
Abstract
Background:
Alzheimer’s disease (AD) is a chronic neurological disorder associated with mental decline and dementia. Several studies focused on investigating the molecular basis of the disease that led to the identification of several causative genes and risk associated alleles. Replication of these studies and findings from different populations is very important.
Objective:
Molecular assessment of a cohort of 117 familial and sporadic AD cases from Saudi Arabia.
Methods:
Comprehensive screening for point mutations was carried out by direct sequencing of coding regions in the three known AD causative genes: PSEN1, PSEN2, APP, as well as the AD associated gene SORL1. All patients were also genotyped for APOE alleles. In silico 3D protein structure analysis was performed for two novel SORL1 variants.
Results:
We identified a total of eight potential pathogenic missense variants in all studied genes. Five of these variants were not previously reported including four in SORL1 (p.Val297Met, p.Arg1084Cys, p.Asp1100Asn, and p.Pro1213Ser) and one in APP (p.Glu380Lys). The frequency of APOE-ɛ4 allele was 21.37% of total investigated cases. In silico 3D protein structure analysis of two SORL1 novel missense variants (p.Pro1213Ser and p.Arg1084Cys) suggested that these variants may affect the folding of the proteins and disturb their structure.
Conclusions:
Our comprehensive analysis of the open reading frame of the known genes have identified potential pathogenic rare variants in 18/117 cases. We found that point mutations in AD main genes (PSEN1, PSEN2, and APP) were underrepresented in our cohort of patients. Our results confirm involvement of SORL1 in familial and sporadic AD cases.
INTRODUCTION
Alzheimer’s disease (AD) is a complex neurodegenerative disorder associated with progressive memory deterioration and cognitive perturbation with no efficient therapy. The pathogenesis of AD is hallmarked by neuronal loss, accumulation of intraneuronal tau proteins [1, 2], and extracellular neuritic aggregation of amyloid plaques (Aβ) peptides [3, 4]. As age is the first risk factor in AD, early onset (EOAD) occurs at age below 65 in less than 5% of patients [5]. Late onset (LOAD) is the common and major sporadic form of the disease that manifests at age 65 years or older.
The genetic component in EOAD is strong with heritability ranging from 92 to 100% of which approximately 90% of cases are due to autosomal recessive causes [6]. Family history is present in 35 to 62% of EOAD patients [5, 8]. However, in about 10 to 15% of EO familial AD (EO-FAD), the inheritance is shown in autosomal dominant pattern with full penetrance [5, 8]. Monogenic mutations in presenilin 1 (PSEN1; MIM104311) [9, 10], presenilin 2 (PSEN2; MIM600759) [11], and amyloid precursor protein (APP; MIM104760) [12, 13] account for 5 to 10% of EO-FAD [6, 14]. To date, PSEN1 is found to be the most implicated gene with 221 mutations reported as pathogenic in the Alzforum database, followed by 32 pathogenic mutations described in APP, while mutations in PSEN2 are less common with only 19 reported so far (http://www.alzforum.org/mutations).
The genetic architecture of LOAD reflects the heterogeneous and complex form of the disease with heritability ranging from 58 to 79% [15]. Several studies showed the presence of reliable genes implicated in risk for LOAD (reviewed in [16]). The ɛ4 allele of Apolipoprotein E (APOE; MIM107741) (APOE ɛ4) is still described to be the most important genetic risk factor for LOAD [17–19]. In addition, sortilin related receptor gene (SORL1; MIM602005) was primarily described as a risk for AD [20–23]. Rare variants in SORL1 are associated with EO and LO-FAD [24–26]. The relation of SORL1 to AD was confirmed in various populations by meta-analysis [27–30] and genome wide association studies (GWAS) [28, 31].
As genetics for AD in the Saudi population need to be explored, the aim of this study is to determine the spectrum of mutations in Saudi AD patients. We focused on identifying point mutations in PSEN1, PSEN2, APP, and SORL1 and establishing the genotypes of all the patients for APOE alleles to provide a background for molecular basis of AD in the Saudi population.
