
Abstract
Waste clearance from the brain parenchyma occurs along perivascular pathways. Enlargement of the perivascular space (ePVS) is associated with pathologic features of Alzheimer’s disease (AD), although the mechanisms and implications of this dilation are unclear. Fluid exchange along the cerebral vasculature is dependent on the perivascular astrocytic water channel aquaporin-4 (AQP4) and loss of perivascular AQP4 localization is found in AD. We directly measured ePVS in postmortem samples of pathologically characterized tissue from participants who were cognitively intact or had AD or mixed dementia (vascular lesions with AD). We found that both AD and mixed dementia groups had significantly increased ePVS compared to cognitively intact subjects. In addition, we found increased global AQP4 expression of the AD group over both control and mixed dementia groups and a qualitative reduction in perivascular localization of AQP4 in the AD group. Among these cases, increasing ePVS burden was associated with the presence of tau and amyloid-β pathology. These findings are consistent with the existing evidence of ePVS in AD and provide novel information regarding differences in AD and vascular dementia and the potential role of astroglial pathology in ePVS.
Keywords
INTRODUCTION
Previously considered a benign neuroradiological finding or an artifact of tissue fixation, enlargement of the perivascular space (ePVS) is emerging as clinically relevant, as increasing ePVS burden is associated with Alzheimer’s disease (AD) status [1, 2], cerebral small vessel disease [3], and cerebral amyloid angiopathy (CAA) [4]. The frequency and severity of ePVS is significantly greater in AD than in cognitively intact subjects and correlates with amyloid-β (Aβ) load, severity of CAA, and ApoE ɛ4 genotype [5]. Perivascular spaces, also termed the Virchow-Robin spaces, recently have been established as elements of a brain-wide perivascular network termed the “glymphatic system” that facilitates the clearance of solutes including Aβ from the brain parenchyma [6, 7]. Glymphatic function is thought to depend on the expression of astrocytic water channel aquaporin-4 (AQP4) at the perivascular endfoot; deletion of the Aqp4 gene in mice slows interstitial solute clearance [6] and promotes deposition of Aβ [8]. Although several subsequent studies have corroborated a role for AQP4 in maintaining interstitial solute homeostasis [8–12], a recent study by Smith et al. failed to recapitulate the effect of Aqp4 gene deletion on glymphatic pathway function [13]. In addition to the relative contributions of diffusion, perivascular and interstitial bulk flow to perivascular CSF-ISF exchange is a subject of substantial current debate [14, 15], as is the role of anesthesia on glymphatic function [16, 17].
Despite these remaining questions, studies carried out in human subjects suggest a link between AQP4 expression, localization, and the development of AD pathology. Two recent genetic studies demonstrated that single nucleotide polymorphisms in the human AQP4 gene were associated with altered rates of neurocognitive decline in AD [18], and moderated the relationship between sleep disruption and amyloid deposition [19]. As reviewed by Nagelhus and Ottersen [20], AQP4 is highly abundant in the brain and is implicated in numerous pathophysiological processes, and the organized expression of AQP4 at the perivascular domains of astrocytes is of particular relevance from a fluid dynamics perspective. Indirect experimental paradigms as well as modeling studies suggest that AQP4 supports the diffusion of water between perivascular spaces and tissue by forming low-resistance pathways for CSF influx and ISF efflux routes [21, 22]. In histopathological studies in AD subjects, AQP4 expression is increased at cerebrospinal fluid (CSF)-brain interfaces [23]. Our group recently reported that loss of perivascular AQP4 localization surrounding the cortical microvasculature is predictive of AD status and is associated with increasing Aβ burden and neurofibrillary pathology [24]. In the present study, we evaluated whether histologically quantified ePVS surrounding large (greater than 6 μm in diameter) cortical vessels are associated with AD or vascular pathology. In addition, we assessed perivascular AQP4 localization along these vessels as a predictor of increased burden of ePVS. These are the first data to evaluate ePVS as a putative marker of glymphatic insufficiency in the AD brain, with the goal of understanding if ePVS may represent an early marker of glymphatic dysfunction. In addition, these data provide an opportunity to evaluate the relationship between clinical dementia status, histopathological characteristics, and targeted quantification of ePVS, which will inform future studies targeting the mechanism and potential therapeutic target of ePVS in progressive neurodegenerative processes.
