
Abstract
Administration of the recombinant analog of the pancreatic amyloid amylin, Pramlintide, has shown therapeutic benefits in aging and Alzheimer’s disease (AD) models, both on cognition and amyloid-β (Aβ) pathology. However, the neuroprotective mechanisms underlying the benefits of Pramlintide remain unclear. Given the early and critical role of oxidative stress in AD pathogenesis and the known reactive oxygen species (ROS) modulating function of amyloids, we sought to determine whether Pramlintide’s neuroprotective effects involve regulation of oxidative stress mechanisms. To address this, we treated APP/PS1 transgenic mice with Pramlintide for 3 months, starting at 5.5 months prior to widespread AD pathology onset, and measured cognition (Morris Water Maze), AD pathology, and oxidative stress-related markers and enzymes in vivo. In vitro, we determined the ability of Pramlintide to modulate H2O2-induced oxidative stress levels. Our data show that Pramlintide improved cognitive function, altered amyloid-processing enzymes, reduced plaque burden in the hippocampus, and regulated endogenous antioxidant enzymes (MnSOD and GPx1) and the stress marker HO-1 in a location specific manner. In vitro, Pramlintide treatment in neuronal models reduced H2O2-induced endogenous ROS production and lipid peroxidation in a dose-dependent manner. Together, these results indicate that Pramlintide’s benefits on cognitive function and pathology may involve antioxidant-like properties of this compound.
INTRODUCTION
Lifestyle factors like obesity and ramifications stemming from obesity, such as metabolic syndrome and the consequential development of type 2 diabetes mellitus (T2DM), are associated with the development of sporadic Alzheimer’s disease (AD) [1, 2]. T2DM is associated with cognitive decline when compared with age-matched controls [4–7] and is strongly correlated with the development of both mild cognitive impairment (MCI) and AD [2, 3].
AD and T2DM share multiple common characteristics, such as metabolic dysfunction [8], mitochondrial dysfunction [9], inflammation [10, 11], and, relevant to this work, increased oxidative stress production and impaired oxidative stress defenses [10, 12–14]. Similarly, T2DM and AD also each contain a pathological component related to aberrant aggregation of amyloid proteins, namely, amyloid-β (Aβ) in AD and amylin in T2DM [15].
Amylin, or IAPP, is a polypeptide hormone composed of 37 amino acids, synthesized by the β-cells of the pancreas, is co-released with insulin [16] and crosses the blood-brain barrier [17]. Amylin also plays a role in glucose homeostasis by preventing the release of glucagon from the liver, delaying gastric emptying, and signaling satiety [18]. Similar to Aβ in AD, under normal conditions, amylin exists as a soluble monomer; however it undergoes a conformational change in T2DM which leads to insolubility and aberrant aggregation [19]. This aggregation of amylin eventually leads to the formation of amyloid deposits, which have been proposed, to be the main source of toxicity to β-cells, leading to their death and consequent decrease of amylin and insulin synthesis later in T2DM [20].
Importantly, a growing number of studies have highlighted a direct relationship between T2DM and AD through the interaction and/or function of these two amyloids, both at a pathological and neuroprotective level. Some work suggests that both amyloids may exert their toxicity to neurons through a shared receptor [21–25] and/or their aggregation serves as a seeding mechanism for the other disease [26, 27]. Conversely, clinical data show a positive correlation between plasma and cerebrospinal fluid amylin levels and cognitive function in T2DM [28], AD, and MCI patients [29, 30]; pre-clinical studies also demonstrated the neuroprotective benefits of amylin and pramlintide (PRAM), a non-aggregating synthetic analogue derived from rat amylin [31]. Evidence suggests that both amylin and PRAM modulate cognition, neuroplasticity, inflammation [29, 32], and Aβ deposition [33].
Importantly, recent work has shown that while both human amylin and Aβ1–42 dysregulate identical proteins that lead to increased reactive oxygen species (ROS) levels [34]. Rat amylin, which does not aggregate [22, 35], does not share the same cytotoxic properties as its human counterpart or Aβ1–42 [34]. Thus, these data suggest that loss of native function through amylin aggregation could play a role in both AD and T2DM pathogenesis. The above hypothesis is supported by the known antioxidant function of both amylin [36] and Aβ [37, 38] in their native conformational states and by the fact that there is increased production of Aβ in AD [39–42]. Additionally, treatment with non-aggregating forms of amylin such as PRAM in T2DM patients [43, 44] and the accelerated aging mouse model (SAMP8) [29], lead to reduced ROS levels.
Thus, given the native role of amyloids in the regulation of oxidative mechanisms and neuronal function as well as the toxic role of oxidative stress in T2DM and AD, we addressed the ability of PRAM to improved cognitive function and reduced pathology in the APP/PS1 mouse model. Furthermore, we were particularly interested in determining whether the benefits seen are associated with the anti-oxidative properties of PRAM.
MATERIALS AND METHODS
In vivo experiments
Animals
Male and female APP/PS1 mice (B6.Cg-Tg (APPswe, PSEN1dE9)85Dbo/Mmjax) (n = 30) and wild-type (WT) littermates (n = 25) were bred in the animal facility of Kent State University for use in this study. Animals were socially caged (2–4 mice per cage) and maintained on a 12/12-h light-dark cycle (lights on at 08 : 00 h, off at 20 : 00 h) in a temperature-controlled room (72°F±2). Food and water were available ad libitum. The Institutional Animal Care and Use Committee of Kent State University approved all protocols within this study.