MATERIALS AND METHODS
Patients and family history
One hundred and seventeen patients were recruited for this study after obtaining written informed consents following institutional approval of Internal Review Board (IRB) at King Faisal Specialist Hospital and Research Center (RAC#2120020). All patients were reviewed by neurologists with full clinical and family histories recorded and fulfilled criteria for probable AD [32]. Patients were subdivided into familial (Supplementary Table 1) or sporadic (Supplementary Table 2) according to age and family history. Patients’ classification and clinical features are summarized in (Table 1).
Demographic and clinical features of the enrolled patients
AD, Alzheimer’s disease; SP, sporadic; EO, early onset; LO, late onset; EO-FAD, early onset-familial AD; Poss. FAD, possible familial AD, AD-FH, AD with family history; F, female; M, male; Y/N/NR, yes/no/not reported.
Variants screening in AD Mendelian and associated genes
Genomic DNA was extracted from whole blood from patients and quantified using standard methods. The affected individuals were screened for variants in following AD genes: PSEN1, PSEN2, APP, and SORL1 by direct sequencing using ABI Prism ® Big Dye ® Terminator v3.1 Cycle Sequencing Kit as described by the manufacturer (Applied Biosystems, Foster City, CA, USA). Results were exported in one of several formats for visualization and sequence was compared to the reference GenBank sequence using SeqMan 6.1 (DNASTAR Lasergene 10 software package). Primer3 was used for designing primers corresponding to the entire coding sequence including intron/exon boundaries of all four studied genes (The information of primers and polymerase chain reaction (PCR) conditions are available upon request). The PSEN1, PSEN2, APP, APOE, and SORL1 were sequenced in all sporadic and familial cases. Frequency of identified variants was investigated in international databases (ExAC/1000 Genomes Project) and in the local ethnically matching controls (∼2500 exomes) from the Saudi Human Genome Program Database (SHGP) [33, 34].
APOE genotyping
The region within exon 4 including the two target markers rs429358 and rs7412 of APOE was amplified by PCR, followed by sequencing using ABI Prism ® Big Dye ® Terminator v3.1 Cycle Sequencing Kit and compared to the reference GenBank sequence. Then APOE genotyping analysis was done for ɛ2, ɛ3, and ɛ4 alleles.
In silico 3D protein structural analysis
To evaluate the effect of the novel missense variants in SORL1 exons 23 and 26, 3D-models for the wild and mutant SORL1 proteins were predicted using different protein analytical tools; PROSA [35], RAMPAGE [36], and NIH-SAVES server (http://services.mbi.ucla.edu/SAVES/) of which ERRAT application was involved [37]. The prediction of homology-based protein structure was performed using I-TASSER [38]. Protein superimposition and molecular graphics were established using PyMol (Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC) and the protein pathological character and stability for the missense variants were determined, by PMUT [39] and I-Mutant [40], respectively.
RESULTS
Our study revealed a total of 8 rare missense variants (Table 2) and 21 exonic polymorphisms (Supplementary Table 3). We found 5 novel rare heterozygous missense variants in APP and SORL1 (Table 2). Nineteen variants were detected in SORL1 (Table 2 and Supplementary Table 3). Six of the SORL1 variants were localized in the vacuolar protein sorting-10 receptor (VPS10) domain, one in the low-density lipoprotein receptor class A (LDLRA), 6 in the low-density lipoprotein receptor class B (LDLRB) and 6 in the fibronectin 3 (FN3) domain (Fig. 1).
Rare variants identified in the studied genes
Het, heterozygous; Hom, homozygous; FMR, familial related; SP, sporadic; APOE, Apolipoprotein E; SHGP LC, Saudi Human Genome Project Local Control; #: APOE genotyping for patients carrying p.Glu270Lys variant in SORL1: 6 cases are carriers of ɛ3/ɛ4 and 5 cases are carriers of ɛ3/ɛ3; EO, early onset; LO, late onset.