MATERIALS AND METHODS
Subject inclusion
This study consisted of data acquired from 33 subjects from the Oregon Health & Science University (OHSU) Layton Aging and Alzheimer’s Disease Center (OADC) and the affiliated brain tissue repository, the Oregon Brain Bank. Brain autopsy was performed on all participants after consent was obtained from the next of kin and in accordance with Oregon Health and Science University guidelines. Selection of tissue samples was made on the basis of AD or mixed AD/vascular dementia (VaD) diagnosis established in a consensus meeting, wherein all data, including clinical, cognitive, and pathology data are considered by a multi-disciplinary team as previously described [25] in accordance with established criteria [26]. Samples from community dwelling individuals with no known neurological disease were included as controls. Patients who were found to have neocortical or midbrain Lewy bodies, hippocampal sclerosis, macroscopic infarction, or other non-AD pathologic findings were excluded, as were subjects for whom this comprehensive assessment was not available. The final dataset consisted of 16 control and 17 dementia cases. Among the dementia group, 10 were classified as AD, and 7 as mixed AD/VaD. See Table 1 for summary information. Diagnosis of VaD was made on the basis of pathological assessment of macro- and microscopic vascular disease contributing to the dementing illness [27].
Summary participant characteristics by clinical subgroup
**p < 0.01 *p < 0.05. C, control; AD, Alzheimer’s disease; MVL, microvascular lesion; PMI, postmortem interval; AA, amyloid angiopathy; PV, perivascular; PVS:V, ratio of the maximum distance between parenchymal boundaries surrounding the vessel (PVS, Fig. 1, green) and the maximum diameter of the vessel wall (V, Fig. 1, yellow line); (t), targeted assessment on contiguous tissue slice; AQP4, aquaporin-4.
Targeted histopathology and immuno-fluorescence
Immunofluorescence and targeted histopathology was performed in frontal cortical tissue as previously described [24]. Briefly, 6-micron paraffin-embedded sections were labeled with antibodies against AQP4 (Millipore AB3594, 1:800) and glial fibrillary acidic protein (GFAP; Millipore MB360, 1:500) and counterstained with DAPI (Fig. 1A, B). All vessel-wise quantification was completed by a research assistant (EL) blind to clinical group status. For each subject, the perivascular space was evaluated at ten large caliber (mean diameter = 15.36 μm, standard deviation (SD) = 5.52 μm) vessels that cross-sectionally transected the plane of imaging. As sampling was targeted to larger caliber vessels, vessels exceeding 40 μm as well as those less than 6 μm at cross-section were excluded from the analyses. For one subject this resulted in 9 vessels sampled. Based on previously published methods [6, 24], vessels were identified in the DAPI channel, while measurement of regions were generated in the GFAP channel using autofluorescence to distinguish tissue boundaries (Fig. 1B, insert). Vessel size was defined as the maximum diameter between vessel walls (Fig. 1B, insert, yellow line). Perivascular space size was defined as the maximum distance between parenchymal boundaries surrounding the vessel (Fig. 1B, insert, blue line). There was a significant correspondence (r = –0.38, p < 0.05) between the pathologist’s categorical rating of dilated perivascular spaces and the vessel-wise approach described.
Perivascular AQP4 localization was quantified based on previously established methods [6]. Specifically, four independent 50-pixel (40.23 μm) line regions of interest were generated for each vessel, then measured in the AQP4 channel. Fluorescence intensity values were measured along these lines with the first 10 pixels (8.01 μm) of each line defined as the “perivascular signal”, while the remaining 40 pixels (32.18 μm) was defined as the “parenchymal or ‘global’ signal” (Fig. 1, green & orange lines). For each vessel, the “perivascular intensity” was defined as the maximum value in the first 10 pixels for each vector averaged across the four vectors. The average maximum was selected to eliminate the potential for spurious inclusion of pixels inside the PVS lumen, which would introduced error of low values, and the average of the four was to attend to possible high intensity pixels. “Global” AQP4 was defined as the mean of the remaining 160 pixels (40×4 vectors) for each vessel. Perivascular AQP4 localization for each vessel was quantified as “perivascular”/“global” intensity, such that values close to 1 indicate no polarization in AQP4 expression and values greater than 1 indicate elevated relative perivascular AQP4 expression. Mean values were calculated across vessels per sample for ePVS and AQP4 variables.
