
Abstract
Valosin-containing protein (VCP) is involved in multiple cellular activities. Mutations in VCP lead to heterogeneous clinical presentations including inclusion body myopathy with Paget’s disease of the bone, frontotemporal dementia and amyotrophic lateral sclerosis, even in patients carrying the same mutation. We screened a cohort of 48 patients with familial frontotemporal dementia (FTD) negative for MAPT, GRN, and C9orf72 mutations for other known FTD genes by using whole exome sequencing. In addition, we carried out targeted sequencing of a cohort of 37 patients with frontotemporal lobar degeneration with Transactive response DNA-binding protein 43 (TDP-43) subtype from the Netherlands Brain bank. Two novel (p.Thr262Ser and p.Arg159Ser) and one reported (p.Met158Val) VCP mutations in three patients with a clinical diagnosis of FTD were identified, and were absence in population-match controls. All three patients presented with behavioral changes, with additional semantic deficits in one. No signs of Paget or muscle disease were observed. Pathological examination of the patient with VCP p.Arg159Ser mutation showed numerous TDP-43 immunoreactive (IR) neuronal intranuclear inclusions (NII) and dystrophic neurites (DN), while a lower number of NII and DN were observed in the patient with the VCP p.Thr262Ser mutation. Pathological findings of both patients were consistent with FTLD-TDP subtype D. Furthermore, only rare VCP-IR NII was observed in both cases. Our study expands the clinical heterogeneity of VCP mutations carriers, and indicates that other additional factors, such as genetic modifiers, may determine the clinical phenotype.
INTRODUCTION
Valosin-containing protein (VCP), also known as p97, is a member of ATPase Associated with diverse cellular Activities protein family [1, 2]. VCP is composed of an N-terminal domain (CDC48) involved in ubiquitin binding, two D1-and D2 domains that bind and hydrolyze ATP, and a flexible C-terminal region [1, 3]. These structures can be assembled into a homohexamer [3, 4]. VCP is involved in multiple cellular processes including protein degradation via ubiquitin proteasome system [5], cell division, nuclear envelope formation [6], endoplasmic reticulum associated protein degradation (ERAD) [7], aggresome formation [8], Golgi apparatus assembly, autophagy, mitophagy, endosomal trafficking, cell cycle, and DNA repair [9].
The classical phenotype of mutations in VCP is inclusion body myopathy with Paget’s disease of the bone and frontotemporal dementia (IBMPFD) with an autosomal dominant inheritance [10]. IBMPFD is characterized by proximal and distal muscle weakness resembling a limb-girdle dystrophy syndrome, Paget disease of bone and frontotemporal dementia (FTD) [11]. Currently, different mutations in domains linker 1, D1, and predominantly CDC48 are reported to this multisystem disease [12]. The phenotype of these mutations can be variable, even within the same family: patients could solely present with myopathy, FTD, or amyotrophic lateral sclerosis (ALS) [12–15]. Myopathy is the most common presenting features of this disease, and is found in about 91% of the patients, while an FTD phenotype was observed in approximately 30% of patients with VCP mutations [16]. Pathologically, transactive response DNA binding protein of 43 kDa (TDP-43) and p62 immunoreactive (IR) short dystrophic neurites (DN) with neuronal intranuclear inclusions (NII), classified as FTLD-TDP type D, are characteristic for VCP mutations [17, 18]
In this study, we sequenced a cohort of 48 patients using whole exome sequencing (WES) and analyzed 37 pathological confirmed FTLD-TDP cases using targeted sequencing, leading to the identification of three mutations in VCP (including two novel) in three unrelated Dutch patients presenting with FTD. Here, we here describe the clinical and available pathological characteristics of these mutation carriers.