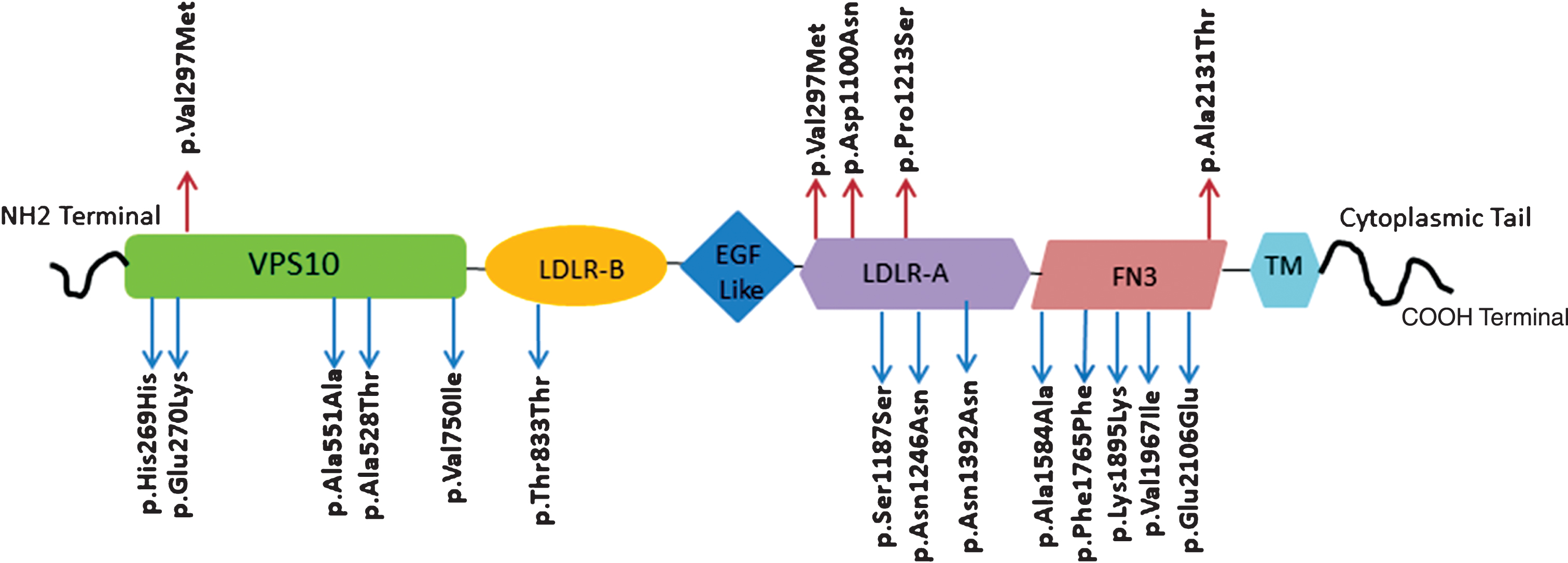
Diagram of SORL1 protein (2214 amino acids). SORL1 domains and the localization of total variants identified in our study (red arrows indicate novel variants and blue arrows indicate reported ones). SORL1, sortilin-related receptor; VPS10, vesicular protein sorting 10 domain; LDLR-B, low-density lipoprotein receptor B; EGF, epidermal growth factor; LDLR-A, low-density lipoprotein receptor A; FN3, fibronectin type3 repeats; TM, transmembrane region.
Demographics and clinical data
Our cohort is composed of 43 EOAD and 74 LOAD patients (Table 1). The familial history is noticed in 40.2% of AD cases (Supplementary Table 1). The mean age is 58.9 for EOAD and 71.2 for LOAD (Supplementary Tables 1 and 2). Fifty-four patients of our cohort were females and 63 were males (Table 1). The age and gender distribution of AD in Saudi patients (Table 1) are similar to other studied populations (http://www.alz.co.uk/research/statistics). The clinical phenotypes of our patients with detected rare variants showed the presence of typical amnestic, behavioral and neurological presentations of the disease (Supplementary Table 4). Worsening of cognition, impairment in learning, and brain atrophy are the most common clinical features detected in these studied cases (Supplementary Table 4).
