau), and phospho-tau (pTau) were measured in 18 patients with AD and 10 controls. Patients with AD presented decreased levels of ACE when compared with controls. We found a significant positive correlation between ACE and Aβ42 levels among patients. Our results strengthen the hypothesis that ACE is associated with Aβ pathology in AD.
Keywords
INTRODUCTION
Although the renin-angiotensin system (RAS) is traditionally regarded as a system implicated in blood pressure and fluid homeostasis, accumulating evidence has expanded the physiological and pharmacological functions of this system [1]. The reconceptualization of the RAS resulted from the discovery of new functional pathways of the system in tandem with the findings of RAS components in unlikely places, such as renin in the brain [2]. Currently, besides the ‘systemic’ RAS, it is assumed that there are different ‘local’ RAS, including a brain RAS as all its components can be found in the central nervous system. Studies have revealed that the RAS is involved in brain development, water and food intake, maintenance of the blood-brain barrier, and also in more complex functions, including motor, cognitive, and emotional control [2].
The pathological hallmarks of Alzheimer’s disease (AD) are the progressive abnormal accumulation of amyloid-β (Aβ) in the intercellular space and neurofibrillary tangles composed by aggregates of hyperphosphorylated tau protein inside neurons. These changes are accompanied by neuronal damage and death [3]. Although amyloid plaques and neurofibrillary tangles are believed to interfere with neuron-to-neuron communication and to contribute to neurodegeneration in AD, interventions targeting them have been limited in modulating the clinical trajectory of the disease or modifying its natural course [4]. While Aβ and tau remain strong candidates for AD therapy, additional mechanisms may be involved in the development or progression of the disease.
Epidemiological and clinical data indicate that the use of antihypertensive drugs targeting the RAS, i.e., angiotensin-converting enzyme (ACE) inhibitors and angiotensin II type 1 (AT1) receptor antagonists, may have beneficial effects for AD. For instance, a prospective study showed the potential of these medications (mainly AT1 receptor antagonists) in reducing the incidence and progression of AD and other dementias [5]. In addition, a six-month treatment of hypertension in patients with AD with the AT1 receptor antagonist telmisartan resulted in more beneficial effects in cognition and in cerebral blood flow than non-RAS antihypertensive drugs such as amlodipine [6]. A meta-analysis reported that the use of drugs targeting the RAS was significantly associated with a reduced risk of AD [7]. These findings were corroborated by experimental studies showing that AT1 receptor antagonists were capable of preventing or attenuating stress-induced cognitive impairment in rodents [8–11]. Experimental evidence also proved the potential of AT1 receptor antagonists in reducing AD-related pathological changes. Telmisartan reduced both intracellular Aβ and extracellular senile plaque accumulation after transient middle cerebral artery occlusion in the stroke-resistant spontaneously hypertensive rat strain [12]. Losartan, another AT1 receptor antagonist, protected cognition and cerebrovascular reactivity when given preventively to human amyloid precursor protein transgenic mice. Losartan also exerted preventive and restorative effects on AD hallmarks [13]. In view of all these findings, researchers are currently performing clinical trials to evaluate the efficacy of antihypertensives targeting the RAS in reducing AD-related pathology [14] and to investigate the mechanisms underlying their protective effects [15].
The evidence of direct involvement of RAS components in the pathophysiology of AD is still limited. Therefore, the current study was undertaken to compare the cerebrospinal (CSF) levels of RAS components – Angiotensin (Ang) II, Ang-(1-7), angiotensin-converting enzyme (ACE) and ACE2 – in AD patients and controls. In addition, we sought to evaluate whether putative alterations in the levels of RAS components were associated with CSF biomarkers of AD pathology.