Drug treatments
All mice were aged to 5.5 months prior to the beginning of treatment. APP/PS1 mice were randomly assigned to saline (N = 15) or PRAM treatment (N = 15), while all WT mice received saline (N = 25). Drugs were delivered through Alzet mini-osmotic pumps (Durect, CA) delivering 0.15 μL/h steadily for 6 weeks (model 2006) and were replaced every 6 weeks over the course of 18 weeks. Pramlintide acetate (Anaspec, CA) was made at a concentration of 421.9 μM in sterile, MilliQ H2O and delivered at the desired dose of 6 μg/day. This dose was shown to be maximally therapeutic in humans and has also been used in rodent models to improve cognitive function and reduce pathology [29]. Saline pumps were made with a 0.89% NaCl saline solution was made in ultrapure MilliQ H2O and sterile-filtered. Prior to filling pumps, all solutions were brought to room temperature. All pumps were filled in a sterile hood using sterile techniques and manufacturer’s instructions.
Surgical pump implantations
Mice were transferred onto a heating pad and fitted with a nosecone to continue anesthesia with 3% isoflurane at 1 L/min. A 1 cm long incision was made horizontally on the dorsal neck region using dissecting scissors, the pumps were inserted and subcutaneously and the incision was closed using clip wound closures and removed 2 weeks post-surgery (Stoelting, IL). Pumps were changed every 40 days using this same procedure (Fig. 1).
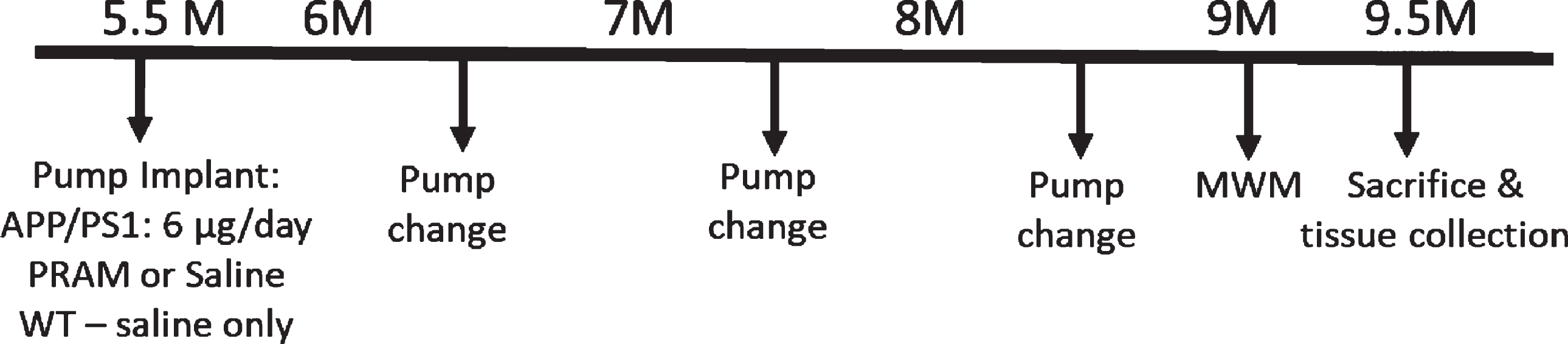
Experimental timeline for in vivo study. Male (n = 15) and female (n = 15) APP/PS1 and their WT male (n = 13) and female (n = 12) littermates were aged until 5.5 months until a 6-week Alzet mini-osmotic pump was surgical implanted subcutaneously and replaced every 40 days 2 times. A week before sacrifice, mice were subjected to the Morris Water Maze swim task.
Behavioral testing
Morris Water Maze (MWM) was used to test spatial reference memory using an adapted protocol [45]. Briefly, mice were placed into an MWM pool (Med Associates, VT) with a tank diameter of 100 cm and were tracked using behavior software (Noldus Ethovision XT 10). The water colored with white tempera non-toxic white paint to conceal the platform (10 cm diameter) located in the northwest (NW) corner, 5 mm under the water surface. Temperature was maintained at during testing 22°C. Mice were placed into the pool surrounded by distal visual cues, at four different locations to reduce location biases. Each mouse performed four trials per day. Mice that did not reach the platform in 60 s were gently guided toward it and all mice were made to remain on the platform for 15 s prior to their rescue. Mice were trained for 6 days followed by a probe trial as the last trial on day 6. For the probe trial, the platform was removed and mice were given 60 s to explore the maze. The amount of time spent in the target quadrant that had contained the platform (NW) was calculated. A visual platform test was also performed on day 7 to identify potentially blind mice for exclusion in the behavioral task. Animals that floated in four consecutive trials during two days of training or were not able to swim for the duration of the trial were also excluded from the study.
Tissue preparation
Animals were sacrificed by cervical dislocation and temporal cortex and hippocampi dissected, homogenized in 1X cell lysis buffer (Cell Signaling Technologies, Danver, MA) supplemented with 1mM PMSF, and stored at -80°C for future use. Aβ plaque load was visualized in coronal sections from APP/PS1 mice. Briefly, mice were transcardially perfused with 4% paraformaldehyde made in 1X PBS before brains were removed. Perfused brains were subsequently for 4 h in 4% paraformaldehyde and then transferred to 30% sucrose made in 1X PBS until brains sunk for cryoprotection. PFA fixed brains were then frozen in OTC and immersed in 2-Methylbutane (Fisher Scientific, MA) and sliced at 40 μM using a cryostat (Leica CM 1950). Sections were stored at –20°C in polyethylene glycol until stained with thioflavin S.