Novel variants
We identified 5 novel rare missense heterozygous variants: one in APP (p.Glu380Lys) in a sporadic LOAD patient and 4 in SORL1 (p.Val297Met, p.Arg1084Cys, p.Pro1213Ser, and p.Asp1100Asn) in familial AD cases (Table 2). The missense variants p.Pro1213Ser and p.Arg1084Cys were identified in EO-FAD patients (FM02-FM03/Supplementary Table 1). The p.Pro1213Ser variant was detected in the proband and one unaffected sister of 53 years old (Fig. 2A, B). The p.Arg1084Cys variant was found in the proband and two family members (affected brother of 63 years old and unaffected son of 21 years old) (Fig. 2C, D). As for p.Asp1100Asn variant, patient’s brother diagnosed with AD and the familial history indicates the possibility of censoring effect [41, 42] (Supplementary Table 1, FM36) due to early death of patient’s father prior to onset of disease symptoms (Supplementary Figure 2).
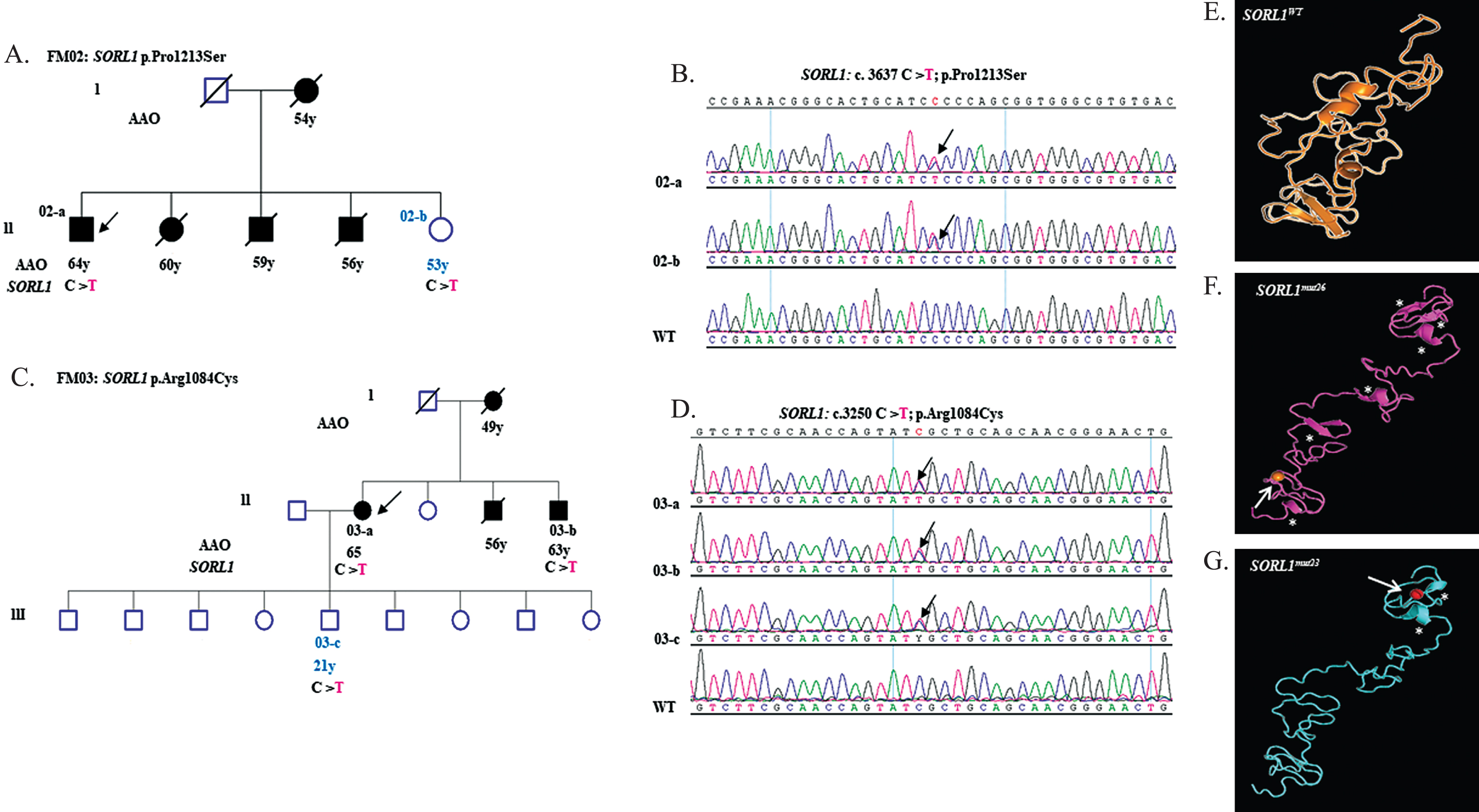
Genetic characterization of SORL1 variants p.Pro1213Ser and p.Arg1084Cys. A) Pedigree of FM02 with an SORL1 exon 26 (p.Pro1213Ser) variant. B) Sequence chromatogram of SORL1 exon 26 amplicon displaying heterozygous c.3637 C>T mutation in individuals 02-a, 02-b compared to WT. C) Pedigree of FM03 with an SORL1 exon 23 (p.Arg1084Cys) variant. D) Sequence chromatogram of SORL1 exon 23 amplicon displaying heterozygous c.3250 C>T mutation in individuals 03-a, 03-b and 03-c compared to WT. E, F, and G show prediction of functional impact of p.Pro1213Ser and p.Arg1084Cys mutations. E) Ribbon demonstration of structural models of LDLR-A domain from 1078 to 1235 amino acid harboring SORL1WT.
We also considered Combined Annotation Dependent Depletion (CADD) score and minor allele frequency (MAF) in international databases (ExAc/1000 genomes) as they are important in completing the assessment of potential pathogenicity of the novel variants identified here [43]. The novel p.Arg1084Cys variant is expected to increase AD risk by 12-fold and is