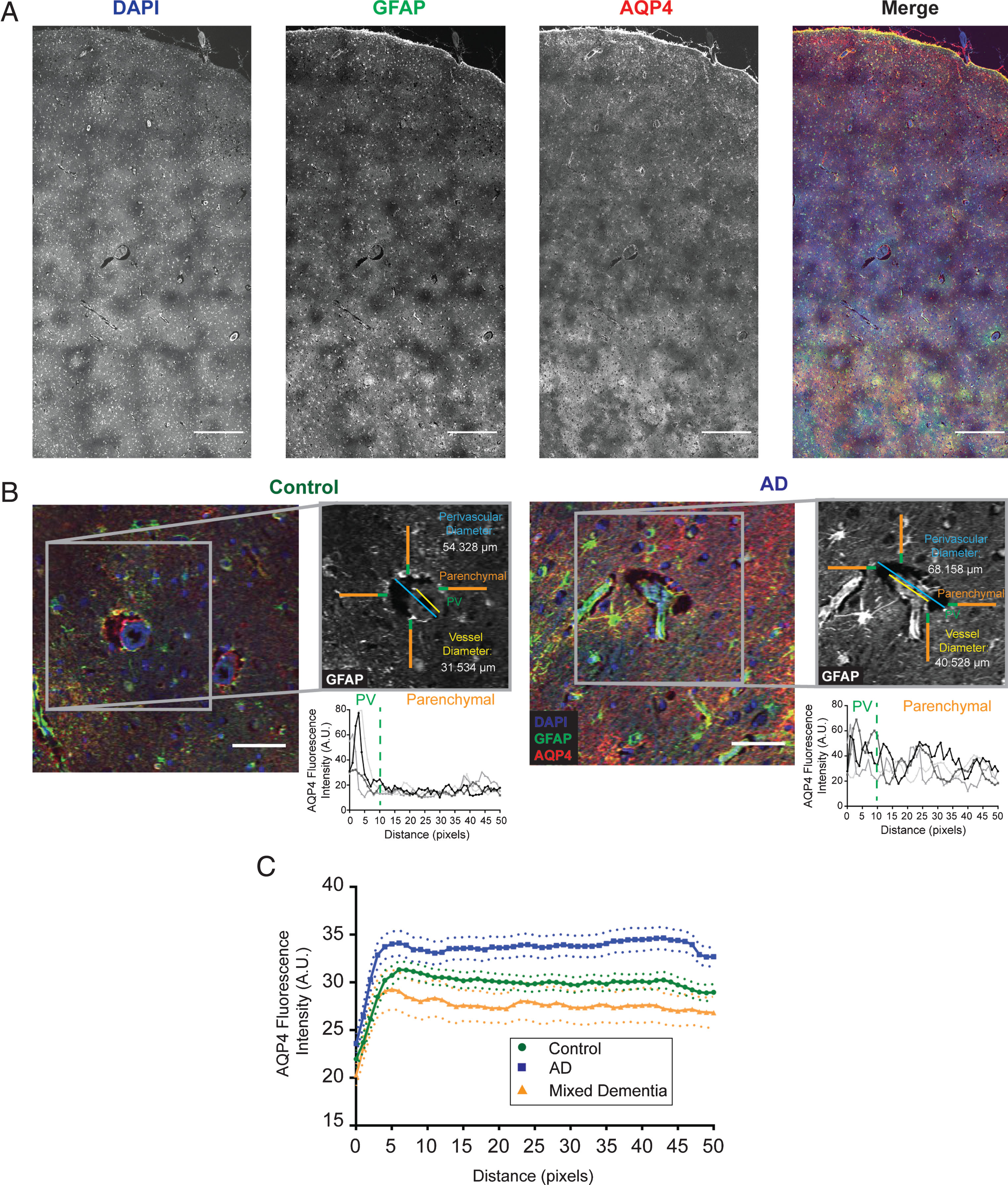
Standardized analysis of perivascular dilation and perivascular AQP4 localization. Representative image of large field images (A) as well as individual vessels (B) used for quantification. Example images are shown for both control (B, left) and Alzheimer’s disease (B, right) along with the associated methodology for analysis of the perivascular compartment (inset). Regions of interest were generated using the GFAP channel (grey), then measured using the AQP4 channel. The yellow line represents the vessel diameter measurement, while the perivascular dilation measurements is shown in blue. Orange and green lines represent the perivascular AQP4 quantifications with green representing signal defined as “perivascular” while the orange segment is the “parenchymal” signal. Plots are representative of AQP4 intensity measurements for each of the linear regions of interest. Blue = DAPI, green = GFAP, and red = AQP4. Scale bars = 500 μm (A) and 50 μm (B). C) Plots showing the average intensity along the 50 pixel linear ROIs for control (green), Alzheimer’s disease (blue), and mixed vascular and Alzheimer’s disease (orange) cases for all vessels measured. Dotted lines represent the 95% confidence intervals for each category.
In contiguous slices, H&E stained slides were examined and coded by a pathologist blind to study group for cerebrovascular lesion severity. Specifically, these were given a categorical rating (0–3) on each of the following; perivascular space dilation, arteriosclerosis, perivascular hemosiderin, and myelin pallor, according to the criteria published by Deramecourt et al. [28]. Case-based pathology data obtained from assessment of the entire brain obtained at autopsy by a neuropathologist included Aβ plaque density, Braak tangle stage, neocortical Lewy bodies, atherosclerosis, arteriolosclerosis, deep microvascular lesions, cerebral amyloid angiopathy, and cortical microvascular lesions, were assembled from the data repository of the OADC.