Aβ plaque staining and quantification
Fixed brain slices were mounted on a slide and immersed in 1% Thioflavin S solution diluted in MilliQ water for 5 min. Slides were then moved into 70% ethanol for 5 min. Slides were washed in PBS, then allowed to completely dry and cover slipped on VECTRASHIELD mounting media. Brains were then imaged as 10x z-stacks with 2 μM thickness using the Olympus FV500 confocal microscope. For each slice, the entire hippocampus and 3 temporal cortical fields were imaged. Images were stitched together to create a single field and plaques were quantified using Fiji (Image J) to obtain particle count, area, average size, and percent area.
Soluble Aβ measurements
Aβ (1–40, 1–42, and total) from frozen hippocampal and cortical tissues measured by sandwich ELISA as previously described [46, 47]. Briefly, tissues were extracted first in 1X cold PBS, then 2% sodium dodecyl sulfate (SDS) and last, 70% formic acid (FA). Antibodies Ab42.5 (human sequence Aβ1–16 for Aβ1–40 capture) and 2.1.3 (end specific for Aβ1–42) were used in each brain fraction (PBS, SDS, and FA) all containing 1:200 Protease Inhibitor Cocktail Set III (EMD Millipore).
Western blotting
Tris-acrylamide gels were made at 4% for stacking gel and 10% for resolving gel. 15 μg of protein was separated using an SDS-PAGE Bio-Rad mini protein system (Bio-Rad, CA). Blots were blocked in 10% nonfat milk for 1 h at room temperature. Primary antibodies [HO-1 (Rabbit, 1 : 2000, abcam), Glutathione Peroxidase (Rabbit, 1 : 1000, abcam), BACE-1 (Rabbit, 1 : 1000, abcam), ADAM10 (Rabbit, 1 : 1000, abcam), and GAPDH (mouse, 1:25,000, Sigma-Aldrich)] were incubated overnight at 4°C in 1% milk made in TBST. Secondary antibodies (1 : 1000) Ms IgG HRP-conjugated or Rb IgG HRP-conjugated (Cell Signaling Technologies, Danvers, MA) were incubated for 1 h at room temperature in 1% milk made in TBST. Proteins were visualized using ECL (Millipore) and a chemiluminescent developer (Syngene Pxi 6 Touch). Images were quantified using Image J software.
In vitro experiments
SH-SY5Y (ATCC® CRL-2266™, Manassas, VA), where purchased and differentiated with retinoic acid following previous protocols [48, 49]. Cells used where all treated within 4–7 passages. Cells were plated accordingly for in vitro assays per manufacturer’s instructions. Doses were based on preliminary study data, studies showing antioxidant function at these doses [36, 51] and/or and kept below 10 μM given recent data suggesting the ability of amylin to activate downstream TRPV4 receptors and induce high levels of intracellular Ca+2 and cytotoxicity [50, 51]. All assays were conducted in triplicate and replicated at least twice in two independent experiments.
DCF assay
The dichlorofluorescein (DCF) assay was performed using differentiated SH-SY5Y as described in [52]. Following standard retinoic acid differentiation media was changed to serum/phenol-free Optimem (Life Technologies) for 16–18 h. Treatments included: no treatment, DCF alone, DCF + FeSO4, DCF + FeSO4 + H2O2, and PRAM + DCF + FeSO4 + H2O2. Four PRAM doses (1 nM, 10 nM, 100 nM, 250 nM) were applied 24 h prior to exposure of an oxidative insult (5 mM H2O2) as described in [36] showing DCF changes by amylin in β-cells of the pancreas. On the experimental day, media was changed and PRAM was re-added to each well for 1 h to ensure stable levels of PRAM prior to the oxidative insult. Cells were rinsed twice and 20 μM H2DCF-DA (Molecular Probes) in Optimem was added to cells for 1 h at 37°C. Cells were washed twice again and H2O2 (Acros Organics, NJ) at final concentration of 5 mM was added to all wells in addition to 8 μM FeSO4. This solution was allowed to incubate with cells for 30 min. The florescence was read at 485 nm excitation wavelength and 530 nm emission wavelength using a microplate reader (Molecular Devices) at 37°C. Levels of florescence were expressed relative to untreated controls.
TBARS assay
SH-SY5Y cells were plated in 96 well plates (5 ×104) and differentiated following standard retinoic acid differentiation. PRAM doses (500 nM, 1000 nM, 5000 nM) and incubation times were chosen based and previous work identifying similar antioxidant effects of amylin in pancreatic cell cultures [36]. After 1 h of PRAM incubation, 5 mM H2O2 was added into the wells and allowed to incubate for 30 min. Supernatant was then collected for TBARS assay, which was carried out using manufacturer’s instructions (R&D Systems, MN). MDA levels were measured colorimetrically through its reaction with thiobarabituric acid (TBA) at an absorbance of 535 nm using a microplate reader (Molecular Devices) for all treatment groups. Data is portrayed as MDA % change (TBARS % change).