considered to be strongly damaging because of its CADD score (>30) and absence in ExAc/1000 genomes databases [43]. In the same manner, the pathogenicity of our novel SORL1 variants (p.Val297Met, p.Pro1213Ser, p.Asp1100Asn) are expected to range from mildly to moderately damaging, therefore may contribute to the risk of AD since their CADD scores are between 10 to 30 and their absence in the ExAC/1000 genome databases (Table 2).
Previously reported variants
Three reported rare heterozygous missense variants were detected: one in each of PSEN1, PSEN2, and SORL1 (Table 2). In PSEN1, a missense variant p.Tyr195Cys [44] was identified in a familial EOAD case (FM30; Supplementary Table 1). In PSEN2, one HGMD-listed missense variant p.Val139Met [45] was identified in a sporadic EOAD case (SP13; Supplementary Table 2). A previously HGMD-listed missense variant p.Glu270Lys in SORL1 was detected in 7 familial EOAD cases and in 4 sporadic LOAD cases (Table 2).
Exonic polymorphisms
Six missense and 15 synonymous polymorphisms were identified in the coding regions of our studied genes (Supplementary Table 3). Fourteen of these variants were present in SORL1, 6 in PSEN2, and 1 in APP. All these polymorphisms were previously reported except for one novel SORL1 missense variant (p.Ala2131Thr) that was considered neutral in all the prediction tools (Supplementary Table 3). Both SORL1 missense p.Ala528Thr and synonymous p.Ser1187Ser variants were reported in HGMD. The p.Ala528Thr was described as disease-associated polymorphism with additional supporting functional evidence [26] and p.Ser1187Ser as disease-associated-polymorphism that may alter the gene expression [22]. The SORL1 single nucleotide polymorphisms identified in our study (Supplementary Table 3) were localized in three of SORL1 domains (VPS10, LDLRA and FN3) (Fig. 1). Some of these variants were reported in various populations [20, 46–48] and described to be associated with LOAD (Supplementary Table 5) [20, 49].