Statistical approach
The distinction between AD and mixed dementia with AD is commonly not made in clinical cohort studies, and participants with mixed dementia are often reported as part of the AD cohort, including the larger cohort from which this subset was taken [24]. As such, we evaluated the sample in two ways; initial assessment as dementia (which included AD and mixed AD/VaD) compared to control, and subsequently as three distinct groups (AD, mixed AD/VaD, control). VaD case status was made if the histopathological assessment by a neuropathologist indicated vascular pathology was significant enough to have likely contributed to their cognitive status. Accordingly, the mixed dementia group had significantly higher atherosclerosis, and microvascular lesion scores than the cognitively intact and AD groups (see Table 1).
As an additional level of analysis, we tested specific pathology variables for which group differences were observed as potential within-subject determinants of dilation of the perivascular space and of perivascular AQP4 expression. Group differences in demographic, clinical, and pathological characteristics were assessed via t-test (for two group models) and ANOVA (three group models) for continuous variables and via Mann-Whitney U or Kruskal-Wallis, respectively, for ordinal variables, including pathological ratings. Multiple comparisons within models were Bonferroni corrected. Assessment of ePVS and specific pathological variables was made by dichotomized as “presence” or “absence” of the pathology noted. One subject’s AQP4 expression data were excluded from analyses due to extreme (high) outlier values. The statistical significance or α in these models was set at p < 0.05. Unless otherwise noted, data are reported as mean±standard deviation.
RESULTS
Clinical group comparison
Dementia: Control
Dementia and control groups did not differ with respect to age, sex, years of education, or postmortem interval (Table 1). The dementia group had a significantly greater neuritic plaque burden and Braak stage (p < 0.01) than the control group. Summary statistics revealed no group-wise difference in vascular pathology variables between dementia and control groups, although median scores for deep and cortical microvascular lesion load were higher in the dementia group (p = 0.1). Targeted histology found significantly increased incidence of amyloid angiopathy (p < 0.05) and myelin pallor (p < 0.01) in the dementia group compared to control.
Vessel-wise outcomes: There was significantly greater perivascular space dilation in the dementia group relative to control (p < 0.01, Fig. 2A). While no significant group differences were observed for perivascular AQP4 localization, a significant increase in global AQP4 expression was observed in the dementia group (p < 0.01, Fig. 3A).
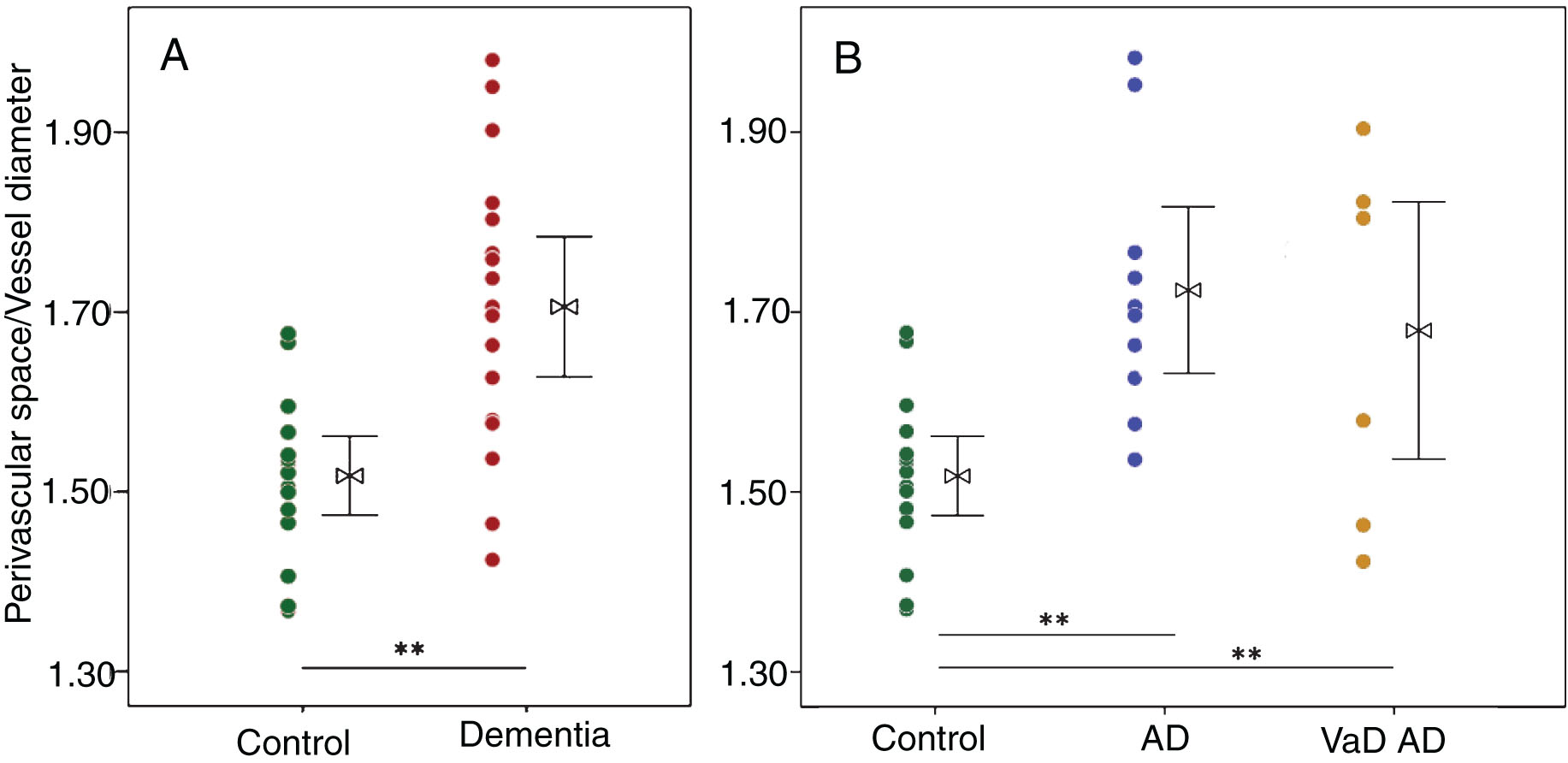
Enlargement of the perivascular space of large caliber (>6 μm) vessels, defined as direct assessment of the ratio of perivascular space to vessel diameter, is significantly increased in dementia cases over aged control (A). When assessed by dementia subgroup, the perivascular space is enlarged in both Alzheimer’s (AD, n = 10) and vascular (VaD, n = 7) dementia cases over aged control (n = 16) (B). t-test (for two group model) and ANOVA with Bonferroni correction (three group model), *p < 0.05, **p < 0.01.