Statistical analysis
All in vivo testing and analyses were carried out by an investigator blind to treatment, cell culture data was analyzed blind to treatment. Experiments were powered based on previous published data in the field and experiments in the laboratory at was set at 80%. Outliers were detected using the Grubb’s test and excluded from the data analysis if significant. Normality and homogeneity of variance were verified to ensure normality prior to parametric statistical analyses. Statistical significance was determined using a One-Way ANOVA with Tukey’s post hoc analysis for any experiments with more than two groups. Independent samples T-test were used to statistically analyze differences between treated and untreated transgenic mice. A p < 0.05 was deemed as statistically significant. Data is expressed as mean and standard error mean (mean±SEM). Sex differences were determined for variables where power was sufficient. There were no sex differences for cognitive function. Due to increased death of female APP/PS1 (of all groups) the female n number was too small to identify relevant sex differences for biochemical measurements. Based on these analyses all statistical analyses were carried out grouping sexes.
RESULTS
In vivo/ex vivo studies
Pramlatide treatment rescues spatial learning and memory
MWM was used to determine differences in spatial hippocampal memory between saline treated WT, saline treated APP/PS1 (Tg + SAL), and PRAM treated APP/PS1 (Tg + PRAM) mice. A One-Way ANOVA analysis identified a significant difference between subjects group (F1,2 = 7.806; p = 0.002) and a within subjects significant difference across days (F1,5 = 18.574; p = 0.0001). However, there were no statistical differences in the day*group interaction (F1,5 = 1.523; p = 0.136). These data indicate that while there was an overall group difference in performance across groups for the task, the rate of learning across days was not different between groups. Tukey’s post-hoc multiple comparisons analysis demonstrated that Tg + SAL mice performed significantly worse compared to WT animals (p = 0.002) and Tg + PRAM animals (p = 0.026). There were no statistically significant differences between WT and Tg + PRAM treated animals (p = 0.964), suggesting that drug treatment rescued performance in the MWM to WT levels (Fig. 2A).

Pramlintide treatment improves AD-related hippocampal dysfunction. A) MWM training in wild-type (WT + Sal, n = 21 [11 females, 10 males]), saline treated APP/PS1 (Tg + Sal, n = 14 [6 females, 8 males], and pramlintide-treated APP/PS1 mice (Tg + PRAM, n = 12 [4 females, 7 males]). B) Probe trial, % time spent in target quadrant for WT+Sal, Tg+Sal, and Tg+PRAM groups. Results depicted as mean±SEM. Significance: *p<0.05 relative to WT; #p<0.05 relative to Tg + Sal.
Within subjects comparisons across days of training revealed significant group differences for Days 5 (F1,2 = 9.461; p = 0.001) and 6 (F1,2 = 6.047; p = 0.006) of training. Tukey’s post-hoc analyses for Day 5 of training revealed statistically significant differences between Tg + SAL and Tg+PRAM (p = 0.015) and Tg + SAL versus WT control animals (p = 0.001), with Tg + SAL mice showing cognitive deficits. There was no statistical difference between Tg + PRAM and WT (p = 0.935). Tukey’s post-hoc analyses for Day 6 of training revealed similar statistically significant difference between Tg + SAL and Tg+PRAM (p = 0.035), as well as between Tg + SAL and WT (p = 0.005), again indicating a cognitive deficit. Tg + PRAM and WT mice performed similarly (p = 0.998) (Fig. 2A).
In order to determine differences in retention and spatial strategy, a One-Way ANOVA was used to identify differences in the probe trial. An overall significant difference between groups was found (F1,2 = 7.376; p = 0.002). Tukey’s post-hoc analysis revealed a statistically significant difference between WT and Tg + SAL (p = 0.002), and a non-significant trend between Tg + PRAM and Tg + SAL (p = 0.08). There were no statistically significant differences between Tg + PRAM and WT control groups (p = 0.695), as shown in Fig. 2B. This data indicate that PRAM was partially able to rescue spatial memory retention.
Pramlatide treatment reduces soluble Aβ and plaques in the hippocampus
Plaque load. Independent Samples T-test comparisons between PRAM-treated and non-treated APP/PS1 mice revealed that, in the hippocampus, Tg + PRAM mice had a significantly decreased percent area plaque coverage (t1,9 = –2.818, p = 0.022) and plaque number (t1,9 = –2.872, p = 0.024) compared to Tg + SAL mice (Fig. 3A, B). In the cortex, there were no significant differences in plaque count or percent area with plaques between treatments. There were also no significant differences between groups for average plaque size either in the cortex or hippocampus.

Pramlintide reduces soluble and insoluble Aβ levels in the hippocampi of APP/PS1 mice. A) Representative images of Thioflavin S in the hippocampus of saline treated APP/PS1 mice (Tg + Sal, n = 6 [2 females, 4 males]) and PRAM treated APP/PS1 mice (Tg+PRAM, n = 5 [1 females, 4 males]). B) Quantification of % area stained and number of plaques (plaque count). C) Sandwich ELISA for soluble (PBS), membrane bound (SDS), and insoluble [formic acid (FA)] levels of Aβ1–40 and Aβ1–42 in the hippocampus of saline treated (Tg + Sal, n = 4 (all males)) and PRAM treated (Tg + PRAM, n = 4 (all males)) APP/PS1 mice. Results depicted as mean±SEM. Significance: *p<0.05 relative to WT.