METHODS
Subjects
The Dutch FTD cohort included patients with a clinical diagnosis of FTD, ascertained as part of an ongoing genetic-epidemiological study in the Netherlands since 1994 [19]. Detailed clinical information of the patients was obtained by interviewing relatives and reviewing medical records from hospitals or nursing homes. Patients underwent a neurological examination and neuropsychological assessment when possible. Neuroimaging including computed tomography (CT), magnetic resonance imaging (MRI), and single-photo emission computed tomography (SPECT) was also reviewed when available. The diagnosis of behavioral variant of FTD (bvFTD) was established according to the international consensus criteria by Rascovsky [20], and ALS was diagnosed when patients met El Escorial criteria [21]. Pathological diagnosis of frontotemporal lobar degeneration (FTLD) is confirmed by the pathologist of the Netherlands Brain Bank.
In the Dutch FTD cohort, we selected all patients with FTD and with a positive family history for dementia or ALS, but without mutations in MAPT, GRN, and repeat expansion in c9orf72 (n = 48). The mean age at onset was 59.0±9.4 years (range 36.2–73.9), and the mean age at death was 67.3±9.9 years (range 42.5–82.7). Pathological diagnosis of FTLD was confirmed in 13 patients (FTLD-TDP in 12 and 1 FTLD-Tau), and no autopsy was performed in the remaining 35 patients. Additionally, a cohort of 37 patients with FTLD-TDP without mutations in GRN and repeat expansion in c9orf72 from the Netherlands Brain Bank was selected for targeted sequencing (data of family history were incomplete). The mean age at onset of the latter cohort was 59.9±7.4 years (range 45.2–80.5). The mean age at death was 68.3±6.4 (range 54.5–87.4).
DNA was obtained from blood in patients selected for WES and was extracted from cerebellar brain tissue of patients selected for targeted sequencing. The study is approved by the Medical Ethical Committee of the Erasmus Medical Centre and the Netherlands Brain Bank, and informed consent is obtained from all participants.
Genetic and bioinformatic analysis
WES was performed using Nimblegen v2 Seqcap EZ Exome Kit (Roche) for exome capturing. DNA was prepared with the Illumina TruSeq Paired-End Library Preparation Kit according to manufacturer’s instructions, and sequenced on a HiSeq 2000 (Illumina, San Diego, CA). The data was generated at the Human Genomics Facility (HuGeF; http://www.glimdna.org) at Erasmus MC according to an in-house pipeline.
Targeted sequencing was performed using customized exon capture kit. A list of known neurodegenerative disease genes was captured using custom-designed targeted panel created by SureDesign (Agilent Technologies, Santa Clara, CA). DNA library was prepared by NEBNext Ultra DNA Library Prep Kit for Illumina according to manufacturer’s instructions, and paired-end sequencing was performed using Illumina HiSeq 4000 (Illumina, San Diego, CA). Sequences were processed in-house using Mayo Clinic’s DNA analysis pipeline Genome GPS v4.0.
Alignment of the sequencing reads to the human reference genome GRCh37 was performed using Burrows-Wheeler Aligner [22]. After alignment, duplicate reads were marked and removed using Picard (v1.119) [23]. HaplotypeCaller from Genome Analysis Toolkit (GATK) was used for variant calling, and quality control using variant quality score recalibration and hard filters according to GATK best practice. Functional annotations of variant sites were performed using ANNOVAR [24]. Combined Annotation Dependent Depletion (CADD) score (v1.3) was used to predict the pathogenicity of the variants [25]. In both sequencing data, we focused on disease-causing genes in AD and FTD (APP, PSEN1, PSEN2, MAPT, GRN, TARDBP, VCP, SQSTM1, CHMP2B, CHCHD10, FUS, CSF1R, TREM2, UBQLN2, SOD1, OPTN, and TBK1). Non-synonymous or protein truncating variants (including nonsense or frameshift INDELS) with minor allele frequency of <0.01% from the genome Aggregation Database (gnomAD) were retained [26]. Additionally, we used the sequencing data from Genome of the Netherlands (GoNL) and exome data of non-demented individuals from the Rotterdam study (n = 2101) to filter out polymorphism [27, 28].