A) Increased global expression of astrocytic water channel Aquaporin-4 (AQP4) was observed in cases with Alzheimer’s disease (AD, n = 10) over aged control cases (n = 16), but not in cases of mixed dementia consisting of AD and vascular pathologies (n = 7). B) While no significant group differences in perivascular localization of AQP4 expression was observed, the AD group had a qualitative reduction in perivascular localization of AQP4 compared to mixed dementia cases (p = 0.08). ANOVA with Bonferroni correction, *p < 0.05, **p < 0.01.
Alzheimer’s disease: Control: vascular dementia
Assessed by group as control, AD, and mixed dementia, there were no significant differences in sex, years of education, or postmortem interval by group. While the mixed dementia cohort was older than both the control and the AD group (p < 0.01), there was no main effect of age on the primary dependent variable of ePVS (R = 0.073, p = 0.69). Both the mixed dementia group and AD group had a significantly greater neuritic plaque burden and Braak stage (p < 0.01) than the control group, and no significant difference in these measures were observed for the AD compared to the mixed dementia group. As expected, the mixed dementia group had a greater prevalence of atherosclerosis than the AD group (p < 0.05) and a greater prevalence of deep and cortical microvascular lesions (MVL) than both the AD and the control groups (p < 0.01). In addition, the mixed dementia group had a greater prevalence of myelin pallor than control (p < 0.01).
Vessel-wise outcomes
Increased dilation of the perivascular space was found in the AD and mixed dementia groups compared to control groups (p < 0.01, Fig. 2B). A significant group-wise increase in AQP4 expression in the AD group relative to control and VaD groups was observed (p < 0.01, Table 1, Fig. 3B). Perivascular localization of AQP4 did was not significantly differ across groups, although a qualitative reduction was found for the AD group relative to mixed dementia (p = 0.08, Fig. 3B).
Histopathological predictors of enlarged perivascular space burden
Presence or absence of the pathology variables for which significant group differences were observed (above) were tested as predictors of ePVS. Cases with tau pathology (Braak staging greater than 0) had significantly increased dilation of the perivascular space (k = 11.5, p < 0.01) over those with a score of 0, and cases with neocortical neuritic plaque (CERAD C score greater than 0) had significantly increased perivascular space dilation (k = 4.6, p < 0.05) over those with a CERAD C score of 0. Among vascular features, only presence of deep, but not cortical, microvascular lesions (k = 2.5, p = 0.12) and CAA (k = 2.3, p = 0.13) were qualitatively associated with enlargement of the perivascular space, although these findings did not meet statistical threshold (Fig. 4). No statistically significant differences in AQP4 localization were observed with respect to pathology features, although a qualitative reduction was observed for cases with noted CAA (k = 3.1, p = 0.08).