APOE genotyping
We determined the genotype and frequencies of APOE alleles in our studied cohort. The analysis showed that the most frequent allele was ɛ3 (77.78%) and ɛ2 had the lowest percentage (0.85%). The frequency of ɛ4 allele was 21.37% (Supplementary Table 6). APOE ɛ4 frequency was higher in LOAD patients (13.26%) compared to EOAD cases (8.11%) (Supplementary Tables 1 and 2). Furthermore, APOE ɛ4 frequency was higher in FAD cases (11.97%) (Supplementary Table 1) than in sporadic ones (9.40%) (Supplementary Table 2). Interestingly, we showed in Table 2 that part of EOAD patients with SORL1 rare variants are carriers of APOE ɛ4. The p.Pro1213Ser variant coincided with APOE ɛ4/ɛ4. The variants p.Val297Met and p.Arg1084Cys were detected in patients with APOE ɛ3/ɛ 4. As for.p.Glu270Lys variant, APOE ɛ3/ɛ4 was found in 6 patients carrying this variant in heterozygous state.
In silico 3D protein structural analysis
We examined the impact of the rare novel identified variants p.Arg1084Cys and p.Pro1213Ser on structure and function of SORL1. These two variants were selected for in silico 3D protein structural analysis due to their occurrence in EO-FAD patients. Additional considerations were taken for choosing p.Arg1084Cys and p.Pro1213Ser variants: damaging prediction analysis, conservation of native amino acids in various vertebrate species (Supplementary Figure 1A, B) and positions respectively within the first and fourth complementary repeats of low density lipoprotein (LDL) receptor class A (LDLR-A) conserved domain (Fig. 1). The SORL1 protein region (1079–1235 amino acids) containing the two novel variants was used for the structural modeling. The wild type SORL1 (Fig. 2E) was shown in a coiled form with the presence of two helices and two beta sheets. The two SORL1 variants caused evident reduction in protein folding and changes in secondary structure components (alpha helix and beta sheet) (Fig. 2F, G). For the p.Pro1213Ser substitution, we noticed doubling of beta sheets and presence of three shortened helices (Fig. 2F). In the presence of p.Arg1084Cys variant, the protein structure showed complete loss of both beta sheets and appearance of two shortened alpha helices (Fig. 2G). The p.Arg1084Cys variant was localized between these shortened helices (Fig. 2G).
DISCUSSION
In the present study, we focused on investigating the genetic basis of AD in 117 familial and sporadic Saudi patients. We carried out sequencing of APP, PSEN1, PSEN2, and SORL1 in addition to genotyping of APOE alleles. Our results showed the presence of 5 novel rare missense variants and 3 previously reported ones. We also found 6 missense and 15 synonymous polymorphisms.
Identifying AD causative mutations is essential for disease management and carrier testing. Frequency of mutations in AD known genes was shown to vary from approximately 17 to 77% depending on diagnostic pattern, studied populations and cohort sizes [5, 50–53]. Variants in APP, PSEN1, and PSEN2 were underrepresented in our cohort of patients, (5/117) carried a missense variant in one of these genes (Table 2 and Supplementary Table 3).
The majority of the reported APP pathogenic variants are clustered in exons 16 and 17 (http://www.alzforum.org/mutations) that codes for the Aβ peptide [12]. Several APP variants were observed outside Aβ regions in certain sporadic cases and some of them were suggested to be rare benign polymorphisms or described to have unclear pathogenicity [41, 55]. Our APP p.Glu380Lys variant is located in exon 9 and further functional studies are needed to confirm any possible pathogenic role for this APP variant. The previously reported PSEN1, p.Tyr195Cys is situated in transmembrane 4 domain of PSEN1 where conformational changes were found in association with altered Aβ42 production [44].