General characteristics and cerebrospinal fluid levels of angiotensin-converting enzyme (ACE), ACE2, amyloid-β, phosphorylated Tau (pTau) and Tau protein (hTau) in Alzheimer’s disease (AD) patients and controls
MMSE, Mini-Mental State Examination; IATI, Innotest Amyloid Tau Index. aStudent’s t test; bFisher’s exact test; cMann-Whitney test. Significant values are highlighted in
METHODS
Subjects and biological samples obtaining
This study included 18 patients with probable AD diagnosed according to the National Institute on Aging – Alzheimer’s Association (NIA-AA) workgroup criteria [16]. Patients were recruited from the Cognitive and Behavioral Neurology Clinic of the Hospital das Clínicas, Universidade Federal de Minas Gerais, Brazil. A control group composed by 10 subjects undergoing lumbar puncture for anesthetic purpose was also enrolled in the same hospital. These subjects had no history of neuropsychiatric disorders. All subjects, or their relatives when appropriate, provided written informed consent before admission to the study. The Research Ethics Committee of the Universidade Federal de Minas Gerais, Brazil approved this study.
CSF samples were obtained by lumbar puncture on the same day of the clinical interview. The samples were centrifuged at 1,800 g for 10 min within 30 min after collection. Cell-free CSF samples were stored at – 80°C until analysis.
Biochemical analyses
Samples were thawed and CSF levels of AD-related pathology biomarkers and RAS components were measured by enzyme-linked immunosorbent assay (ELISA), as routinely performed in our laboratory [17].
Aβ40, Aβ42, total tau (hTau), and phosphorylated tau 181 (pTau) were measured using the standardized commercially available ELISA kit (Innotest ®), according to the procedures supplied by the manufacturer (Innogenetics/Fujirebio, Belgium). Ang II, Ang-(1-7), ACE, and ACE2 levels were also measured by ELISA following the manufacturer’s procedures (MyBioSource, USA).
All kits applied the sandwich ELISA technique, except for ACE measurement whose kit applied the competitive ELISA method. The assays were performed by a person blind to the clinical status of the subjects. Concentrations were expressed as pg/mL. The sensitivity of the assays was 65.0 pg/mL for Aβ42, 1.8 pg/mL for Aβ40, 34.0 pg/mL for hTau, 13.0 pg/mL for pTau, 1.0 pg/mL for ACE and ACE2; 2.0 pg/mL for Ang-(1– 7); and 18.75 pg/mL for Ang II.
Statistical analysis
Associations between dichotomous variables were assessed with the Fisher’s exact test. All continuous variables were tested to assess whether they follow a Gaussian distribution using the Shapiro-Wilk normality test. Two groups (patients with AD versus controls) were compared using the Student’s t test or the Mann– Whitney U test when data were determined to follow or not a normal distribution, respectively. Spearman’s correlation analyses were performed to examine the relationship between CSF levels of RAS components and AD-related pathologybiomarkers.
All statistical tests were two-tailed and were performed using a significance level of α= 0.05. Statistical analyses were performed using SPSS software version 25.0 (SPSS Inc., Chicago, IL, USA), as well as GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA).
RESULTS
Clinical features and CSF levels of the evaluated biomarkers are shown in Table 1. Patients and controls presented similar age and sex distributions.
All patients with AD met the CSF criteria for biomarker-based diagnosis, [i.e., Innotest ® Amyloid Tau Index (IATI) <0.8; hTau/Aβ42 ratio >0.52 and pTau/Aβ42 ratio >0.08] [18, 19]. Accordingly, we found decreased levels of Aβ40 and Aβ42, and increased levels of hTau and pTau in the CSF of patients AD in comparison with controls (Table 1).
Ang II and Ang-(1-7) were not detected in the CSF samples. Patients with AD presented decreased levels of ACE (Fig. 1A), but similar levels of ACE2 (Fig. 1B) when compared with controls. Among patients with AD, we found a moderate positive correlation (rho = 0.4946, p = 0.0369) between ACE levels and Aβ42 levels (Fig. 1C). The same correlation was not observed in controls.