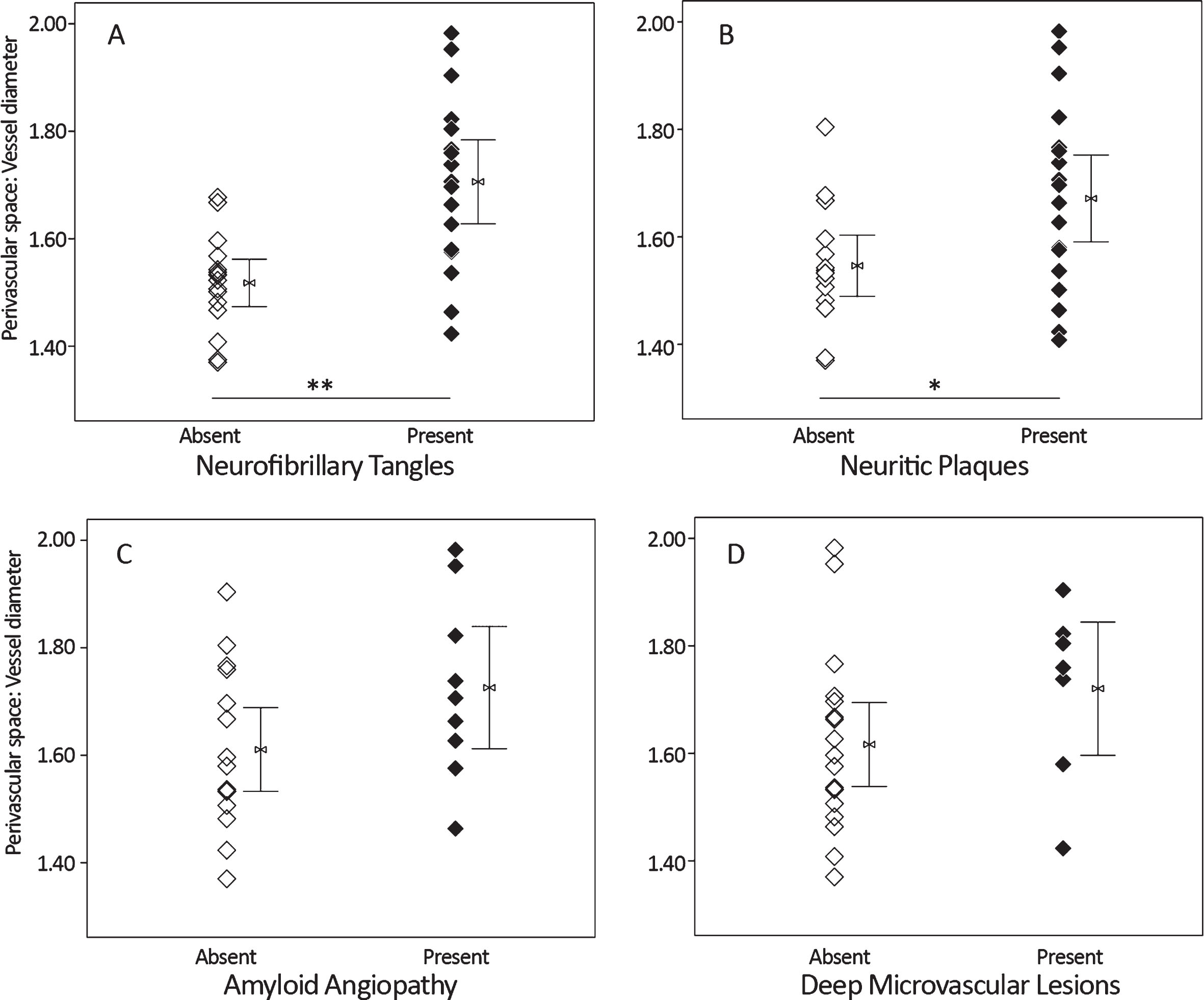
In a cohort of control, Alzheimer’s disease (AD), and mixed (AD with vascular) dementia samples (n = 33), presence of tau (A) and amyloid plaque (B) assessed at autopsy is associated with significantly increased perivascular space dilation. Among cerebrovascular risk factors, perivascular space dilation was qualitatively increased in cases with cerebral amyloid angiopathy (C) and deep microvascular lesions (D). Mann-Whitney U, *p < 0.05, **p < 0.01.