Soluble and insoluble Aβ fractions. Independent Samples T-test was also used to determine differences in soluble (PBS), membrane bound (SDS), and insoluble (FA) fractions of Aβ1–40 and Aβ1–42 in the in the hippocampus and cortex of PRAM-treated and saline-treated animals (Fig. 3C). In the hippocampus, Tg + PRAM mice had significantly lower levels of insoluble (FA) Aβ1–40 (t1,6 = –2.051, p = 0.046) and Aβ1–42 (t1,6 = –2.631, p = 0.039) compared to Tg + SAL animals. A trend toward lower soluble (PBS) Aβ1–42 in the Tg + PRAM compared to Tg + SAL treated mice (t1,6 =–2.061, p = 0.085) was also identified (Fig. 3C). In the cortex (data not shown), there were no significant differences between groups other than reduced levels of membrane bound (SDS) Aβ1–40 (t1,6 = 2.9220, p = 0.027) in the Tg + SAL group when compared to the Tg + PRAM group. A non-significant trend toward lower insoluble (FA) Aβ1–42 in the PRAM-treated compared to saline-treated mice was also observed (t1,6 = –2.064, p = 0.083).
Pramlatide treatment alters APP processing and oxidative stress markers
Independent Samples T-test analysis demonstrated a statistically significant increase in α-secretase (ADAM 10) expression in the cortex of Tg + PRAM versus Tg + SAL mice (t1,9 = 4.277, p = 0.022) and hippocampus (t1,9 = 5.032, p = 0.001). Interestingly, β-secretase expression (BACE1) was significantly increased by PRAM treatment in the hippocampus (t1,9 = 3.413, p = 0.008) but not in the cortex of APP/PS1 mice (p = 0.759) (Fig. 4).
Western blots for oxidative-stress related markers such as heme-oxygenase-1 (HO-1), glutathione peroxidase (GPx), and manganese superoxide dismutase (MnSOD) expression in the cortex and hippocampus of Tg + SAL and Tg + PRAM mice revealed region-specific differences. Independent groups T-test analyses identified a significant decrease in HO-1 expression in the cortex of Tg + PRAM treated compared to TG + SAL mice (t1,9 = –3.407; p < 0.01) while there was a significant increase in HO-1 expression in the hippocampus (t1,9 = 2.310; p = 0.046). GPx expression was significantly increased in the hippocampi of Tg + PRAM animals compared to controls (t1,9 =3.658; p = 0.008) but not in the cortex, despite a strong trend (p = 0.07). Lastly, significant increases in MnSOD expression in the Tg + PRAM mice were observed when compared to Tg + SAL in the hippocampus (T1,9 =2.93; p = 0.017) but not in the cortex (p = 0.58) (Fig. 4).
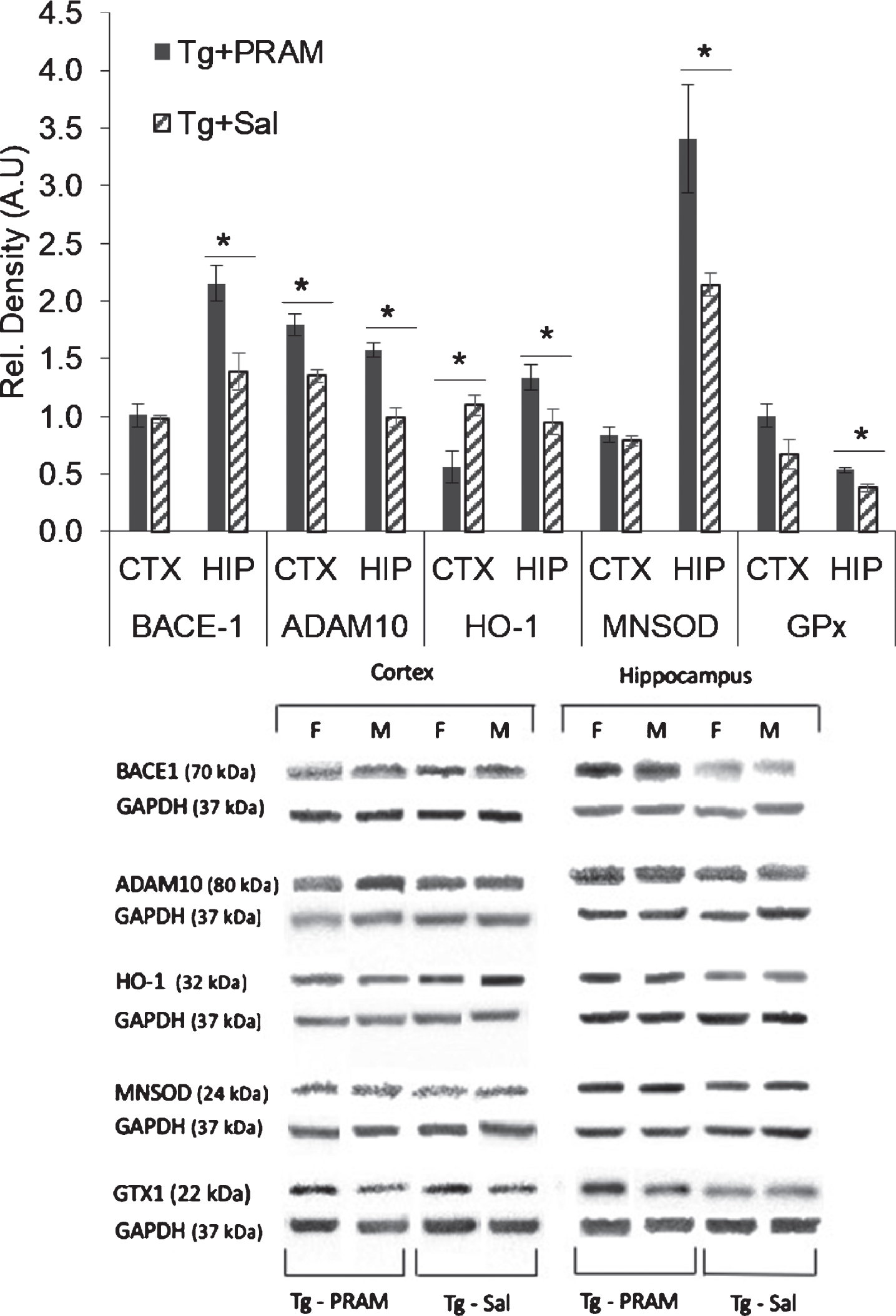
Pramlintide treatment regulates APP processing and oxidative stress in a region-specific manner in APP/PS1 mice. Western blot quantification of BACE1 (β-secretase) and ADAM10 (α-secretase) expression in cortex and hippocampus in saline treated APP/PS1 mice (Tg + Sal, n = 6 [3 females, 3 males]) and PRAM treated APP/PS1 mice (Tg + PRAM, n = 5 [2 females, 3 males]). Western blot quantification of heme oxygenase 1 (HO-1) and MNSOD expression in cortex and hippocampus (Hip) in saline treated APP/PS1 mice (Tg + Sal, n = 6 [3 females, 3 males]) and PRAM treated APP/PS1 mice (Tg + PRAM, n = 5 [2 females, 3 males]). Results depicted as mean±SEM *p<0.05 relative to WT; #p<0.05 relative to Tg + Sal.