All candidate variants were validated by Sanger sequencing, and sequenced on an ABI3730xl genetic analyzer (Applied Biosystems, CA, USA).
Histology and immunohistochemistry
Brain autopsy was performed in the p.Thr262Ser and p.Arg159Ser VCP mutation carriers by the Netherlands Brain Bank. Immunohistochemical staining was performed using the following antibodies: Hyperphosphorylated tau (AT8, 1:40, Innogenetics), amyloid-β protein (1:100, DAKO), p62 (1:200, BD Biosciences Pharmingen), phospho (p) TDP-43 (1:100, Biotech) and VCP (ab11433-50; 1:500, Abcam). The pathological diagnosis of FTLD was made by neuropathologist (A.J.M.R.). TDP-43 pathology was classified in subtypes according to the algorithm described by Mackenzie et al. [18].
RESULTS
Genetic analysis
In the WES cohort, we found three rare nonsynonymous variants after filtering: two in VCP (p.Thr262Ser and p.Met158Val), and one in TARDBP (p.Ile383Val). In the targeted sequencing data, we found one additional variant in VCP (p.Arg159Ser) and one variant in APP (p.G119R). Sanger sequencing confirmed all variants. The variant in APP gene was most likely not pathogenic due to its location (exon 4). The variant in TARDBP had been reported previously [29]. All three variants in VCP were unknown in gnomAD, GoNL and exome data from the Rotterdam Study, and two of these (p.Thr262Ser and p.Arg159Ser) have not been reported previously (Table 1).
Summary of clinical characteristics of VCP mutation carriers
alanguage problems early in the disease process; CADD, Combined Annotation Dependent Depletion; bvFTD, behavioral variant frontotemporal dementia; ALS, Amyotrophic Lateral Sclerosis; PDB, Paget disease of the bone; IBM, Inclusion Body Myopathy; LGMD, Limb-Girdle Muscular Dystrophy; HSP, Hereditary Spastic Paraplegia; IBMPFD, Inclusion Body Myopathy with Paget’s disease of the bone and frontotemporal dementia.
Clinical features of VCP mutations carriers
Patient 1 (p.Met158Val) - At 41 years old, this patient presented with reduced empathy, loss of interest in grooming, reduced verbal output, and increased aggressiveness. Prominent difficulties with finding words and naming objects were noticed early in the disease process. He was also obsessive in collecting and disassembling devices, and was ticking on couches. Neurological examination one year after disease onset showed word finding problems and short-term memory impairment. Fasciculations or muscle weakness were absent. Routine blood tests showed a normal calcium, phosphate, and alkaline phosphatase. Neuropsychological assessment revealed lack of disease insight, semantic deficits, executive dysfunction, and to a lesser extent impairment of memory and visual constructive function. MRI showed cortical atrophy and moderate bilateral temporal atrophy. The clinical diagnosis was bvFTD with pronounced semantic deficits. Patient died at the age of 46. No autopsy was performed. Family history showed no dementia in his both parents at age of 64, but dementia with behavioral changes in his grandfather on father’s side at the age of 55 (1A), and dementia in his 79-year-old grandmother. Both grandparents on mother’s side developed dementia at old age.
Patient 2 (p.Arg159Ser) – At 56 years old, this female patient developed personality changes and lost interest in her hobbies. She became egoistic and aggressive towards her husband, and she had transiently suicidal thoughts. There were no memory complaints and her orientation was normal. Her spontaneous speech diminished, and finally she became mute. CT-scan at 59 years old showed enlarged ventricles with cortical atrophy. The patient died at the age of 62 due to cachexia. Brain autopsy was performed. Family history revealed that her mother developed dementia at the age of 58 years (Fig. 1B).