Although there were no main effects of age on perivascular space dilation, given the group-wise differences in age, and to confirm findings with dichotomized variables (above), we tested the effect of age on ePVS and semi-continuous (categorical) pathology ratings. In univariate analyses of variance (ANOVA), categorical Braak stage score remained significantly associated with ePVS (F = 15.2, p < 0.01) while no significant effect of age was observed. Similarly, categorical rating of neuritic plaque density remained significantly associated with ePVS (F = 4.7, p < 0.05), while the effect of age was not significant. In univariate ANOVA with age and presence of deep MVL, while age was significant in the model (F = 7.4, p < 0.05), presence of deep MVL remained significant as well (F = 5.6, p < 0.05).
DISCUSSION
The brain perivascular space is well demonstrated to be a route of fluid flow serving to maintain brain homeostasis [6, 29], and recent studies have shown clearance of interstitial wastes including Aβ and tau by way of the perivascular compartment [6]. There is mounting evidence that dilation of this space is associated with clinical and diagnostic features of amyloid and vascular pathology. Postmortem evaluation has linked enlarged perivascular spaces (ePVS) with vessel-bound amyloid in the form of CAA [4, 5] and with increased amyloid burden within the parenchyma [5]. Further in vivo work employing MRI has identified a strong correlation between radiologically visible (and thus enlarged) PVS and AD [1, 2], cerebral small vessel disease [3], and CAA [4].
In the present study, we found that enlargement of the perivascular space, assessed in postmortem tissue, is associated with the clinical-pathological diagnosis of AD and mixed dementia, which consists of both AD and vascular contributions. In addition, we found that ePVS are increased in cases with neurofibrillary tangle burden, most prominently, and also in cases with neocortical neuritic plaques. Cerebrovascular pathology was not as strongly associated with increased ePVS burden, although among cerebrovascular variables, there was a qualitative increase in ePVS in cases with CAA and deep MVL. These findings are generally consistent with the literature describing AD and cerebrovascular pathology to be associated with dilation of the perivascular space, as above. Our observation of tauopathy being most prominent in ePVS burden remains relatively unexplored in existing literature. As this is a major pathologic hallmark of AD progression, this may be a surrogate marker of AD severity. There is emerging evidence for tau clearance along the PVS, including AQP4 mediated clearance mechanisms; loss of perivascular AQP4 was associated both with increased phosphorylated tau immunoreactivity in experimental animal models and with greater neurofibrillary pathology in human AD subjects [10, 30].
While enlargement of the perivascular space increasingly is recognized as pathologically relevant, particularly in the setting of AD and VaD, the underlying mechanism remains unclear. The astrocytic water channel AQP4 has a clear role in glymphatic pathway function, including perivascular CSF-interstitial fluid (ISF) exchange and Aβ and tau clearance from the brain [6, 31]. Consistent with previous work [24], the present study found significantly increased global AQP4 expression in AD relative to control. In addition, we found that, when assessed as AD and mixed (AD/vascular) dementia sub-groups, this increased global expression was specific to the AD group; global expression of AQP4 was increased in AD over both control and mixed dementia groups while global AQP4 expression in mixed dementia cases was statistically equivalent to control cases (Figs. 1B, 3A). In contrast to our prior study, which identified reduced perivascular AQP4 localization surrounding the small-caliber cerebral microvasculature in AD subjects [24], this study of larger vessels found only a qualitative reduction in perivascular localization of AQP4 in the AD group (Fig. 3B). We assessed the possibility of these disparate findings as a reflection of insufficient power to detect a significant difference in the present dataset. When analyzed in an independent samples t-test between dementia subgroups, rather than an ANOVA, the difference between the dementia sub-groups meets statistical significance (t(15) = –2.27, p < 0.05). Post-hoc power analysis of mean differences between AD and mixed dementia group (Table 1) at alpha = 0.05 gives 13.3% power. These results may indicate a type two error in these data and there is a true difference in perivascular localization of AQP4 in the AD group relative to mixed dementia. However, without statistically significant difference from control, we must retain the null hypothesis for the current data. Future studies, such as those employing animal models sufficient to tease apart the role of astrocytopathy in “pure” amyloid, tau, or vascular pathology to symptomatic disease onset are certainly warranted.
In the setting of our previous work, in which the same global increase, as well as significantly reduced perivascular localization of AQP4 was found in the small vessels of AD subjects these data may also suggest that pathological features of perivascular spaces of larger caliber vessels are distinct from those of the microvasculature, perhaps reflecting their association with Virchow-Robin spaces rather than the microvascular basal lamina. A possible model for this could be that glymphatic dysfunction in the small vessels impedes clearance of interstitial waste products [6, 24], which may result in the accumulation of these proteins. Deposition of protein aggregates may cause reactive astrogliosis, including global upregulation of AQP4, consistent with our finding of increased global AQP4 expression in AD but not mixed dementia (Fig. 3). Additional work evaluating the expression pattern of AQP4 in small caliber vessels proximal to larger arteries and veins would elucidate these mechanistic considerations.