In vitro studies
Pram decreases H2O2-induced intracellular ROS production and lipid peroxidation
The ability of PRAM to regulate H2O2-induced oxidative stress parameters was evaluated through DCF and TBARS assays in differentiated SH-5Y5Y cells. DCF assays were statistically analyzed via One-way ANOVA. Analyses revealed a statistically significant difference between groups in the DCF assay (F1,5 = 7.638; p = 0.001). Post-hoc analysis identified a significant increase in intracellular ROS production by H2O2 administration compared to untreated control cells (p < 0.001) and control and H2O2 + 1 nM (p = 0.023) and H2O2 + 10 nM PRAM doses (p = 0.034). Importantly, pre-incubation with PRAM at 100 nM (p < 0.0001) and 250 nM (p < 0.0001) was able to significantly reduce DCF fluorescence compared cells treated with H2O2 alone (Fig. 5A).
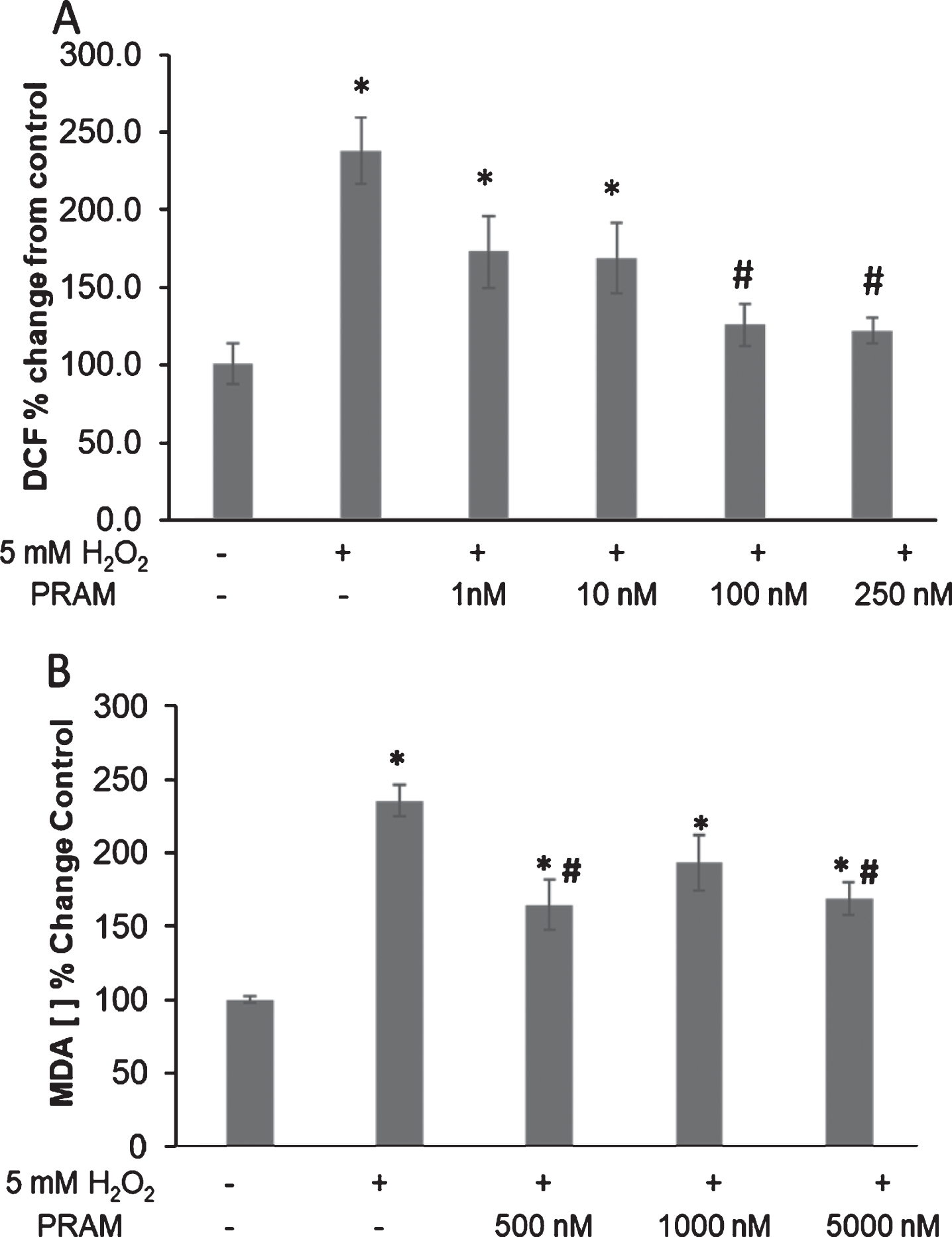
Pramlintide reduces H2O2-mediated endogenous oxidative stress production and damage in differentiated SHSY5Y. A) Change in relative DCF fluorescence. Doses: H2O2 alone and 1, 10, 100, & 250 nM PRAM + H2O2. Each treatment group was run in n = 6 and the experiment was repeated 3 times. B) Change in MDA concentration. Doses: H2O2 alone and 500, 1000, & 5000 nM PRAM + H2O2 . Results depicted as percent change from untreated control as mean±SEM. *p<0.05, relative to untreated control; #p<0.05, relative to H2O2 alone. The treatment groups were run in triplicate and repeated 3 times.