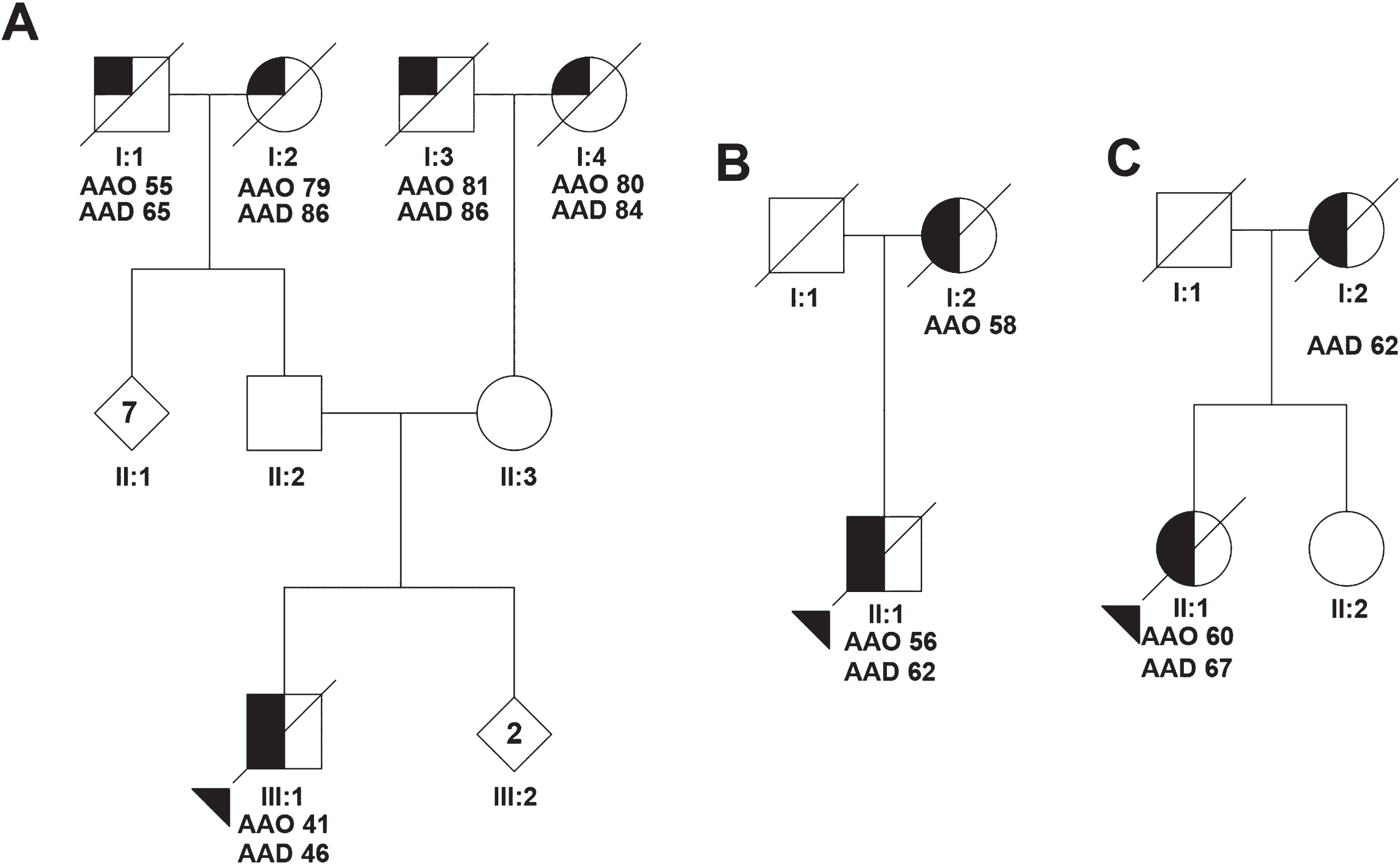
Pedigree of the VCP mutation carriers. Pedigrees showing family members of p.Met158Val mutation carrier (A), the p.Arg159Ser mutation carrier (B), and the p.Thr262Ser mutation carrier (C). Symbols with a diagonal line represent deceased individuals. The proband is indicated with the triangle. Numbers in the symbols indicate the number of individuals. Half-filled symbols represent individuals with Frontotemporal dementia and a quarter-filled symbols represent individuals with uncharacterized dementia. AAO, Age at onset; AAD, Age at death.