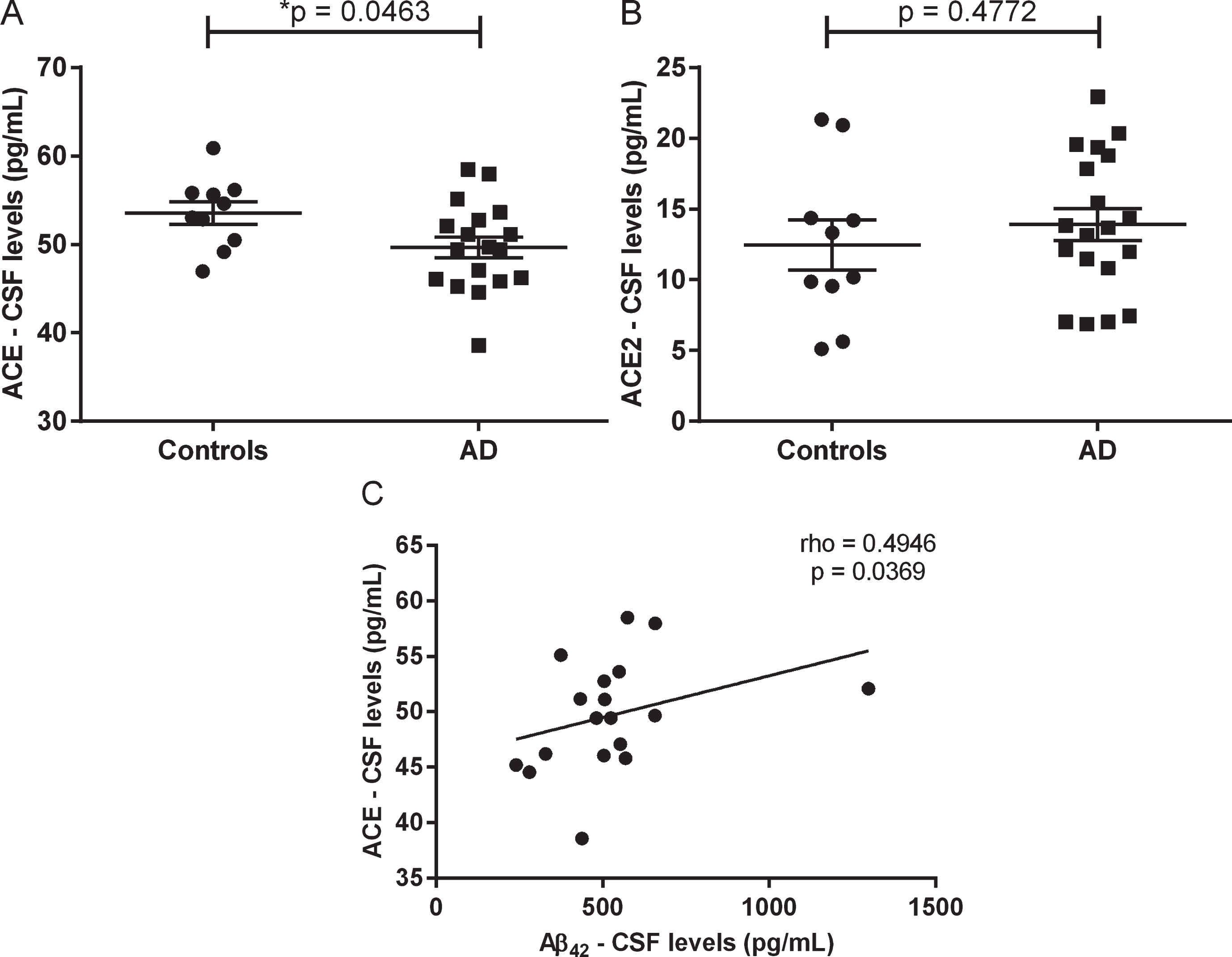
Cerebrospinal fluid levels of angiotensin-converting enzyme (ACE) and ACE2 in patients with Alzheimer’s disease (AD) and controls. Patients with AD presented decreased levels of ACE (A), but similar levels of ACE2 (B) when compared with controls. Among patients with AD, ACE levels correlated positively with Aβ42 levels (C). Horizontal bars (A & B) represent the mean and the standard error of the mean. p-values from Student’s t test (A & B). A p-value < 0.05 was considered significant.
DISCUSSION
The purpose of this study was to evaluate whether RAS components are involved in AD pathophysiology. We found decreased CSF levels of ACE in patients with AD in comparison with controls. In addition, lower ACE levels were associated with lower Aβ42 levels, an indicative of increased Aβ accumulation in the brain. Our results support the hypothesis that the RAS is involved in AD pathological changes.