Six of our identified SORL1 variants were located in the VPS10 domain. VPS10 binds to the synthesized Aβ and orient it toward lysosomes degradation [56, 57]; this binding was found to be disturbed by SORL1 p.Gly511Arg variant associated with FAD [57]. The rare variant p.Val297Met identified in this study was found to be situated close to the previously reported variant p.Glu270Lys and in the same VPS10 domain as the common variant p.Ala528Thr (Fig. 1). The p.Glu270Lys and p.Ala528Thr variants were found in our patients (Table 2 and Supplementary Table 3). Both variants were previously shown to generate more Aβ42 and to increase AβPP protein levels on the cell surface indicating failure in orientation of the full length of AβPP towards the retromer-recycling endosomes pathway [26]. LDLRA domain of SORL1 was shown to bind AβPP leading to its protection from being processed into Aβ [58, 59]. The two novel SORL1 variants (p.Arg1084Cys and p.Asp1100Asn) identified in this study are located in this domain (Fig. 1).
Prediction of SORL1 protein modeling by in silico analysis suggested that both p.Arg1084Cys and p.Pro1213Ser variants affected protein folding (Fig. 2F, G) which might disrupt its activity as it is generally known that the proper biological function of proteins is correlated to the correct folding of their constituent amino acids (reviewed in [60]). Further functional studies are needed to explore the possible role of these novel SORL1 variants in the pathogenesis of AD.
While PSEN1, PSEN2, and APP are associated mainly with FAD form of the disease, APOE is associated with the sporadic form. A 3–5 folds increase in AD risk was estimated for APOE ɛ4 heterozygous carriers and a 10–15-fold for homozygous ones [17]. The ɛ4 allele frequency is about 13.7% in general populations and 36.7% in AD patients [61]. The ɛ4 allele frequency needs to be investigated in normal controls from the Saudi population and to be determined in larger cohort of Saudi AD patients since it was found at 21.3% in our AD studied cases. A recent study proposed the involvement of a rare SORL1 genetic variant in increasing the penetrance of AD in a family with multi-generation of homozygous APOE ɛ4 carriers [62]. The novel heterozygous SORL1 rare variants (p.Arg1084Cys, p.Pro1213Ser, and p.Val297Met) were present in APOE ɛ4 carriers suggesting a possible relation between these SORL1 variants and APOE ɛ4 allele in the pathology of AD in these patients.
Our focused study of AD yielded 8 rare missense variants in PSEN1, PSEN2, APP, and SORL1. In silico 3D structural assessment and pathogenicity prediction tools suggested significant evidence for pathogenicity. However, functional evaluation is needed for clarifying the possible role(s) of these variants in the development of AD. Our results showed a low frequency (5/117) of patients carrying possible pathogenic variants in AD causative genes (PSEN1, PSEN2, and APP). The lack of mutations in the remaining diagnosed familial and sporadic cases, suggests the involvement of other genes or mechanisms for the development of this heterogeneous disorder. Several GWAS studies have identified risk genes (ABCA7, BIN1, CD33, CD2AP, CLU, CR1, EPHA1, MS4A6A/MS4A4E, and PICALM) for AD [63–66]. Furthermore, rare variants in TREM2 and PLD3 are associated with AD [67–70]. In this regard, these genes can be taken in consideration in future Saudi AD genetic studies.
Footnotes
ACKNOWLEDGMENTS
This work was supported by King Abdulaziz City for Science and Technology (KACST). KACST’s grant number is 13-MED684-20. The authors would like to thank Dr. Dorota Monies and the sequencing core facility members for their help in DNA Sequencing. We thank Dr. Dana Bakheet and biobanking, DNA extraction and oligosynthesis unit for their help in DNA extraction. We thank the Saudi Human Genome Program (SHGP) and KACST for providing us with MAF's from the SHGP database. We thank Drs Walid Nouh and Hanan Katlan from the laboratory of Al Habib Medical Center for assisting in blood collection from Alzheimer’s patients.