Statistical analysis of TBARS assays in differentiated SHSY5Y cells (Fig. 5B) revealed a significant difference between groups (F1,4 = 13.722; p < 0.001) via one-way ANOVA. Tukey’s post-hoc analysis indicated a significant increase in lipid peroxidation between all H2O2 treated groups (with and without PRAM treatment) compared to untreated controls (p < 0.01–0.001). Significant decreases in MDA concentration were seen in the 500 nM PRAM + H2O2 (p = 0.007) and 5000nM PRAM + H2O2 (p = 0.018) treatments when compared to H2O2 alone. There were no significant differences across PRAM dosages.
DISCUSSION
Here we show that continuous and chronic administration of PRAM ameliorates cognitive deficits in MWM test. These findings are in accordance with an independent study showing the ability of intraperitoneal (i.p.) and/or ICV delivered amylin or PRAM to improve function in other AD mouse models [33]. Importantly, our work was carried out using a dose within the therapeutic range administered to humans, therefore highlighting its translational therapeutic applicability.
Notably, treatment with PRAM reduced Aβ plaque burden in APP/PS1 mice. However, unlike previous work [33], this study revealed a reduction in Aβ plaque burden in the hippocampus but not the cortex. These changes were also reflected in the soluble fraction measurements, where hippocampal levels of insoluble (FA) Aβ1–40 and/or Aβ1–42 were more effectively reduced by our treatment than in the cortex. Also of note, a non-significant trend was observed for the soluble (PBS) Aβ1–42 fraction in the hippocampus. Together, these data suggest that PRAM may increase the clearance of Aβ in the brain, thus reducing plaque formation, an assertion supported by previous reports demonstrating a dose-dependent increase in Aβ1–42 in efflux into the periphery [33].
Differences between our study and others [33] may stem from treatment protocol differences (i.p. versus continuous s.c. infusion), length of treatment, peptide used (amylin versus PRAM), transgene, or staining methods. However, a potential contributor to these differences may also be the timing of treatment initiation in relation to the temporal pattern of plaque deposition and regional expression across the different mouse models. In our model, plaque deposition begins 4.5 months of age in the cortex, and progresses to the hippocampus at 6.5 months of age [53]. As our treatment began when the mouse was aged at 5.5 months, when cortical plaque deposition has been established, this may indicate that PRAM can prevent plaque pathology but not modify existing plaques. This hypothesis is supported in the work using Tg2576 models showing widespread reduction in pathology [33] at relevant ages for that mouse. In Tg2576 mice, initial plaque deposition occurs at 10 months with substantial plaque burden at 12 months [54]. Therefore, the initiation of treatment at 9 months of age would occur prior to initial plaque deposition and yield more widespread reductions in pathology. This is further validated by our Aβ fraction measurements demonstrating more efficient reductions of Aβ1–42 in the hippocampus versus the cortex, where only a trend was observed for FA soluble (fibrilized) Aβ1–42.
To address potential effects of PRAM on APP processing, we determined expression levels of amyloidogenic β-secretase (BACE1) and non-amyloidogenic α-secretase (ADAM-10) enzymes in cortex and hippocampi of our APP/PS1 animals. Interestingly, PRAM increased both BACE1 and ADAM-10 in the hippocampus, where both soluble Aβ fractions and plaque load were significantly reduced. Conversely, ADAM-10 but not BACE1 was significantly increased in the cortex, yet we saw no significant difference in plaque load or soluble/insoluble Aβ levels. Previous work has demonstrated that amylin but not PRAM has inhibitory BACE1 properties; however, to our knowledge, there are no reports of upregulation of these enzymes by either PRAM or amylin treatment. While the significance of these data is unclear, it is possible that PRAM drives a generalized enzymatic response, perhaps, associated with efflux of soluble Aβ species into the periphery regions but only in areas that are actively undergoing Aβ deposition. This aspect has been previously associated with administration of PRAM and amylin [33]. Certainly, our data do not provide a clear relationship between APP cleavage enzyme expression and Aβ deposition in relation to PRAM treatment. However, our data do suggest that PRAM treatment may have a more robust effect on regulating α-secretase expression than β-secretase, given that both cortex and hippocampus shows increases in ADAM-10 expression. These data potentially highlight a novel role of PRAM through increasing α-secretase activity and cleavage of APP toward the non-amyloidogenic pathway. Time course studies starting therapy at different ages will help clarify and further validate the precise nature of interactions between Aβ processing, Aβ deposition, and the role of PRAM and timing of treatment onset in these processes.