Patient 3 (p.Thr262Ser) – At age 60, this female patient presented with reduced empathy, childish behavior, withdrawal from social contact, and loss of interest in grooming (Table 1). She spent money on charities and gambling, and was drinking excessive amounts of alcohol. She developed memory problems and became disoriented. She also lost her skills in playing violin at professional level. Apathy, wandering, and reduced verbal output were observed at the age of 64 years. She was admitted in the hospital with epileptic seizures, which was treated with anti-epileptics. Neurological examination five years after disease onset revealed reduced verbal output with echolalia. Signs of myopathy or bone disease, muscle weakness, or fasciculations were absent. At neuropsychological testing, she had impairment in fluency, executive tasks, and semantic paraphasias. Routine blood and cerebrospinal fluid examination revealed no abnormalities, with normal alkaline phosphatase and phosphate. CT scan showed atrophy of the frontal lobes and SPECT showed frontal hypoperfusion. The clinical diagnosis of bvFTD was established. In the end stage of her disease, epileptic seizures increased in frequency, and she died at the age of 67. Brain autopsy was performed. Family history revealed that her mother presented with hyperorality, roaming, and disorientation suggestive for the clinical diagnosis of bvFTD; she died at 62 years of age (Fig. 1C).
Neuropathological findings of p.Arg159Ser and p.Thr262Ser carriers
Macroscopically, prominent frontal atrophy was observed in both cases. Histological examination showed neuronal loss with gliosis in upper three neocortical layers, cornu amonnis (CA) 1, subiculum, and parahippocampus, and in the insula and thalamus. Staining with pTDP-43 and p62 antibodies showed moderate to numerous NII and short DN in all layers of the frontal and temporal cortex, and to a lesser extent in the parietal cortex (Fig. 2A-E), but none were detected in the dentate gyrus, CA regions of the hippocampus and occipital cortex. Abundant DN and NII were found in the neocortex of p.Arg159Ser case (Fig. 2C, D), while much lesser DN and NII were observed in p.Thr262Ser (Fig. 2A, B). Overall, round or lentiform-shaped NII of variable size and DN were found in the upper cortical layers, and less frequently in the deeper layers. A few TDP-43-IR neuronal cytoplasmic inclusions (NCI) were also found in the cortical regions of both cases, and were sporadically found in the medulla oblongata and spinal cord of p.Thr262Ser case. Both cases can be classified as FTLD-TDP type D [18]. VCP immunostaining revealed low numbers of NII scattered in the upper layer of the cortical regions (Fig. 2F), but no NCI were found. Amyloid-β immunostaining showed a few senile plaques and sporadically neuritic plaques in p.Arg159Ser carrier corresponding with Thal 2, Braak 2 and Consortium to Establish a Registry for AD (CERAD) 0 [30]. In p.Thr262Ser carrier, only a few hyperphosphorylated tau depositions without plaques were observed corresponding with Braak 1.

Immunohistochemistry of the brains from two VCP mutations carriers. Lentiform neuronal intranuclear inclusions (NII) positive for TDP-43 immunohistochemical staining were observed in frontal cortex of p.Thr262Ser mutation carrier (A). Some rare TDP-43 positive NII were also found in the brainstem of this case (B). In contrast, numerous TDP-43 immunoreactive NII and dystrophic neurites (DN) were observed in all layers of the frontal (C) and temporal cortices (D) of p.Arg159Ser mutation carrier. The NII were also immunoreactive for p62 (E). Staining of VCP revealed rare NII (F). The arrows indicate the pathological findings. Scale bar is 20μm.