Our data confirm previous studies’ results showing that ACE activity or levels are decreased in CSF of patients with AD in comparison with controls [20–22]. Similar abnormalities in ACE activity were described in CSF samples of patients with AD, Parkinson’s disease, and progressive supranuclear palsy, suggesting that reduced ACE activity in CSF may be a non-specific marker of neurodegeneration [21]. Also in line with the current findings, lower CSF ACE levels were associated with lower CSF Aβ1 - 42 in patients with AD [22]. Decreased ACE levels/activity may be associated with AD pathophysiology by a mechanism involving Aβ metabolism. ACE was found to significantly inhibit Aβ aggregation in vitro in a dose-response manner [23]. These findings might represent a biological link between ACE abnormalities and AD pathophysiology.
However, there are conflicting results in the literature. One study did not find difference between AD patients and controls regarding ACE activity in CSF samples [24]. Not only the ACE activity but also ACE levels were found to be similar in the CSF of patients with AD and controls [25]. Increased ACE levels and activity have also been described in CSF samples from patients with mild cognitive impairment and AD in comparison with controls [26, 27]. This apparent discrepancy can be partly explained by different methods of ACE measurement or sample characteristics. Studies advocating that decreased ACE levels/activity are associated with increased AD risk are based on the fact that ACE inhibits Aβ aggregation. On the other hand, studies supporting the opposite believe that higher ACE activity might increase the risk of AD by enhancing blood pressure with subsequent development of small vessel cerebral disease. However, since increased CSF ACE activity was associated with a reduced risk of global brain atrophy, high ACE may have protective effects on the brain [28]. Either way, altered ACE activity/level seems to be implicated in AD pathophysiology.
Results from genetic studies may help solving the inconsistencies regarding ACE activity/levels in AD. In this regard, genetic association studies have provided further evidence that decreased ACE levels/activity contribute to AD. A meta-analysis concluded that the Alu insertion allele in the ACE gene confers an increased risk for AD. A single-nucleotide polymorphism, A-262T, located in the ACE promoter was also associated with AD. Moreover, there is an equivalence between the alleles that are associated with AD and those that are associated with reduced levels of circulating ACE [29].
We are aware of our study limitations, which include the cross-sectional design and the relatively small sample size. Larger studies with longitudinal follow-up might help to advance the understanding of RAS in AD pathophysiology. Studying patients in different disease stages and the association between RAS components levels and neuropsychiatric symptoms would also be very informative in this regard. Another limitation is the use of medications. Four out of 10 controls (40%) and 7/18 (38%) patients with AD were in use of anti-hypertensive drugs, which cannot be terminated for the sake of scientific research for obvious ethical considerations. Although the observed findings might be caused or influenced by their ongoing treatments, we did not find difference in plasma levels of RAS components when we compared patients in use of vs. patients not using anti-hypertensives. The strict criteria in the selection of controls and patients, and the analysis of RAS components and AD-related markers together can be regarded as strengths of the current study.
In conclusion, CSF levels of ACE are reduced in AD, correlating with Aβ42 levels. Our results strengthen the hypothesis that ACE is associated with Aβ pathology in AD and may stimulate the development of disease-modifying strategies for AD focusing on the RAS.
Footnotes
ACKNOWLEDGMENTS
The authors would like to acknowledge the participation of volunteers in this study and are indebted to their caregivers for their magnificent support. They also would like to thank the Surgery Service at the Hospital das Clínicas / UFMG for the help in obtaining CSF samples from controls. This study was funded by the Brazilian agencies FAPEMIG, CNPq and CAPES. NPR is a Huntington’s disease Society of America (HDSA) fellowship recipient. EPFR is a GBHI UCSF Memory and Aging Center fellowship recipient. The Neuropsychiatry Program is funded by the Department of Psychiatry & Behavioral Sciences - UTHealth. LCS, ACSS, PC and ALT are funded by CNPq (bolsa de produtividade em pesquisa). Authors’ disclosures available online (![]() ).
).