Our observation of increased ePVS burden in AD and mixed dementia (Fig. 2), but disparate AQP4 expression levels between groups suggests potentially divergent etiologies of ePVS by pathological sub-type. In the context of perivascular waste clearance, these data suggest that ePVS may reflect failed clearance of parenchymal amyloid in AD, whereas in primary vascular pathology, amyloid may be more successfully cleared from the parenchyma, reducing its propensity to serve as a catalyst for astrocytosis. In the case of CAA, amyloid is deposited in the vessel wall, and resultant sclerosis of the vessel wall results in stagnation of perivascular fluid and dilation of the PVS [29]. Alternatively, changes in AQP4 expression or localization may have different consequences in the setting of vascular pathology. Aqp4 deletion or loss of perivascular AQP4 localization is protective in experimental stroke models, owing to reduced formation of cytotoxic edema [32, 33]. Of course, the myriad roles that AQP4 serves in the maintenance of brain homeostasis (reviewed in [20]) cannot be fully addressed in the scope of this study. Future work assessing focal vascular pathology in ePVS would provide needed information in this respect.
The nature of postmortem tissue analysis is also important to consider with regard to these findings. Tissue fixation artifacts may result in retraction that would alter perivascular diameter measurements. Furthermore, though all measurements were made in a blinded fashion, these were also made by hand. Thus, it is also important to consider sample bias in the measurements of both perivascular and vessel diameters. The precise definition of the perivascular space also remains a point of contention. Here we define this as the space between the vessel and the astrocytic endfoot domain, a space supported by structural study [6, 34]. However, other groups have defined the perivascular compartment using ultrastuctural studies to be between the leptomeningeal layer adjacent to the side of tunica media facing side the parenchyma and the leptomeningeal layer adjacent to the glia limitans (astrocyte end feet) [35]. It will be important to follow up this work with additional tissue staining to elucidate the microstructural anatomy of these dilated spaces. Such stains, including smooth muscle actin to determine if the vessels around which the ePVS are observed are of arterial (as opposed to venous) origin also would inform the mechanistic underpinnings of ePVS.
Given the observations that these spaces are present in the setting of traumatic brain injury [36] and multiple sclerosis [37], it will be important to focally assess the contribution of inflammation and blood-brain barrier permeability to the ePVS and their clinical relevance. In addition, future studies aimed at quantifying the presence and activity of perivascular macrophages and the integrity and functionality of the endothelia and tight junction proteins, as well as ion transporters and channels focal and proximal to the site of dilation will be important next steps in fully characterizing these features. Whether age-related changes in the spaces surrounding intraparenchymal vessels or the microvasculature contribute to development of AD pathology remains an important but unresolved question. One clear limitation to this study is the limited tissue sampling. Regions of tissue were obtained from frontal cortex, a region that is relatively spared in tau and amyloid pathology until very advanced stages of the disease. In addition, ePVS are observed on clinical imaging most predominately in the subcortical white matter and in the basal ganglia [38], regions not sampled in the present study. Future work tracking MRI visible ePVS longitudinally and targeting histopathology to regions of high ePVS burden is certainly warranted. Tissue sampling was also limited by the extent to which focal pathology could be quantified; category ratings of pathology were used to evaluate correlates of ePVS and only AQP4 was evaluated at the field level. While these findings recapitulate more targeted histopathological observations of ePVS in that they are associated with both vascular and total amyloid load [5] and provide novel information informing the potential role of astroglial biology in ePVS, there is much additional work needed to fully characterize the clinical and mechanistic relevance of ePVs. A clear next step will be to expand the scope of the present study to include focal quantification of additional likely contributors to ePVS. The current data show that ePVS burden are increased in cases with AD and vascular pathology, they also show that the increased expression of astroglial protein AQP4 is specific to primary AD cases (Fig. 3). Quantifying the presence of amyloid and/or tau pathology immediately proximal or distal to the dilated space, and the extent to which such pathology co-localizes with evidence of astrocytopathy, specifically, AQP4 localization, will inform the underlying mechanism and potential clinical relevance of ePVS.
Footnotes
ACKNOWLEDGMENTS
This project was supported by: the NIH (P30AG00817 (ELB, RW, LCS, JAK), P30AG00817–28 (ELB), P30AG024978 (JAK), P30 NS061800, and R01AG054456 (JJI), K01AG059842 (ELB)), the Oregon Alzheimer’s Disease Research Center Pilot Grant (ELB, JJI), and the Paul G. Allen Family Foundation (JJI, ELB). The authors also gratefully acknowledge the research participants and tissue donors.