DISCUSSION
In this study, we identified two novel and one reported mutation in the VCP gene in patients with bvFTD, and we provided neuropathological features of the two novel mutations (p.Thr262Ser and p.Arg159Ser) carriers. The initial clinical presentation of behavioral problems (including semantic deficits in one) without any signs of motor neuron disease or myopathy indicated the clinical diagnosis of bvFTD. Neuropathological findings in two patients showed numerous TDP43-IR NII and short DN compatible with FTLD-TDP type D and rare VCP-IR NII.
The pathogenicity of the three VCP mutations is supported by the fact that mutations at the same codon have been reported previously (Table 1) [13, 31]. Furthermore, the mutations in the present study are not present in population-matched controls, and were identified in the CDC48 and D1 domain, wherein most of the pathogenic VCP mutations have been reported [12]. The mutation p.Met158Val has previously been identified in a patient with sporadic ALS [13], but it has not been reported in FTD before. The pathogenicity of this mutation is supported by increased translocation of TDP-43 from the nucleus to cytoplasm compared to those transfected with wild-type VCP in transfected SH-SY5Y cells and HEK293T cells [13]. Mutations in codon 159 and 262 have been reported in patients with variable phenotypes including FTD, ALS, and IBMPFD [17, 31–33]. The pathological findings of FTLD-TDP type D in our mutations carriers is consistent with previously reported findings of VCP mutation carriers [17, 31].
The clinical presentation of pure FTD without any signs of Paget disease of the bone or myopathy in all three patients is in contrast to the classical phenotype of IBMPFD associated with mutations in VCP. Distinct phenotypes including pure FTD or ALS have been described in other mutations at the same codons [13, 32], suggesting that additional genetic or environmental factors may determine the clinical phenotype. Another possibility might be a preclinical state of IBM or Paget disease, however, normal values of alkaline phosphatase in two patients made Paget disease unlikely. Furthermore, no muscle weakness or fasciculations were reported in neurological examinations ruling out IBM or ALS. The variation in age at onset and the unaffected parents of the proband in pedigree A also supported the presence of possible genetic modifiers in VCP mutations carriers.
The presence of VCP-IR NII in both cases indicated the involvement of mutant VCP in protein degradation. The neuropathological findings of numerous TDP43-IR NII and DN in both cases were similar to previously reported VCP mutations at the same codons [17, 31]. However, the distribution and the severity of the neuronal inclusions and DN differed between the two cases, with a lower number of NII and DN in the p.Thr262Ser carrier. These differences were unlikely to be related to the disease duration as the disease duration was shorter in the p.Arg159Ser carrier. A possible explanation is that the different location of the mutations may explain the extent of the pathology. More studies are needed to elucidate the disease mechanism of VCP mutations.
The frequency of 3.5% of VCP mutations could not be compared to other studies, as we screened a highly biased cohort of those patients without any other known mutations. Our findings indicated that it is worthwhile to screen for VCP mutations, especially after excluding the most common mutations for FTD. In our Dutch FTD cohort, the frequency of VCP mutations was 4.2% in those with a positive family history. A limitation of the present study is that clinical information of the latest stages of the disease is unavailable. Patients were deceased many years ago, and were not followed until dead. At last, co-segregation analysis in families could not be performed due to the lack of DNA samples from relatives.
In conclusion, we report three VCP mutations, of which two novel mutations, in three Dutch patients with bvFTD. Pathological findings and previously reported mutations at the same codon support the pathogenicity of these mutations. Our findings expand the genetic and clinical spectrum of VCP mutations, and further underline the clinical heterogeneity. Future studies are warranted to investigate the involvement of genetic modifiers in the disease process of VCP carriers.