Oxidative stress has been shown to be a major component AD [55–58] and may even precede AD development. [41, 56]. Therefore, an imbalance between increased ROS and decreased antioxidant defenses may be a fundamental event in AD development [59–61]. Critically, it has been established that insulin resistance and diabetes lead to increases in oxidative stress directly [62–65] or indirectly through increasing inflammation [66, 67]. Incidentally, PRAM administration in T2DM patients reduces the levels of serum markers of oxidative stress [43, 44] and can also form a complex with Cu (II) [68] that can mimics antioxidant enzymes to reduce ROS [69, 70]. Importantly, previous work has shown the ability of PRAM to reduce lipid peroxidation damage and inflammatory markers and stress related responses HO-1 in a mouse model of accelerated aging [29].
HO-1 is an inducible enzyme that degrades the pro-oxidant heme, leading to the formation of the powerful antioxidants biliverdin and bilirubin [71, 72]. It is upregulated by numerous factors, including oxidative stress [73, 74], and is upregulated in AD [75, 76]. On the other hand, HO-1 has also been shown to protect against Aβ toxicity in the hippocampus [75] and Aβ-related mitochondrial dysfunction [77]. Our data shows that while expression of HO-1 was reduced by PRAM treatment in the cortex, the expression of HO-1 was significantly increased by PRAM in the hippocampus. These seemingly paradoxical effects of PRAM treatment in different brain regions remains puzzling, however, a plausible explanation may lie in the differential make-up of the cortex versus the hippocampus, the timing of and plateau of pathology, and the ability of PRAM to regulate inflammatory states [29, 78]. As discussed above, in our AD model, the onset of treatment would coincide with initial plaque deposition in the hippocampus but not the cortex. Therefore, PRAM ability to reduce HO-1 in areas were plaque formation has plateaued could be reflective of its general anti-inflammatory function while the activation of HO-1 in the hippocampus could be reflective of a more acute inflammatory response pattern that can foster clearance of soluble Aβ before it has a chance to fibrillize [79]. Future studies will need to more closely identify the role of amylin regulation inflammatory aspects within an AD milieu to fully clarify these data.
Our work also shows that the antioxidant enzymes MnSOD and GPx were significantly increased in hippocampus, but not cortex of PRAM treated mice when compared to saline treated APP/PS1 mice. As discussed previously, patients with MCI and AD exhibit decreased total antioxidant capability [60] and increased production of O2 •– [80] which stimulates the amyloidogenic pathway [81]. With the substantial upregulation of MnSOD to combat this free radical, this may be a potential mechanism by which PRAM exerts its benefits both at a functional and pathology level. Furthermore, the significant increases of GPx in hippocampus by PRAM treatment further bolsters the antioxidant defenses against ROS.
To delve deeper into identifying whether antioxidant regulation may underlie the neuroprotective benefits of PRAM, we carried out a variety of in vitro assays, which showed that PRAM treatment reduced intracellular ROS production (DCF assay) and lipid peroxidation (TBARS) in the response of an oxidative insult (H2O2). Effectiveness of PRAM on lipid peroxidation was seen at higher doses than its ability to quench intracellular ROS production. This is likely due to the fact that for the TBARS assay incubation was carried out for 1 h versus 24 h for the DCF assay. While future studies should replicate the time-course and doses used in the DCF assay for lipid peroxidation measurements, these findings support previous in vivo data demonstrating the ability of PRAM to reduce 4-HNE levels [29]. Intracellular ROS production and lipid peroxidation could be reduced through the ability of PRAM to increase endogenous antioxidant enzymes as shown in in vivo work. An additional intriguing possibility, based on work in pancreatic cell lines [36], is that PRAM may itself become an activated antioxidant after forming complexes with Cu (II). This would also explain the decreases in ROS and, to a greater extent, lipid peroxidation. These latter aspects should be explored in future studies.
Two aspects that remain unanswered by this work but that should form the basis of future studies are the ability of amylin to regulate mitochondrial health and the specific mechanism of action of amylin. In the present study, we found increases in GPx, known to protect mitochondria [82] and MnSOD (located within the mitochondria [83]) in the hippocampi of PRAM treated mice. Mitochondrial dysfunction precedes the increase in ROS and oxidative damage [84]. Thus, a possibility to explore is that PRAM may act directly on the mitochondria health to yield its benefits. In support of this hypothesis, recent work demonstrates that PRAM treatment reduced intracellular of calcium release, rescued mitochondrial membrane potential loss, inhibited the mitochondrial mediated apoptosis pathways, and increased ATP production under both hypoxic and normoxic conditions [50]. Additionally, it is unclear whether PRAM treatment in our in vivo study is providing benefits by directly through one or a number of its native receptors or whether its effects are driven through its ability to regulate insulin sensitivity peripherally and improve general metabolic tone (which also could reduce pathology and ROS) and should also be clarified. This is particularly relevant given the conflicting data showing that both inhibition of the receptor [21–25] and its activation through amylin or PRAM [29, 33] produce benefits in AD models. A recent study showing that while Aβ and human amylin disrupt LTP, pre-treatment with PRAM can rescue this decrease to wild-type levels in a mouse model of AD [85].